

Analysis of Secondary Metabolite Synthesis Potential of Streptomyces fradiae sf106 Based on the Whole Genome and Non-Target Metabolomics and Exploration of the Biosynthesis of Tylosin
 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Strain and Culture Conditions
2.2. Genomic DNA Extraction
2.3. Library Construction and Genome Sequencing
2.4. Genome Assembly and Annotation
2.5. Metabolite Extraction
2.6. (UHPLC–MS/MS) Analysis
2.7. Data Preprocessing and Annotation
3. Results
3.1. Systematic Evolution Analysis of Streptomyces fradiae sf106
3.2. The Genomic Overview and Annotation of Streptomyces fradiae sf106
3.3. Analysis of Mobile Genetic Elements
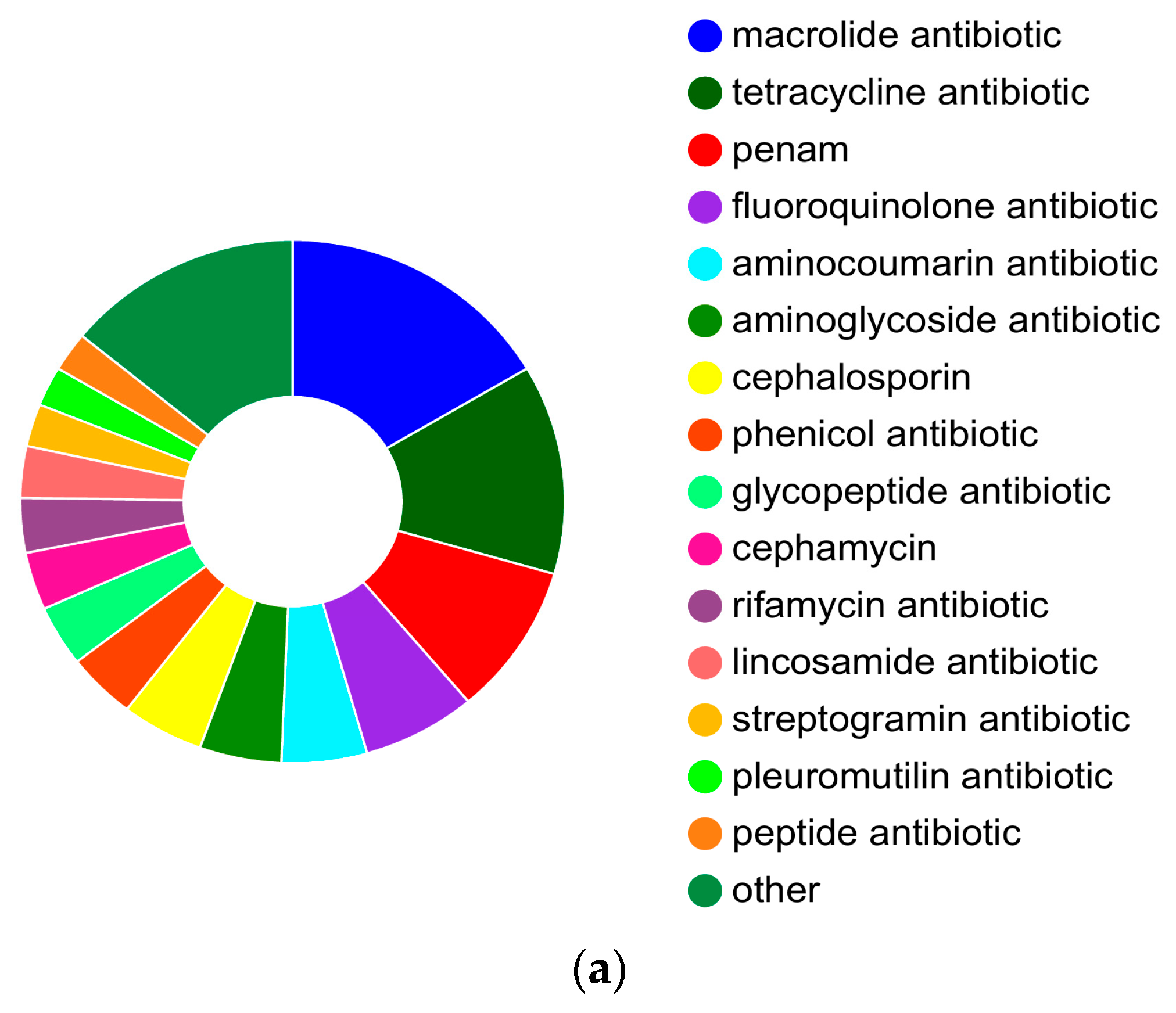
3.4. Prediction of Antibiotic Resistance Genes
3.5. Metabolic System Analysis
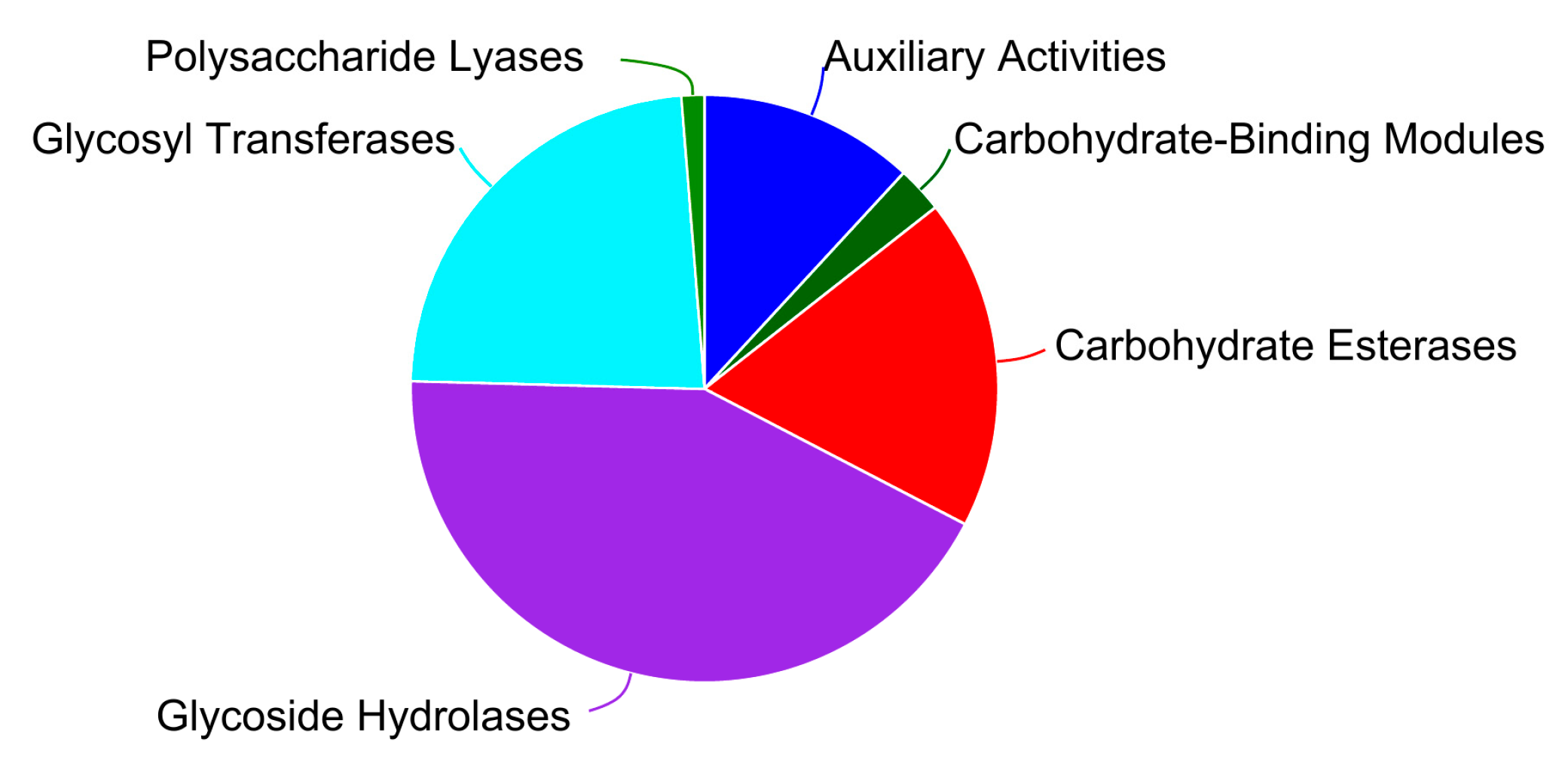
3.5.1. Carbohydrate-Active Enzyme Annotation
3.5.2. Secondary Metabolite Analysis
3.6. Preprocessing of Metabolite Expression Data
3.7. Metabolites KEGG Compounds Classification
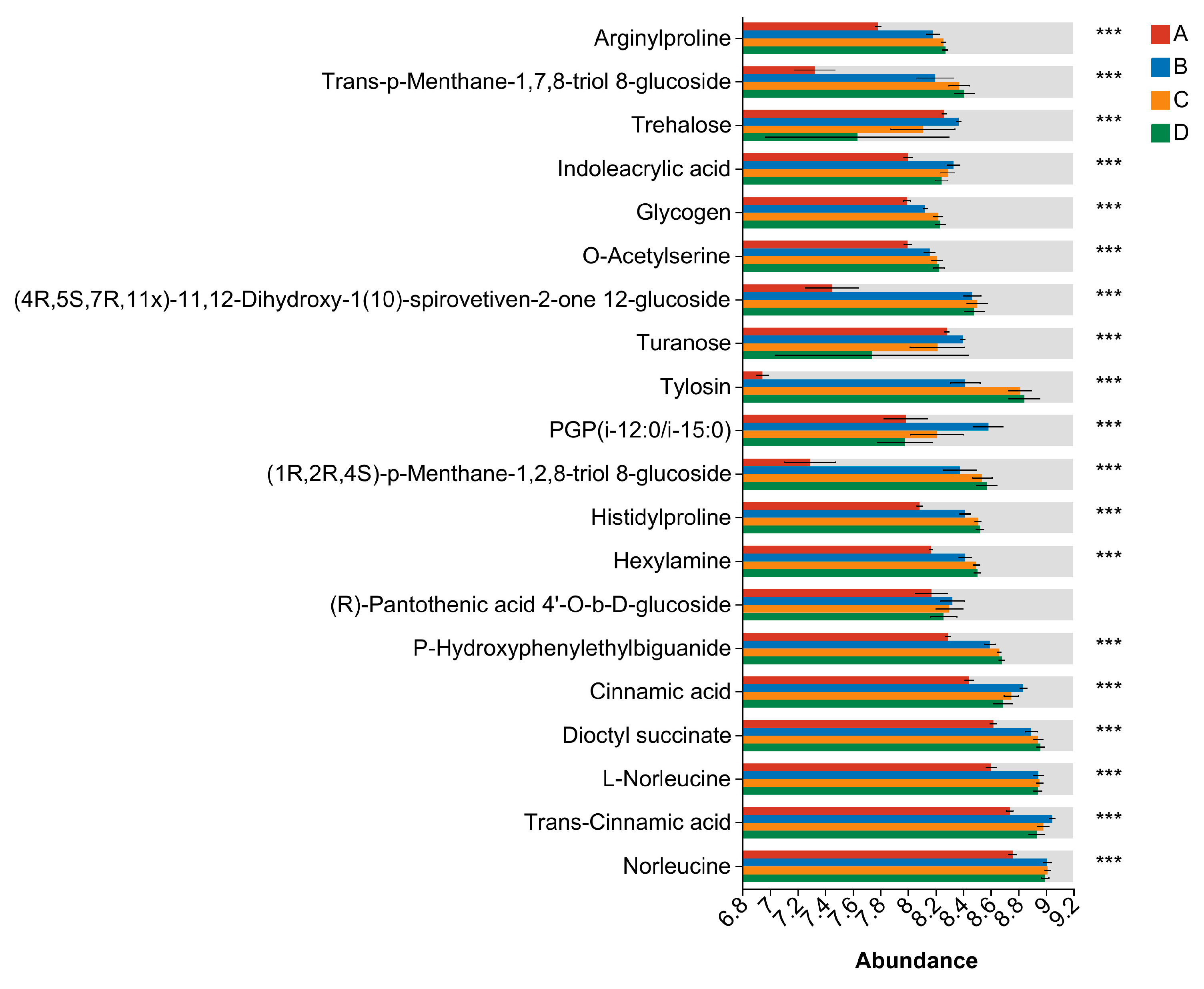
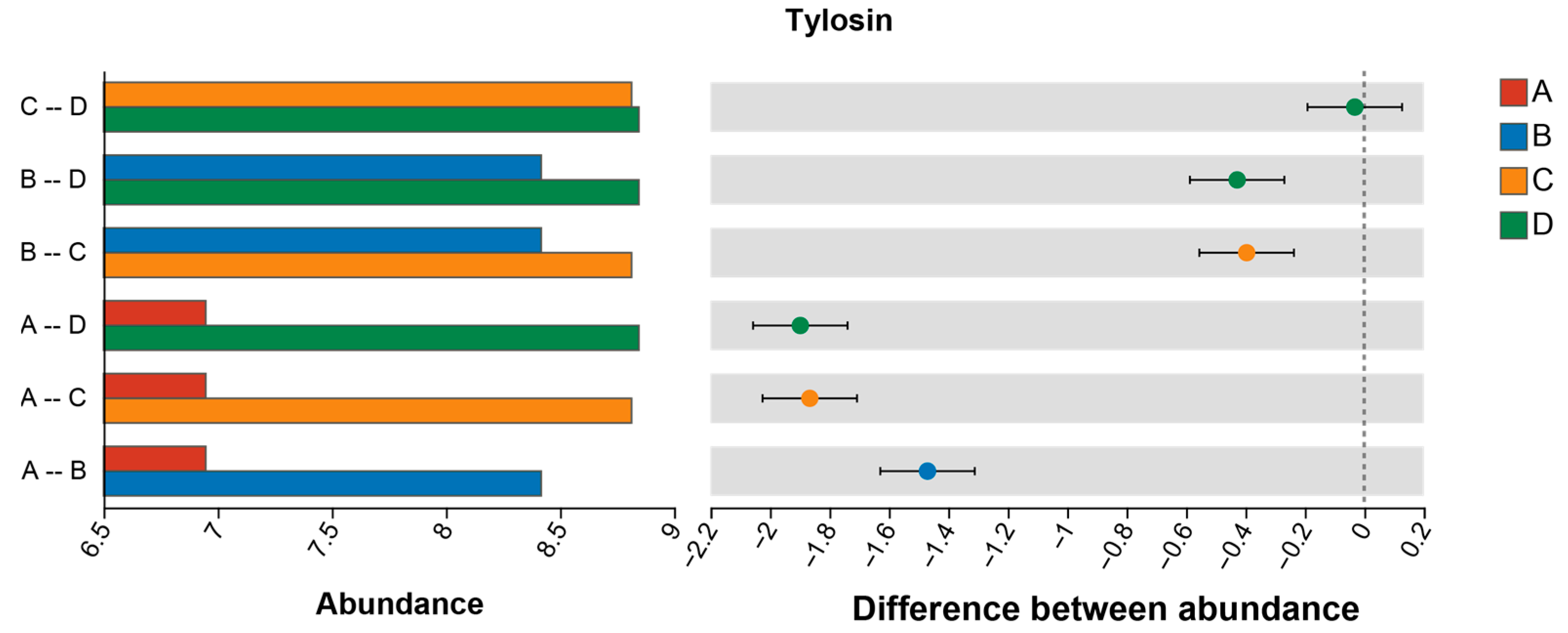
3.8. Differential Metabolite Analysis
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Zhang, H.; Wang, Y.; Pfeifer, B.A. Bacterial hosts for natural product production. Mol. Pharm. 2008, 5, 212–225. [Google Scholar] [CrossRef] [PubMed]
- Aigle, B.; Lautru, S.; Spiteller, D.; Dickschat, J.S.; Challis, G.L.; Leblond, P.; Pernodet, J.L. Genome mining of Streptomyces ambofaciens. J. Ind. Microbiol. Biotechnol. 2014, 41, 251–263. [Google Scholar] [CrossRef]
- Alam, K.; Mazumder, A.; Sikdar, S.; Zhao, Y.M.; Hao, J.; Song, C.; Wang, Y.; Sarkar, R.; Islam, S.; Zhang, Y.; et al. Streptomyces: The biofactory of secondary metabolites. Front. Microbiol. 2022, 13, 968053. [Google Scholar] [CrossRef] [PubMed]
- Weber, J.M.; Wierman, C.K.; Hutchinson, C.R. Genetic analysis of erythromycin production in Streptomyces erythreus. J. Bacteriol. 1985, 164, 425–433. [Google Scholar] [CrossRef]
- Yin, S.; Wang, W.; Wang, X.; Zhu, Y.; Jia, X.; Li, S.; Yuan, F.; Zhang, Y.; Yang, K. Identification of a cluster-situated activator of oxytetracycline biosynthesis and manipulation of its expression for improved oxytetracycline production in Streptomyces rimosus. Microb. Cell Factories 2015, 14, 46. [Google Scholar] [CrossRef] [PubMed]
- Paulus, C.; Zapp, J.; Luzhetskyy, A. New Scabimycins A-C Isolated from Streptomyces acidiscabies (Lu19992). Molecules 2021, 26, 5922. [Google Scholar] [CrossRef]
- Charousová, I.; Medo, J.; Hleba, L.; Javoreková, S. Streptomyces globosus DK15 and Streptomyces ederensis ST13 as new producers of factumycin and tetrangomycin antibiotics. Braz. J. Microbiol. 2018, 49, 816–822. [Google Scholar] [CrossRef]
