
Based on the Co-Evolution of lncRNAs-Microbiota and Metabolites in Rumen Epithelium to Analyze the Adaptation Characteristics of Tibetan Sheep to Nutrient Stress in the Cold Season

 , , , , , and
, , , , , and
Abstract
:1. Introduction
2. Methods
2.1. Experimental Design and Sample Collection
2.2. Observation of Rumen Epithelial Tissue Morphology and Determination of VFAs, Microbiota, and Metabolites
2.3. Construction of lncRNAs Libraries
2.4. Prediction of LncRNAs
2.5. Identification of DE lncRNAs
2.6. Functional Enrichment of DE lncRNAs
2.7. Prediction of LncRNAs Target Genes
2.8. Construction of lncRNA-mRNA Targeting Network
2.9. RT-qPCR Analysis
2.10. Data Analysis
3. Results
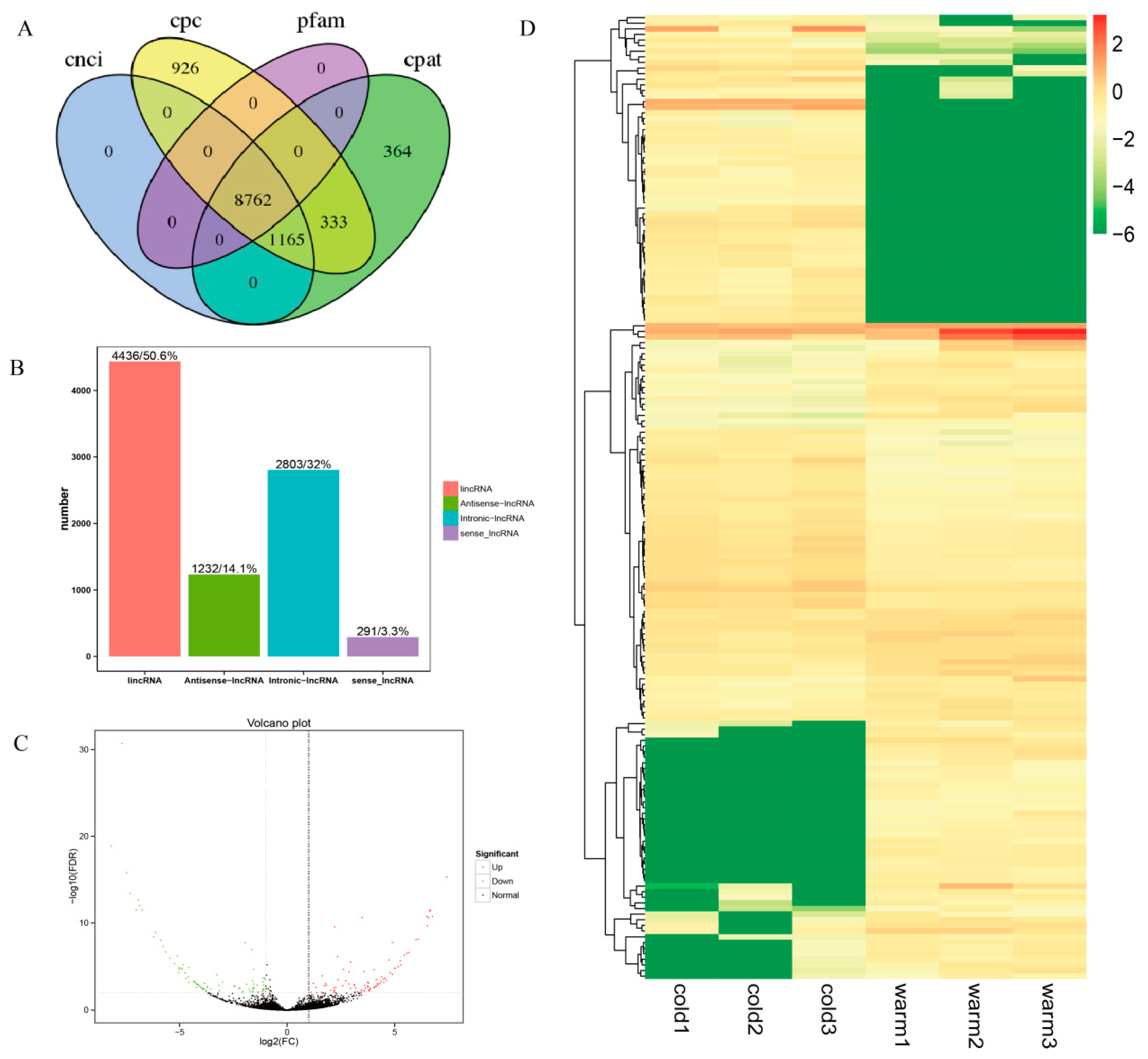
3.1. Overview of Sequencing Data of lncRNAs in Tibetan Sheep Rumen Epithelium Tissue
3.2. Identification and DE lncRNAs in Tibetan Sheep Rumen Epithelium
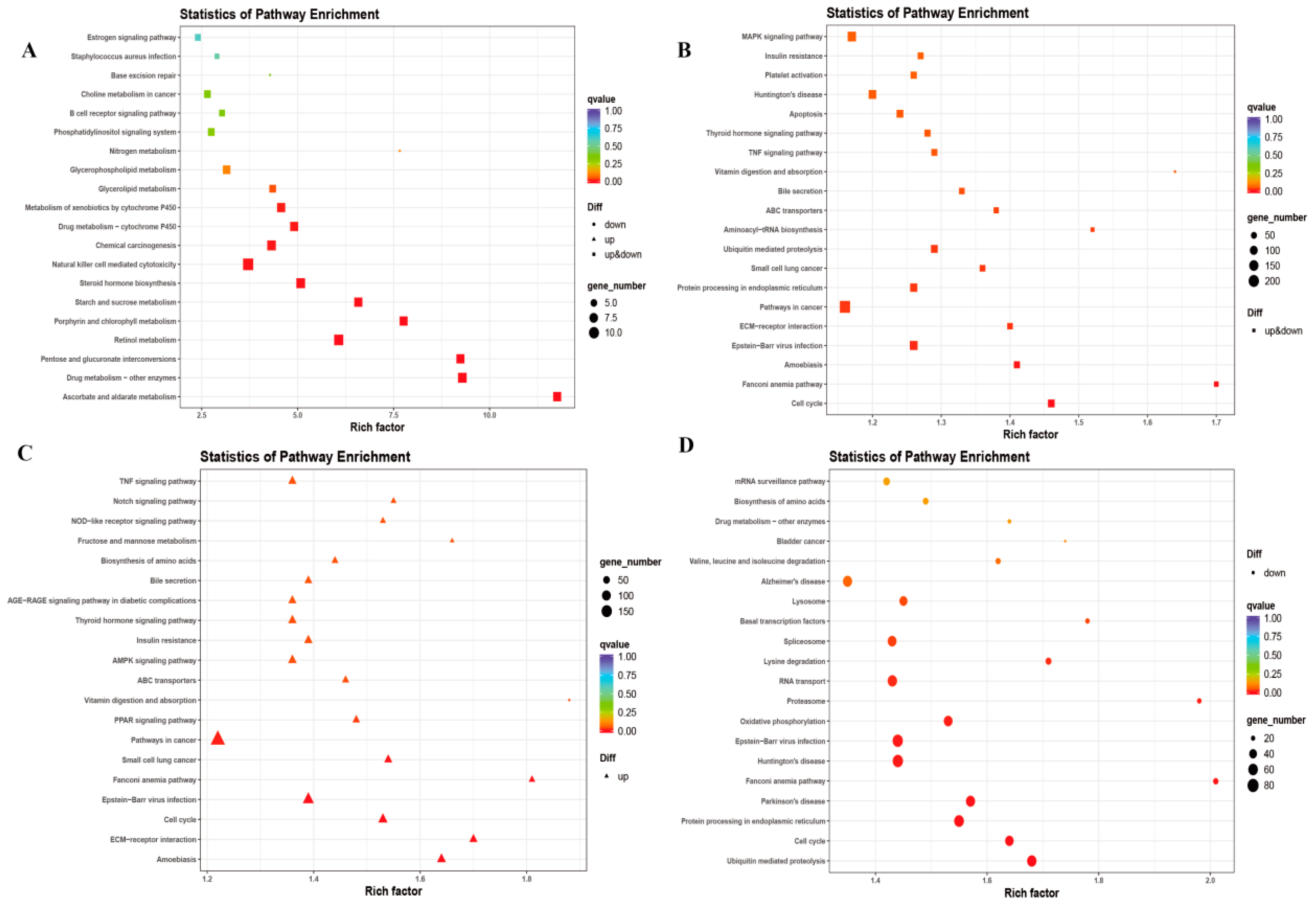
3.3. Functional Characteristics of DE lncRNAs
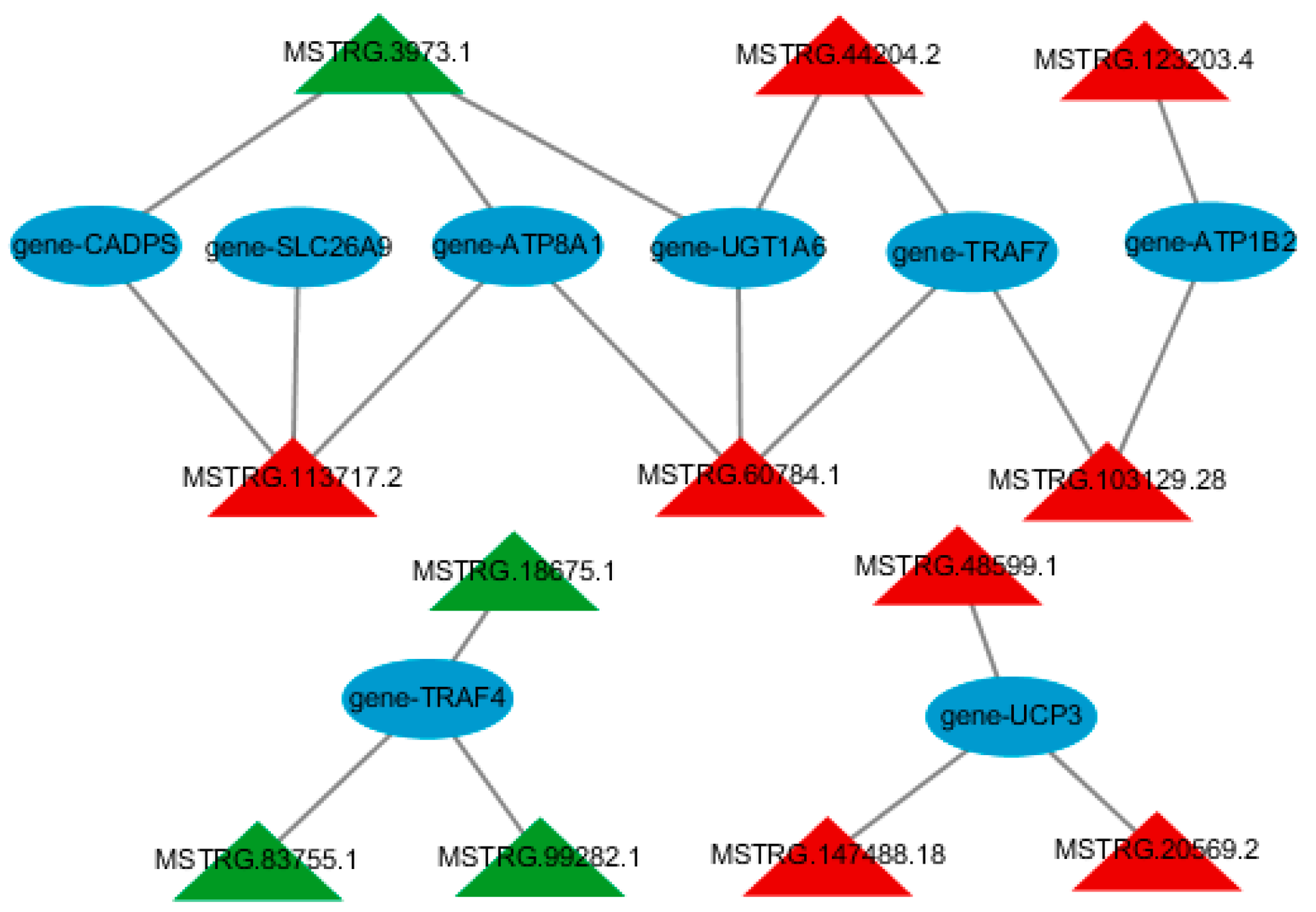
3.4. Construction of lncRNA-mRNA Regulatory Network
3.5. The DE lncRNA Was Verified by RT-qPCR
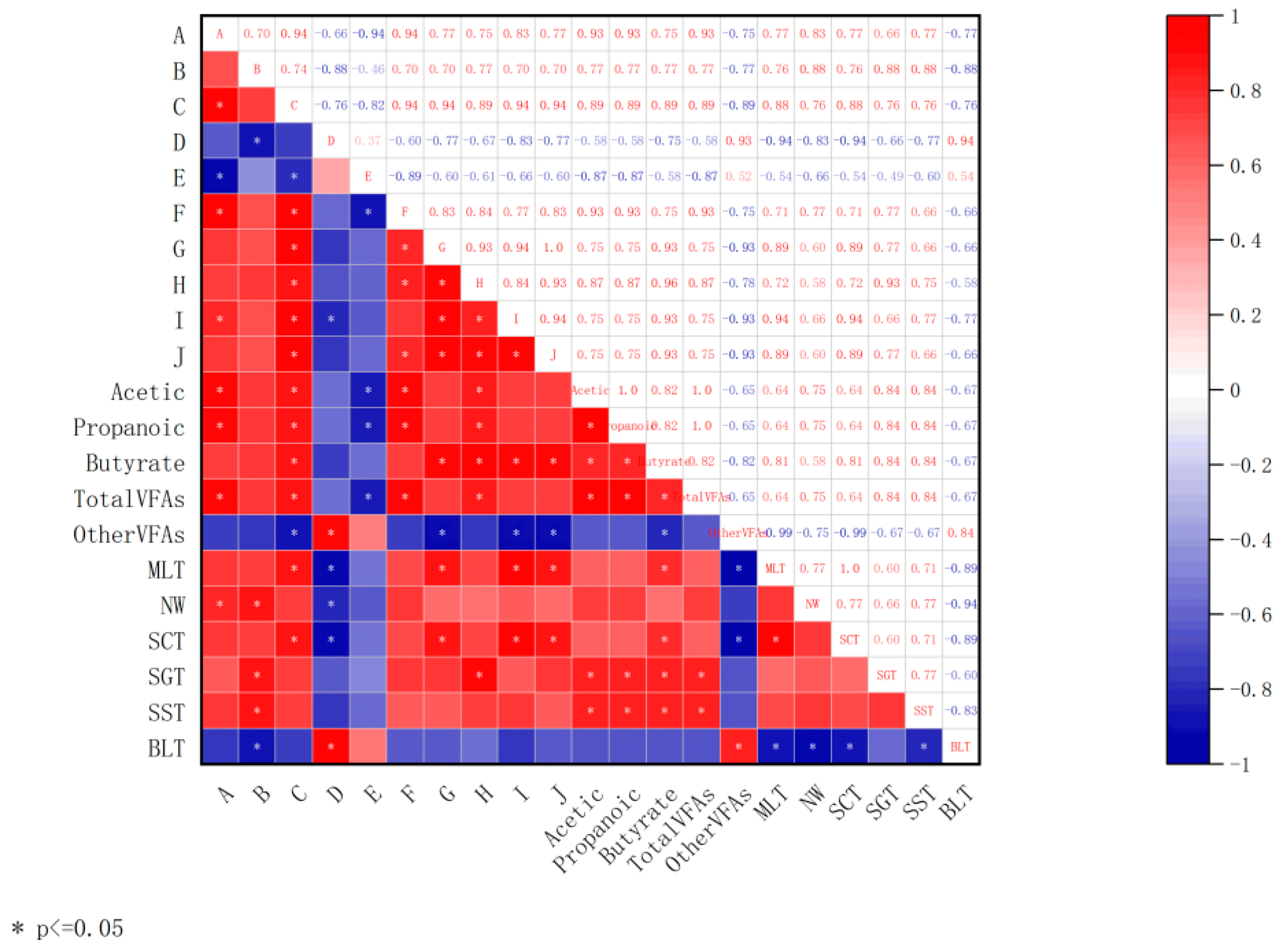
3.6. Association Analysis of lncRNAs with VFAs and Tissue Morphological Characteristics in Rumen Epithelium
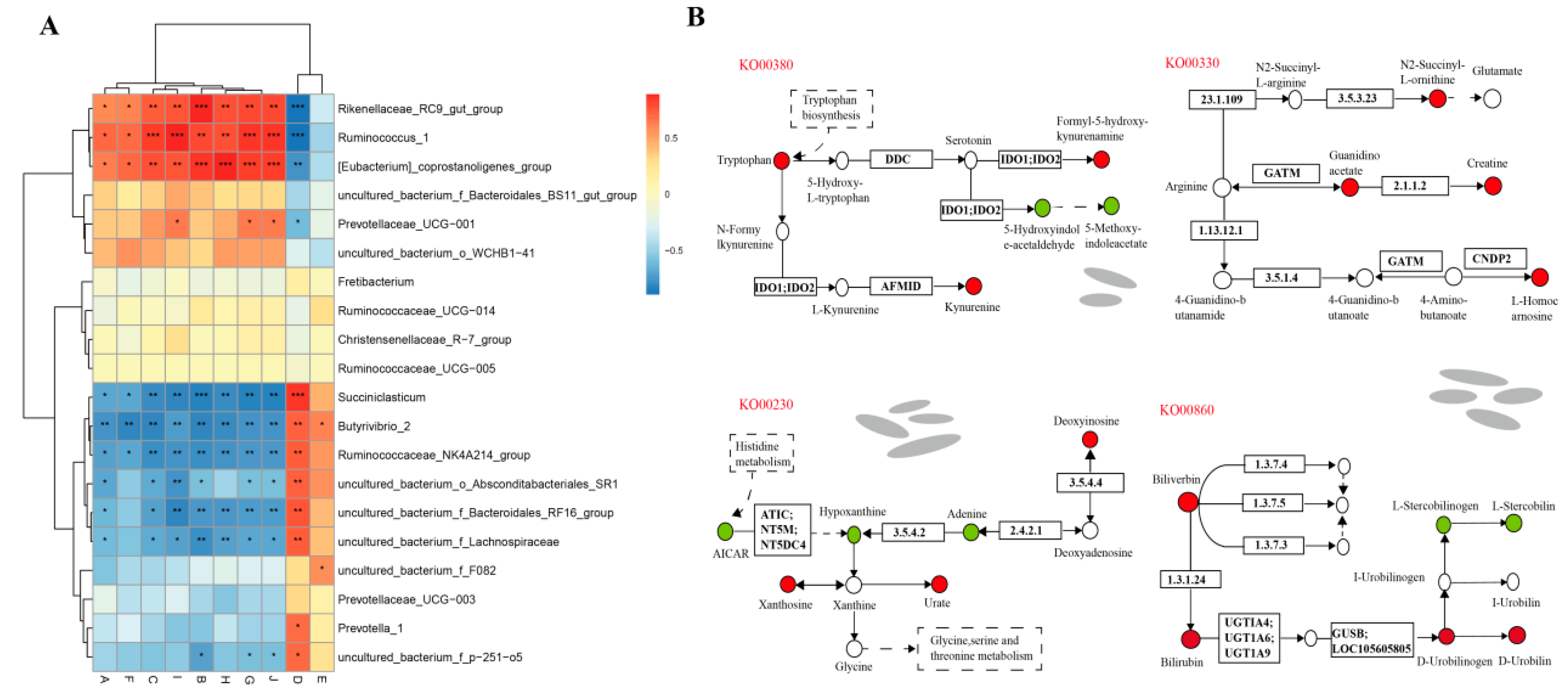
3.7. Analysis of lncRNAs-Microbiota-Metabolite Interaction in Rumen Epithelium Tissue
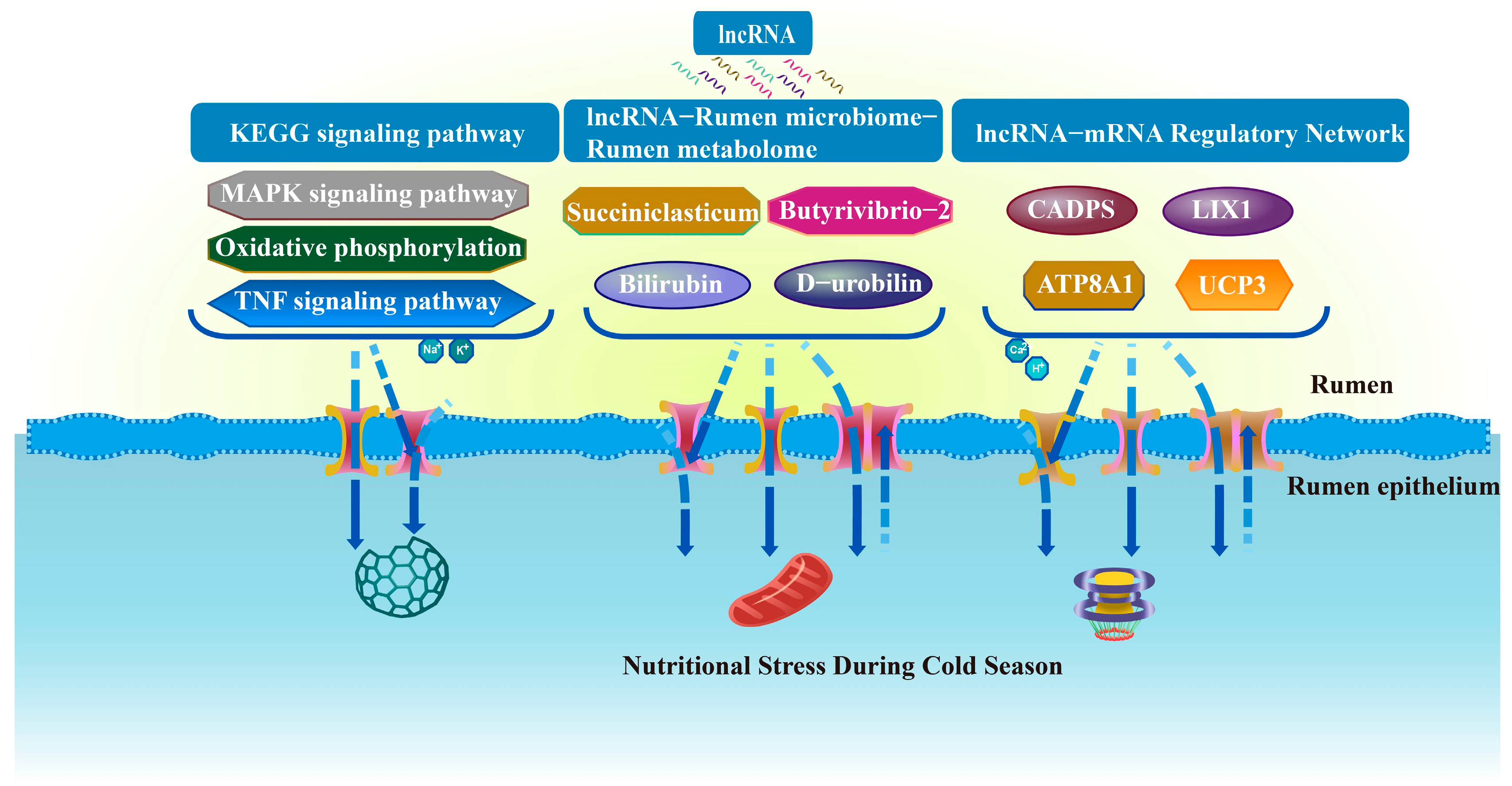
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Pina, L.F.; Balocchi, O.A.; Keim, J.P.; Pulido, R.G.; Rosas, F. Pre-Grazing Herbage Mass Affects Grazing Behavior, Herbage Disappearance, and the Residual Nutritive Value of a Pasture during the First Grazing Session. Animals 2020, 10, 212. [Google Scholar] [CrossRef] [PubMed]
- Backhed, F.; Ding, H.; Wang, T.; Hooper, L.V.; Koh, G.Y.; Nagy, A.; Semenkovich, C.F.; Gordon, J.I. The gut microbiota as an environmental factor that regulates fat storage. Proc. Natl. Acad. Sci. USA 2004, 101, 15718–15723. [Google Scholar] [CrossRef] [PubMed]