- Chakraborty, P. Gene cluster from plant to microbes: Their role in genome architecture, organism’s development, specialized metabolism and drug discovery. Biochimie 2022, 193, 1–15. [Google Scholar] [CrossRef]
- Zhao, J.; Grant, S.F. Advances in whole genome sequencing technology. Curr. Pharm. Biotechnol. 2011, 12, 293–305. [Google Scholar] [CrossRef]
- Baltz, R.H. Natural product drug discovery in the genomic era: Realities, conjectures, misconceptions, and opportunities. J. Ind. Microbiol. Biotechnol. 2019, 46, 281–299. [Google Scholar] [CrossRef]
- Daubin, V.; Szöllősi, G.J. Horizontal Gene Transfer and the History of Life. Cold Spring Harb. Perspect. Biol. 2016, 8, a018036. [Google Scholar] [CrossRef] [PubMed]
- Bertelli, C.; Laird, M.R.; Williams, K.P.; Lau, B.Y.; Hoad, G.; Winsor, G.L.; Brinkman, F.S.L. IslandViewer 4: Expanded prediction of genomic islands for larger-scale datasets. Nucleic Acids Res. 2017, 45, W30–W35. [Google Scholar] [CrossRef] [PubMed]
- Fouts, D.E. Phage_Finder: Automated identification and classification of prophage regions in complete bacterial genome sequences. Nucleic Acids Res. 2006, 34, 5839–5851. [Google Scholar] [CrossRef] [PubMed]
- Bland, C.; Ramsey, T.L.; Sabree, F.; Lowe, M.; Brown, K.; Kyrpides, N.C.; Hugenholtz, P. CRISPR recognition tool (CRT): A tool for automatic detection of clustered regularly interspaced palindromic repeats. BMC Bioinform. 2007, 8, 209. [Google Scholar] [CrossRef] [PubMed]
- Siguier, P.; Gourbeyre, E.; Chandler, M. Bacterial insertion sequences: Their genomic impact and diversity. FEMS Microbiol. Rev. 2014, 38, 865–891. [Google Scholar] [CrossRef]
- Zhao, L.; Beyer, N.J.; Borisova, S.A.; Liu, H.W. Beta-glucosylation as a part of self-resistance mechanism in methymycin/pikromycin producing strain Streptomyces venezuelae. Biochemistry 2003, 42, 14794–14804. [Google Scholar] [CrossRef]
- Kang, H.S.; Kim, E.S. Recent advances in heterologous expression of natural product biosynthetic gene clusters in Streptomyces hosts. Curr. Opin. Biotechnol. 2021, 69, 118–127. [Google Scholar] [CrossRef]
- Shen, B.; Du, L.; Sanchez, C.; Edwards, D.J.; Chen, M.; Murrell, J.M. Cloning and characterization of the bleomycin biosynthetic gene cluster from Streptomyces verticillus ATCC15003. J. Nat. Prod. 2002, 65, 422–431. [Google Scholar] [CrossRef]
- Gomez-Escribano, J.P.; Bibb, M.J. Engineering Streptomyces coelicolor for heterologous expression of secondary metabolite gene clusters. Microb. Biotechnol. 2011, 4, 207–215. [Google Scholar] [CrossRef]
- Peng, Q.; Gao, G.; Lü, J.; Long, Q.; Chen, X.; Zhang, F.; Xu, M.; Liu, K.; Wang, Y.; Deng, Z.; et al. Engineered Streptomyces lividans Strains for Optimal Identification and Expression of Cryptic Biosynthetic Gene Clusters. Front. Microbiol. 2018, 9, 3042. [Google Scholar] [CrossRef]
- Yin, S.; Li, Z.; Wang, X.; Wang, H.; Jia, X.; Ai, G.; Bai, Z.; Shi, M.; Yuan, F.; Liu, T.; et al. Heterologous expression of oxytetracycline biosynthetic gene cluster in Streptomyces venezuelae WVR2006 to improve production level and to alter fermentation process. Appl. Microbiol. Biotechnol. 2016, 100, 10563–10572. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Deng, Z.; Liu, T. Streptomyces species: Ideal chassis for natural product discovery and overproduction. Metab. Eng. 2018, 50, 74–84. [Google Scholar] [CrossRef] [PubMed]
- Tao, W.; Yang, A.; Deng, Z.; Sun, Y. CRISPR/Cas9-Based Editing of Streptomyces for Discovery, Characterization, and Production of Natural Products. Front. Microbiol. 2018, 9, 1660. [Google Scholar] [CrossRef]
- Makitrynskyy, R.; Tsypik, O.; Bechthold, A. Genetic Engineering of Streptomyces ghanaensis ATCC14672 for Improved Production of Moenomycins. Microorganisms 2021, 10, 30. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Ye, R.; Zhang, H.; Liu, Y. Amplification of lmbB1 gene in Streptomyces lincolnensis improves quantity and quality of lincomycin A fermentation. Prep. Biochem. Biotechnol. 2020, 50, 529–537. [Google Scholar] [CrossRef]
- Laureti, L.; Song, L.; Huang, S.; Corre, C.; Leblond, P.; Challis, G.L.; Aigle, B. Identification of a bioactive 51-membered macrolide complex by activation of a silent polyketide synthase in Streptomyces ambofaciens. Proc. Natl. Acad. Sci. USA 2011, 108, 6258–6263. [Google Scholar] [CrossRef]
- An, J.S.; Shin, B.; Kim, T.H.; Hwang, S.; Shin, Y.H.; Cui, J.; Du, Y.E.; Yi, J.; Nam, S.J.; Hong, S.; et al. Dumulmycin, an Antitubercular Bicyclic Macrolide from a Riverine Sediment-Derived Streptomyces sp. Org. Lett. 2021, 23, 3359–3363. [Google Scholar] [CrossRef]
- Marchbank, D.H.; Ptycia-Lamky, V.C.; Decken, A.; Haltli, B.A.; Kerr, R.G. Guanahanolide A, a Meroterpenoid with a Sesterterpene Skeleton from Coral-Derived Streptomyces sp. Org. Lett. 2020, 22, 6399–6403. [Google Scholar] [CrossRef]
- Sun, J.; Zhao, G.; O’Connor, R.D.; Davison, J.R.; Bewley, C.A. Vertirhodins A-F, C-Linked Pyrrolidine-Iminosugar-Containing Pyranonaphthoquinones from Streptomyces sp. B15-008. Org. Lett. 2021, 23, 682–686. [Google Scholar] [CrossRef]
- Zhang, S.Y.; Zhan, Z.J.; Zhang, H.; Qi, H.; Zhang, L.Q.; Chen, S.X.; Gan, L.S.; Wang, J.D.; Ma, L.F. Morindolestatin, Naturally Occurring Dehydromorpholinocarbazole Alkaloid from Soil-Derived Bacterium of the Genus Streptomyces. Org. Lett. 2020, 22, 1113–1116. [Google Scholar] [CrossRef]