- Remond, D.; Ortigues, I.; Jouany, J.P. Energy substrates for the rumen epithelium. Proc. Nutr. Soc. 1995, 54, 95–105. [Google Scholar] [CrossRef] [PubMed]
- Carballo, O.C.; Khan, M.A.; Knol, F.W.; Lewis, S.J.; Stevens, D.R.; Laven, R.A.; McCoard, S.A. Impact of weaning age on rumen development in artificially reared lambs1. J. Anim. Sci. 2019, 97, 3498–3510. [Google Scholar] [CrossRef] [PubMed]
- Bond, J.J.; Donaldson, A.J.; Coumans, J.; Austin, K.; Ebert, D.; Wheeler, D.; Oddy, V.H. Protein profiles of enzymatically isolated rumen epithelium in sheep fed a fibrous diet. J. Anim. Sci. Biotechnol. 2019, 10, 5. [Google Scholar] [CrossRef]
- Bell, T.; Newman, J.A.; Silverman, B.W.; Turner, S.L.; Lilley, A.K. The contribution of species richness and composition to bacterial services. Nature 2005, 436, 1157–1160. [Google Scholar] [CrossRef]
- Goodman, D. theory of diversity-stability relationships in ecology. Q. Rev. Biol. 1975, 50, 237–266. [Google Scholar] [CrossRef]
- Kinross, J.M.; von Roon, A.C.; Holmes, E.; Darzi, A.; Nicholson, J.K. The human gut microbiome: Implications for future health care. Curr. Gastroenterol. Rep. 2008, 10, 396–403. [Google Scholar] [CrossRef]
- Rothe, M.; Blaut, M. Evolution of the gut microbiota and the influence of diet. Benef. Microbes 2013, 4, 31–37. [Google Scholar] [CrossRef]
- Guo, N.; Wu, Q.; Shi, F.; Niu, J.; Zhang, T.; Degen, A.A.; Fang, Q.; Ding, L.; Shang, Z.; Zhang, Z.; et al. Seasonal dynamics of diet-gut microbiota interaction in adaptation of yaks to life at high altitude. Npj Biofilms Microbiomes 2021, 7, 38. [Google Scholar] [CrossRef]
- Wilson, A.C.; Duncan, R.P. Signatures of host/symbiont genome coevolution in insect nutritional endosymbioses. Proc. Natl. Acad. Sci. USA 2015, 112, 10255–10261. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Xu, D.; Wang, L.; Hao, J.; Wang, J.; Zhou, X.; Wang, W.; Qiu, Q.; Huang, X.; Zhou, J.; et al. Convergent Evolution of Rumen Microbiomes in High-Altitude Mammals. Curr. Biol. 2016, 26, 1873–1879. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.M.; Chen, Y.Q. Principles and innovative technologies for decrypting noncoding RNAs: From discovery and functional prediction to clinical application. J. Hematol. Oncol. 2020, 13, 109. [Google Scholar] [CrossRef] [PubMed]