- Yang, Q.J.; Zhao, J.R.; Hao, J.; Li, B.; Huo, Y.; Han, Y.L.; Wan, L.L.; Li, J.; Huang, J.; Lu, J.; et al. Serum and urine metabolomics study reveals a distinct diagnostic model for cancer cachexia. J. Cachexia Sarcopenia Muscle 2018, 9, 71–85. [Google Scholar] [CrossRef] [PubMed]
- Alves, P.C.; Hartmann, D.O.; Núñez, O.; Martins, I.; Gomes, T.L.; Garcia, H.; Galceran, M.T.; Hampson, R.; Becker, J.D.; Silva Pereira, C. Transcriptomic and metabolomic profiling of ionic liquid stimuli unveils enhanced secondary metabolism in Aspergillus nidulans. BMC Genom. 2016, 17, 284. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Lv, J.; Yu, Z.; Wang, Z.; Li, Y.; Li, M.; Deng, Z.; Xu, Q.; Cui, F.; Zhou, W. Integrated metabolomics and transcriptomic analysis of the flavonoid regulatory networks in Sorghum bicolor seeds. BMC Genom. 2022, 23, 619. [Google Scholar] [CrossRef] [PubMed]
- Gandecha, A.R.; Large, S.L.; Cundliffe, E. Analysis of four tylosin biosynthetic genes from the tylLM region of the Streptomyces fradiae genome. Gene 1997, 184, 197–203. [Google Scholar] [CrossRef]
- Merson-Davies, L.A.; Cundliffe, E. Analysis of five tylosin biosynthetic genes from the tyllBA region of the Streptomyces fradiae genome. Mol. Microbiol. 1994, 13, 349–355. [Google Scholar] [CrossRef] [PubMed]
- Baltz, R.H.; Seno, E.T. Genetics of Streptomyces fradiae and tylosin biosynthesis. Annu. Rev. Microbiol. 1988, 42, 547–574. [Google Scholar] [CrossRef]
- Baltz, R.H.; Seno, E.T. Properties of Streptomyces fradiae mutants blocked in biosynthesis of the macrolide antibiotic tylosin. Antimicrob. Agents Chemother. 1981, 20, 214–225. [Google Scholar] [CrossRef]
- Khaliq, S.; Rashid, N.; Akhtar, K.; Ghauri, M.A. Production of tylosin in solid-state fermentation by Streptomyces fradiae NRRL-2702 and its gamma-irradiated mutant (gamma-1). Lett. Appl. Microbiol. 2009, 49, 635–640. [Google Scholar] [CrossRef]
- Fan, J.; Yao, Z.; Yan, C.; Hao, M.; Dai, J.; Zou, W.; Ni, M.; Li, T.; Li, L.; Li, S.; et al. Discovery of a highly efficient TylF methyltransferase via random mutagenesis for improving tylosin production. Comput. Struct. Biotechnol. J. 2023, 21, 2759–2766. [Google Scholar] [CrossRef]
- Dai, P.; Qin, Y.; Li, L.; Li, H.; Lv, L.; Xu, D.; Song, Y.; Huang, T.; Lin, S.; Deng, Z.; et al. Enhancing tylosin production by combinatorial overexpression of efflux, SAM biosynthesis, and regulatory genes in hyperproducing Streptomyces xinghaiensis strain. Synth. Syst. Biotechnol. 2023, 8, 486–497. [Google Scholar] [CrossRef]
- Yao, Z.; Fan, J.; Dai, J.; Yu, C.; Zeng, H.; Li, Q.; Hu, W.; Yan, C.; Hao, M.; Li, H.; et al. A High-Throughput Method Based on Microculture Technology for Screening of High-Yield Strains of Tylosin-Producing Streptomyces fradiae. J. Microbiol. Biotechnol. 2023, 33, 831–839. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Li, D.; Wang, K.; Wei, B.; Zhang, J.; Li, J. An efficient method for targeted cloning of large DNA fragments from Streptomyces. Appl. Microbiol. Biotechnol. 2023, 107, 5749–5760. [Google Scholar] [CrossRef]
- Delcher, A.L.; Bratke, K.A.; Powers, E.C.; Salzberg, S.L. Identifying bacterial genes and endosymbiont DNA with Glimmer. Bioinformatics 2007, 23, 673–679. [Google Scholar] [CrossRef] [PubMed]
- Borodovsky, M.; Mcininch, J. Genmark: Parallel gene recognition for both DNA strands. Comput. Chem. 1993, 17, 123–133. [Google Scholar] [CrossRef]
- Wishart, D.S.; Feunang, Y.D.; Marcu, A.; Guo, A.C.; Liang, K.; Vázquez-Fresno, R.; Sajed, T.; Johnson, D.; Li, C.; Karu, N.; et al. HMDB 4.0: The human metabolome database for 2018. Nucleic Acids Res. 2018, 46, D608–D617. [Google Scholar] [CrossRef]
- Montenegro-Burke, J.R.; Guijas, C.; Siuzdak, G. METLIN: A Tandem Mass Spectral Library of Standards. Methods Mol. Biol. 2020, 2104, 149–163. [Google Scholar] [CrossRef]
- Vatlin, A.A.; Bekker, O.B.; Lysenkova, L.N.; Shchekotikhin, A.E.; Danilenko, V.N. Bioinformatics analysis of genes of Streptomyces xinghaiensis (fradiae) ATCC 19609 with a focus on mutations conferring resistance to oligomycin A and its derivatives. J. Glob. Antimicrob. Resist. 2020, 22, 47–53. [Google Scholar] [CrossRef]
- Bekker, O.B.; Vatlin, A.A.; Zakharevich, N.V.; Lysenkova, L.N.; Shchekotikhin, A.E.; Danilenko, V.N. Draft Genome Sequence of Streptomyces xinghaiensis (fradiae) OlgR, a Strain Resistant to Oligomycin A. Microbiol. Resour. Announc. 2019, 8, e01531-18. [Google Scholar] [CrossRef]
- Krzywinski, M.; Schein, J.; Birol, I.; Connors, J.; Gascoyne, R.; Horsman, D.; Jones, S.J.; Marra, M.A. Circos: An information aesthetic for comparative genomics. Genome Res. 2009, 19, 1639–1645. [Google Scholar] [CrossRef]
- Alam, K.; Hao, J.; Zhong, L.; Fan, G.; Ouyang, Q.; Islam, M.M.; Islam, S.; Sun, H.; Zhang, Y.; Li, R.; et al. Complete genome sequencing and in silico genome mining reveal the promising metabolic potential in Streptomyces strain CS-7. Front. Microbiol. 2022, 13, 939919. [Google Scholar] [CrossRef]
- Chen, Y.Y.; Chen, L.Y.; Chen, P.J.; El-Shazly, M.; Peng, B.R.; Chen, Y.C.; Su, C.H.; Su, J.H.; Sung, P.J.; Yen, P.T.; et al. Probing Anti-Leukemic Metabolites from Marine-Derived Streptomyces sp. LY1209. Metabolites 2022, 12, 320. [Google Scholar] [CrossRef] [PubMed]