- Iwakiri, J.; Terai, G.; Hamada, M. Computational prediction of lncRNA-mRNA interactionsby integrating tissue specificity in human transcriptome. Biol. Direct. 2017, 12, 15. [Google Scholar] [CrossRef]
- Statello, L.; Guo, C.J.; Chen, L.L.; Huarte, M. Author Correction: Gene regulation by long non-coding RNAs and its biological functions. Nat. Rev. Mol. Cell Biol. 2021, 22, 159. [Google Scholar] [CrossRef]
- Malmuthuge, N.; Liang, G.; Guan, L.L. Regulation of rumen development in neonatal ruminants through microbial metagenomes and host transcriptomes. Genome Biol. 2019, 20, 172. [Google Scholar] [CrossRef]
- Zhong, T.; Zhao, J.; Zhan, S.; Wang, L.; Cao, J.; Dai, D.; Guo, J.; Li, L.; Zhang, H.; Niu, L. LncRNA-mRNA modules involved in goat rumen development: Insights from genome-wide transcriptome profiling. Front Physiol. 2022, 13, 979121. [Google Scholar] [CrossRef]
- Lu, Z.; Yuan, C.; Li, J.; Guo, T.; Yue, Y.; Niu, C.; Liu, J.; Yang, B. Comprehensive Analysis of Long Non-coding RNA and mRNA Transcriptomes Related to Hypoxia Adaptation in Tibetan Sheep. Front. Vet. Sci. 2021, 8, 801278. [Google Scholar] [CrossRef]
- Liu, X.; Sha, Y.; Dingkao, R.; Zhang, W.; Lv, W.; Wei, H.; Shi, H.; Hu, J.; Wang, J.; Li, S.; et al. Interactions Between Rumen Microbes, VFAs, and Host Genes Regulate Nutrient Absorption and Epithelial Barrier Function During Cold Season Nutritional Stress in Tibetan Sheep. Front. Microbiol. 2020, 11, 593062. [Google Scholar] [CrossRef]
- Liu, X.; Sha, Y.; Lv, W.; Cao, G.; Guo, X.; Pu, X.; Wang, J.; Li, S.; Hu, J.; Luo, Y. Multi-Omics Reveals That the Rumen Transcriptome, Microbiome, and Its Metabolome Co-regulate Cold Season Adaptability of Tibetan Sheep. Front. Microbiol. 2022, 13, 859601. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.; Han, Y.; He, Q. clusterProfiler: An R Package for Comparing Biological Themes Among Gene Clusters. OMICS J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef] [PubMed]
- Groussin, M.; Mazel, F.; Alm, E.J. Co-evolution and Co-speciation of Host-Gut Bacteria Systems. Cell Host Microbe 2020, 28, 12–22. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Liu, H.T. MAPK signal pathways in the regulation of cell proliferation in mammalian cells. Cell Res. 2002, 12, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Farhadi, S.; Shodja, G.J.; Hasanpur, K.; Mohammadi, S.A.; Ebrahimie, E. Molecular mechanisms of fat deposition: IL-6 is a hub gene in fat lipolysis, comparing thin-tailed with fat-tailed sheep breeds. Arch. Anim. Breed. 2021, 64, 53–68. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wahl, R.; Randeva, H.S. Responses of the insulin signaling pathways in the brown adipose tissue of rats following cold exposure. PLoS ONE 2014, 9, e99772. [Google Scholar] [CrossRef]
- Grisouard, J.; Bouillet, E.; Timper, K.; Radimerski, T.; Dembinski, K.; Frey, D.M.; Peterli, R.; Zulewski, H.; Keller, U.; Müller, B.; et al. Both inflammatory and classical lipolytic pathways are involved in lipopolysaccharide-induced lipolysis in human adipocytes. Innate Immun. (Lond. Engl.) 2012, 18, 25–34. [Google Scholar] [CrossRef]
- Newton, K.; Dixit, V.M. Signaling in innate immunity and inflammation. Cold Spring Harb. Perspect. Biol. 2012, 4, a006049. [Google Scholar] [CrossRef]
- Edilova, M.I.; Abdul-Sater, A.A.; Watts, T.H. TRAF1 Signaling in Human Health and Disease. Front. Immunol. 2018, 9, 2969. [Google Scholar] [CrossRef]
- Xu, Y.; Cheng, G.; Baltimore, D. Targeted disruption of TRAF3 leads to postnatal lethality and defective T-dependent immune responses. Immunity 1996, 5, 407–415. [Google Scholar] [CrossRef] [PubMed]
- Wilson, D.F. Oxidative phosphorylation: Regulation and role in cellular and tissue metabolism. J. Physiol. 2017, 595, 7023–7038. [Google Scholar] [CrossRef]