- Williams, S.E.; Back, C.R.; Best, E.; Mantell, J.; Stach, J.E.M.; Williams, T.A.; Race, P.R.; Curnow, P. Discovery and biosynthetic assessment of ‘Streptomyces ortus’ sp. nov. isolated from a deep-sea sponge. Microb. Genom. 2023, 9, mgen000996. [Google Scholar] [CrossRef] [PubMed]
- Van Lanen, S.G.; Shen, B. Microbial genomics for the improvement of natural product discovery. Curr. Opin. Microbiol. 2006, 9, 252–260. [Google Scholar] [CrossRef] [PubMed]
- Tsunematsu, Y. Genomics-directed activation of cryptic natural product pathways deciphers codes for biosynthesis and molecular function. J. Nat. Med. 2021, 75, 261–274. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.; Zhang, W.; Li, Z.; Li, H.; Geng, C.; Huang, X.; Lu, X. Discovery and Characterization of a PKS-NRPS Hybrid in Aspergillus terreus by Genome Mining. J. Nat. Prod. 2020, 83, 473–480. [Google Scholar] [CrossRef]
- Hashimoto, T.; Hashimoto, J.; Kagaya, N.; Nishimura, T.; Suenaga, H.; Nishiyama, M.; Kuzuyama, T.; Shin-Ya, K. A novel oxazole-containing tetraene compound, JBIR-159, produced by heterologous expression of the cryptic trans-AT type polyketide synthase biosynthetic gene cluster. J. Antibiot. 2021, 74, 354–358. [Google Scholar] [CrossRef]
- Zhang, Y.; Yao, T.; Jiang, Y.; Li, H.; Yuan, W.; Li, W. Deciphering a Cyclodipeptide Synthase Pathway Encoding Prenylated Indole Alkaloids in Streptomyces leeuwenhoekii. Appl. Environ. Microbiol. 2021, 87, e02525-20. [Google Scholar] [CrossRef]
- Craney, A.; Ozimok, C.; Pimentel-Elardo, S.M.; Capretta, A.; Nodwell, J.R. Chemical perturbation of secondary metabolism demonstrates important links to primary metabolism. Chem. Biol. 2012, 19, 1020–1027. [Google Scholar] [CrossRef]
- Aharoni, A.; Galili, G. Metabolic engineering of the plant primary-secondary metabolism interface. Curr. Opin. Biotechnol. 2011, 22, 239–244. [Google Scholar] [CrossRef]
- de Sousa, L.P. Mobile Genetic Elements in Pseudomonas stutzeri. Curr. Microbiol. 2020, 77, 179–184. [Google Scholar] [CrossRef]
- Blin, K.; Shaw, S.; Kloosterman, A.M.; Charlop-Powers, Z.; van Wezel, G.P.; Medema, M.H.; Weber, T. AntiSMASH 6.0: Improving cluster detection and comparison capabilities. Nucleic Acids Res. 2021, 49, W29–W35. [Google Scholar] [CrossRef] [PubMed]
- Ziemert, N.; Alanjary, M.; Weber, T. The evolution of genome mining in microbes—A review. Nat. Prod. Rep. 2016, 33, 988–1005. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Genome Size (kb) | GC Content (%) | CDS No. | tRNA No | rRNA No. | KEGG | GO | COG | NR | Pfam | Swiss-Prot |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Streptomyces fradiae sf106 | 7351 | 73 | 6750 | 60 | 20 | 2300 | 3500 | 4950 | 6100 | 4860 | 3920 |