- Song, S.; Huo, J.L.; Li, D.L.; Yuan, Y.Y.; Yuan, F.; Miao, Y.W. Molecular cloning, sequence characterization, and gene expression profiling of a novel water buffalo (Bubalus bubalis) gene, AGPAT6. Genet. Mol. Res. 2013, 12, 4116–4126. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Li, C.; Chen, H.; Li, R.; Chong, Q.; Xiao, H.; Chen, S. Genome-wide scan of selection signatures in Dehong humped cattle for heat tolerance and disease resistance. Anim. Genet. 2020, 51, 292–299. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Cao, C.; Tao, C.; Ye, R.; Dong, M.; Zheng, Q.; Wang, C.; Jiang, X.; Qin, G.; Yan, C.; et al. Cold adaptation in pigs depends on UCP3 in beige adipocytes. J. Mol. Cell. Biol. 2017, 9, 364–375. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Cheong, H.S.; Shin, H.D.; Lee, S.S.; Roh, H.J.; Jeon, D.Y.; Cho, C.Y. Genetic diversity and divergence among Korean cattle breeds assessed using a BovineHD single-nucleotide polymorphism chip. Asian Austral. J. Anim. 2018, 31, 1691–1699. [Google Scholar] [CrossRef] [PubMed]
- Schroder, B.; Vossing, S.; Breves, G. In vitro studies on active calcium absorption from ovine rumen. J. Comp. Physiol. B 1999, 169, 487–494. [Google Scholar] [CrossRef] [PubMed]
- Guerin, A.; Angebault, C.; Kinet, S.; Cazevieille, C.; Rojo, M.; Fauconnier, J.; Lacampagne, A.; Mourier, A.; Taylor, N.; de Santa, B.P.; et al. LIX1-mediated changes in mitochondrial metabolism control the fate of digestive mesenchyme-derived cells. Redox Biol. 2022, 56, 102431. [Google Scholar] [CrossRef]
- Zou, J.; Zhu, X.; Xiang, D.; Zhang, Y.; Li, J.; Su, Z.; Kong, L.; Zhang, H. LIX1-like protein promotes liver cancer progression via miR-21-3p-mediated inhibition of fructose-1,6-bisphosphatase. Acta Pharm. Sin. B 2021, 11, 1578–1591. [Google Scholar] [CrossRef]
- Mao, Y.; Kucuk, B.; Irvine, K.D. Drosophila lowfat, a novel modulator of Fat signaling. Development 2009, 136, 3223–3233. [Google Scholar] [CrossRef]
- Chen, L.; Qiu, Q.; Jiang, Y.; Wang, K.; Lin, Z.; Li, Z.; Bibi, F.; Yang, Y.; Wang, J.; Nie, W.; et al. Large-scale ruminant genome sequencing provides insights into their evolution and distinct traits. Science 2019, 364, eaav6202. [Google Scholar] [CrossRef]
- Jiang, Y.; Xie, M.; Chen, W.; Talbot, R.; Maddox, J.F.; Faraut, T.; Wu, C.; Muzny, D.M.; Li, Y.; Zhang, W.; et al. The sheep genome illuminates biology of the rumen and lipid metabolism. Science 2014, 344, 1168–1173. [Google Scholar] [CrossRef]
- Gaebel, G.; Martens, H.; Suendermann, M.; Galfi, P. The effect of diet, intraruminal pH and osmolarity on sodium, chloride and magnesium absorption from the temporarily isolated and washed reticulo-rumen of sheep. Q. J. Exp. Physiol. 1987, 72, 501–511. [Google Scholar] [CrossRef] [PubMed]
- Meyer, W.; Schoennagel, B.; Kacza, J.; Busche, R.; Hornickel, I.N.; Hewicker-Trautwein, M.; Schnapper, A. Keratinization of the esophageal epithelium of domesticated mammals. Acta Histochem. 2014, 116, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Zhang, D.; Wang, W.; Lv, F.; Pang, X.; Liu, G.; Li, F.; Zhang, X. Transcriptome profiling reveals differential gene expression in the rumen of Hu lambs at different developmental stages. Anim. Biotechnol. 2021, 34, 471–481. [Google Scholar] [CrossRef]
- Qin, Y.; Roberts, J.D.; Grimm, S.A.; Lih, F.B.; Deterding, L.J.; Li, R.; Chrysovergis, K.; Wade, P.A. An obesity-associated gut microbiome reprograms the intestinal epigenome and leads to altered colonic gene expression. Genome Biol. 2018, 19, 7. [Google Scholar] [CrossRef]
- Camp, J.G.; Frank, C.L.; Lickwar, C.R.; Guturu, H.; Rube, T.; Wenger, A.M.; Chen, J.; Bejerano, G.; Crawford, G.E.; Rawls, J.F. Microbiota modulate transcription in the intestinal epithelium without remodeling the accessible chromatin landscape. Genome Res. 2014, 24, 1504–1516. [Google Scholar] [CrossRef] [PubMed]
- Hajam, I.A.; Dar, P.A.; Shahnawaz, I.; Jaume, J.C.; Lee, J.H. Bacterial flagellin-a potent immunomodulatory agent. Exp. Mol. Med. 2017, 49, e373. [Google Scholar] [CrossRef] [PubMed]
- Rhee, S.H.; Im, E.; Riegler, M.; Kokkotou, E.; O’Brien, M.; Pothoulakis, C. Pathophysiological role of Toll-like receptor 5 engagement by bacterial flagellin in colonic inflammation. Proc. Natl. Acad. Sci. USA 2005, 102, 13610–13615. [Google Scholar] [CrossRef]