| Sample Name | GI No. | Is No. | Prophage No. | CRISPR-Cas No. |
|---|---|---|---|---|
| Streptomyces fradiae sf106 | 12 | 5 | 1 | 13 |
| Region | Type | From | To | Most Similar Known Cluster | Similarity | |
|---|---|---|---|---|---|---|
| Region 1 | redox-cofactor | 11,912 | 34,657 | Alanylclavam/2-hydroxymethylclavam/2-fomyloxymethyclavam/clavam-2-carboxylate | Other: Non-NRP beta- lactam | 12% |
| Region 2 | T3PKS, terpene | 367,053 | 406,828 | endophenazine A/endophenazine B | Other: Phenazine | 38% |
| Region 3 | NRPS | 670,555 | 724,534 | scabichelin | NRP | 20% |
| Region 4 | NRPS-like, PKS-like, betalactone, T2PKS, RRE- containing | 1,232,101 | 1,317,375 | LL-D49194α1 (LLD) | Polyketide | 39% |
| Region 5 | lanthipeptide-class-iii | 1,980,221 | 2,002,953 | SapB | RiPP: Lanthipeptide | 100% |
| Region 6 | siderophore | 2,354,686 | 2,365,381 | |||
| Region 7 | ectoine | 2,484,580 | 2,494,984 | ectoine | Other | 100% |
| Region 8 | T3PKS | 3,482,933 | 3,524,030 | flaviolin | Other | 50% |
| Region 9 | T1PKS | 3,545,272 | 3,626,176 | carrimycin | Polyketide | 55% |
| Region 10 | NRPS-like, lassopeptide | 3,657,076 | 3,699,170 | citrulassin B | RiPP | 100% |
| Region 11 | NRPS | 4,128,379 | 4,201,561 | isocomplestatin | NRP | 93% |
| Region 12 | terpene | 4,360,731 | 4,379,029 | |||
| Region 13 | RiPP-like, NRPS-like | 4,422,841 | 4,466,244 | nunapeptin/nunamycin | NRP | 14% |
| Region 14 | lanthipeptide-class-i | 4,599,449 | 4,622,053 | |||
| Region 15 | terpene | 4,728,801 | 4,752,252 | Terpene | 53% | |
| Region 16 | terpene | 4,832,715 | 4,849,274 | |||
| Region 17 | RRE-containing, LAP, thiopeptide | 4,936,167 | 4,971,075 | |||
| Region 18 | siderophore | 5,095,314 | 5,106,980 | desferrioxamine | Other | 83% |
| Region 19 | RiPP-like | 5,186,566 | 5,198,233 | |||
| Region 20 | siderophore | 5,473,885 | 5,488,679 | ficellomycin | NRP | 3% |
| Region 21 | lanthipeptide- class-i | 5,836,957 | 5,862,133 | tetrocarcin A | Polyketide | 4% |
| Region 22 | terpene | 7,023,507 | 7,049,988 | isorenieratene | Terpene | 87% |
| Reference | Similarity Score | Type | Compound(s) | Organism | |
|---|---|---|---|---|---|
| Cluster1 | BGC0001156.1 | 0.15 | Terpene | platencin | Streptomyces platensis |
| Cluster2 | BGC0000674.1 | 0.71 | Terpene | (-)-δ-cadinene | Streptomyces clavuligerus ATCC 27064 |
| Cluster3 | BGC0000426.1 | 0.42 | NRP | sevadicin | Paenibacillus larvae |
| Cluster4 | BGC0000248.1 | 1.65 | Polyketide | naphtocyclinone | Streptomyces arenae |
| Cluster5 | BGC0000551.1 | 0.47 | RiPP | SapB | Streptomyces coelicolor A3(2) |