- Liu, J.H.; Bian, G.R.; Zhu, W.Y.; Mao, S.Y. High-grain feeding causes strong shifts in ruminal epithelial bacterial community and expression of Toll-like receptor genes in goats. Front. Microbiol. 2015, 6, 167. [Google Scholar] [CrossRef]
- Pretheeban, M.; Hammond, G.; Bandiera, S.; Riggs, W.; Rurak, D. Ontogenesis of UDP-glucuronosyltransferase enzymes in sheep. Comp. Biochem. Phys. A 2011, 159, 159–166. [Google Scholar] [CrossRef]
- Muroya, S.; Zhang, Y.; Otomaru, K.; Oshima, K.; Oshima, I.; Sano, M.; Roh, S.; Ojima, K.; Gotoh, T. Maternal Nutrient Restriction Disrupts Gene Expression and Metabolites Associated with Urea Cycle, Steroid Synthesis, Glucose Homeostasis, and Glucuronidation in Fetal Calf Liver. Metabolites 2022, 12, 203. [Google Scholar] [CrossRef]
- Fahmy, K.; Gray, C.H.; Nicholson, D.C. The reduction of bile pigments by faecal and intestinal bacteria. Biochim. Biophys. Acta 1972, 264, 85–97. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, T.; Sato, K.; Akiba, M.; Ohnishi, M. Urobilinogen, as a bile pigment metabolite, has an antioxidant function. J. Oleo Sci. 2006, 55, 191–197. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| LncRNA | Forward (5′ → 3′) | Reverse (5′ → 3′) |
|---|---|---|
| MSTRG.99282.1 | TGCTTAAACTGGCCCCTCTG | CACCACCGAAAGTCCTCCAA |
| MSTRG.118214.1 | GGGCCAGAGACAACTGGAAA | GAATCGTCCAAGGAGACGCA |
| MSTRG.89480.2 | GACGAAAGAAAGGCAGCGTC | CCCAGCTGGTTGTTCCTAGAG |
| β-actin | AGCCTTCCTTCCTGGGCATGGA | GGACAGCACCGTGTTGGCGTAGA |
| Sample | Read Sum | Base Sum | GC (%) | N (%) | Q 20(%) | Q 30(%) |
|---|---|---|---|---|---|---|
| cold1 | 53,859,439 | 16,025,456,748 | 48.37 | 0 | 98.27 | 94.99 |
| cold2 | 60,990,364 | 18,103,396,314 | 48.85 | 0 | 98.34 | 95.04 |
| cold3 | 61,090,369 | 18,095,280,638 | 48.28 | 0 | 98.22 | 94.79 |
| warm1 | 52,304,300 | 15,558,868,816 | 50.68 | 0 | 98.37 | 95.12 |
| warm2 | 54,632,262 | 16,233,111,642 | 51.21 | 0 | 98.38 | 95.17 |
| warm3 | 58,382,319 | 17,397,315,876 | 50.83 | 0 | 98.42 | 95.34 |