| Cluster6 | BGC0001572.1 | 0.34 | Other | desferrioxamine E | Pantoea agglomerans |
| Cluster7 | BGC0000853.1 | 0.64 | Other | ectoine | Streptomyces anulatus |
| Cluster8 | BGC0001310.1 | 0.46 | Terpene | naringenin | Streptomyces clavuligerus ATCC 27064 |
| Cluster9 | BGC0000166.1 | 0.78 | Polyketide | tylactone | Streptomyces fradiae |
| Cluster10 | BGC0001552.1 | 1.16 | RiPP | citrulassin F | Streptomyces avermitilis MA-4680 = NBRC 14893 |
| Cluster11 | BGC0000326.1 | 0.41 | NRP | isocomplestatin | Streptomyces lavendulae |
| Cluster12 | BGC0001910.1 | 0.28 | Terpene | cyslabdan | Streptomyces cyslabdanicus |
| Cluster13 | BGC0000500.1 | 0.41 | RiPP | carnolysin A1, carnolysin A2 | Carnobacterium maltaromaticum |
| Cluster14 | BGC0000544.1 | 0.19 | RiPP | planosporicin | Planomonospora alba |
| Cluster15 | BGC0000648.1 | 0.34 | Terpene | carotenoid | Myxococcus xanthus |
| Cluster16 | BGC0001910.1 | 0.34 | Terpene | cyslabdan | Streptomyces cyslabdanicus |
| Cluster17 | BGC0001146.1 | 1.02 | RiPP | cyclothiazomycin C | Streptomyces sp. NRRL WC-3908 |
| Cluster18 | BGC0001478.1 | 0.58 | Other | desferrioxamine E | Streptomyces sp. ID38640 |
| Cluster19 | No matches found | ||||
| Cluster 20 | BGC0001531.1 | 0.26 | Other | bisucaberin B | Tenacibaculum mesophilum |
| Cluster21 | BGC0001727.1 | 0.23 | RiPP | paenilan | Paenibacillus polymyxa E681 |
| Cluster22 | BGC0000664.1 | 0.42 | Terpene | isorenieratene | Streptomyces griseus subsp. griseus NBRC 13350 |
| Ion Mode | All Peaks | Identified Metabolites | Metabolites in Library | Metabolites in KEGG |
|---|---|---|---|---|
| pos | 6480 | 970 | 697 | 229 |
| neg | 8049 | 885 | 790 | 238 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jia, C.; Ma, X.; Jiang, Y.; Cheng, S.; Yue, S.; Su, J. Analysis of Secondary Metabolite Synthesis Potential of Streptomyces fradiae sf106 Based on the Whole Genome and Non-Target Metabolomics and Exploration of the Biosynthesis of Tylosin. Fermentation 2023, 9, 866. https://doi.org/10.3390/fermentation9100866
Jia C, Ma X, Jiang Y, Cheng S, Yue S, Su J. Analysis of Secondary Metabolite Synthesis Potential of Streptomyces fradiae sf106 Based on the Whole Genome and Non-Target Metabolomics and Exploration of the Biosynthesis of Tylosin. Fermentation. 2023; 9(10):866. https://doi.org/10.3390/fermentation9100866
Chicago/Turabian StyleJia, Chenbo, Xian Ma, Yuting Jiang, Shanshan Cheng, Sijun Yue, and Jianyu Su. 2023. "Analysis of Secondary Metabolite Synthesis Potential of Streptomyces fradiae sf106 Based on the Whole Genome and Non-Target Metabolomics and Exploration of the Biosynthesis of Tylosin" Fermentation 9, no. 10: 866. https://doi.org/10.3390/fermentation9100866