| Sample | Total Reads | Mapped Reads | Uniq Mapped Reads | Multiple Reads |
|---|---|---|---|---|
| cold1 | 107,718,878 | 93,204,904 (86.53%) | 73,478,583 (68.21%) | 19,726,321 (18.31%) |
| cold2 | 121,980,728 | 106,374,890 (87.21%) | 84,457,338 (69.24%) | 21,917,552 (17.97%) |
| cold3 | 122,180,738 | 106,430,676 (87.11%) | 82,687,313 (67.68%) | 23,743,363 (19.43%) |
| warm1 | 104,608,600 | 73,161,590 (69.94%) | 54,951,260 (52.53%) | 18,210,330 (17.41%) |
| warm2 | 109,264,524 | 68,325,730 (62.53%) | 53,242,812 (48.73%) | 15,082,918 (13.80%) |
| warm3 | 116,764,638 | 87,757,254 (75.16%) | 69,574,963 (59.59%) | 18,182,291 (15.57%) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, X.; Guo, X.; Sha, Y.; He, Y.; Shao, P.; Hu, J.; Wang, J.; Li, S.; Hao, Z. Based on the Co-Evolution of lncRNAs-Microbiota and Metabolites in Rumen Epithelium to Analyze the Adaptation Characteristics of Tibetan Sheep to Nutrient Stress in the Cold Season. Fermentation 2023, 9, 892. https://doi.org/10.3390/fermentation9100892
Liu X, Guo X, Sha Y, He Y, Shao P, Hu J, Wang J, Li S, Hao Z. Based on the Co-Evolution of lncRNAs-Microbiota and Metabolites in Rumen Epithelium to Analyze the Adaptation Characteristics of Tibetan Sheep to Nutrient Stress in the Cold Season. Fermentation. 2023; 9(10):892. https://doi.org/10.3390/fermentation9100892
Chicago/Turabian StyleLiu, Xiu, Xinyu Guo, Yuzhu Sha, Yanyu He, Pengyang Shao, Jiang Hu, Jiqing Wang, Shaobin Li, and Zhiyun Hao. 2023. "Based on the Co-Evolution of lncRNAs-Microbiota and Metabolites in Rumen Epithelium to Analyze the Adaptation Characteristics of Tibetan Sheep to Nutrient Stress in the Cold Season" Fermentation 9, no. 10: 892. https://doi.org/10.3390/fermentation9100892
APA StyleLiu, X., Guo, X., Sha, Y., He, Y., Shao, P., Hu, J., Wang, J., Li, S., & Hao, Z. (2023). Based on the Co-Evolution of lncRNAs-Microbiota and Metabolites in Rumen Epithelium to Analyze the Adaptation Characteristics of Tibetan Sheep to Nutrient Stress in the Cold Season. Fermentation, 9(10), 892. https://doi.org/10.3390/fermentation9100892