
Roe Deer Produce Less Methane and Harbor Distinct Gut Microbiota
 and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Experimental Animals and Study Design
2.2. Measurement of Gas Production, Sample Collection, and Analyses
2.3. DNA Extraction, 16S Ribosomal RNA (16S rRNA) Gene Amplification, and Sequencing
2.4. Bioinformatics and Statistical Analyses
2.5. Co-occurrence Network of the Fecal Microbiota, Digestibility, and Gas Yields
3. Results
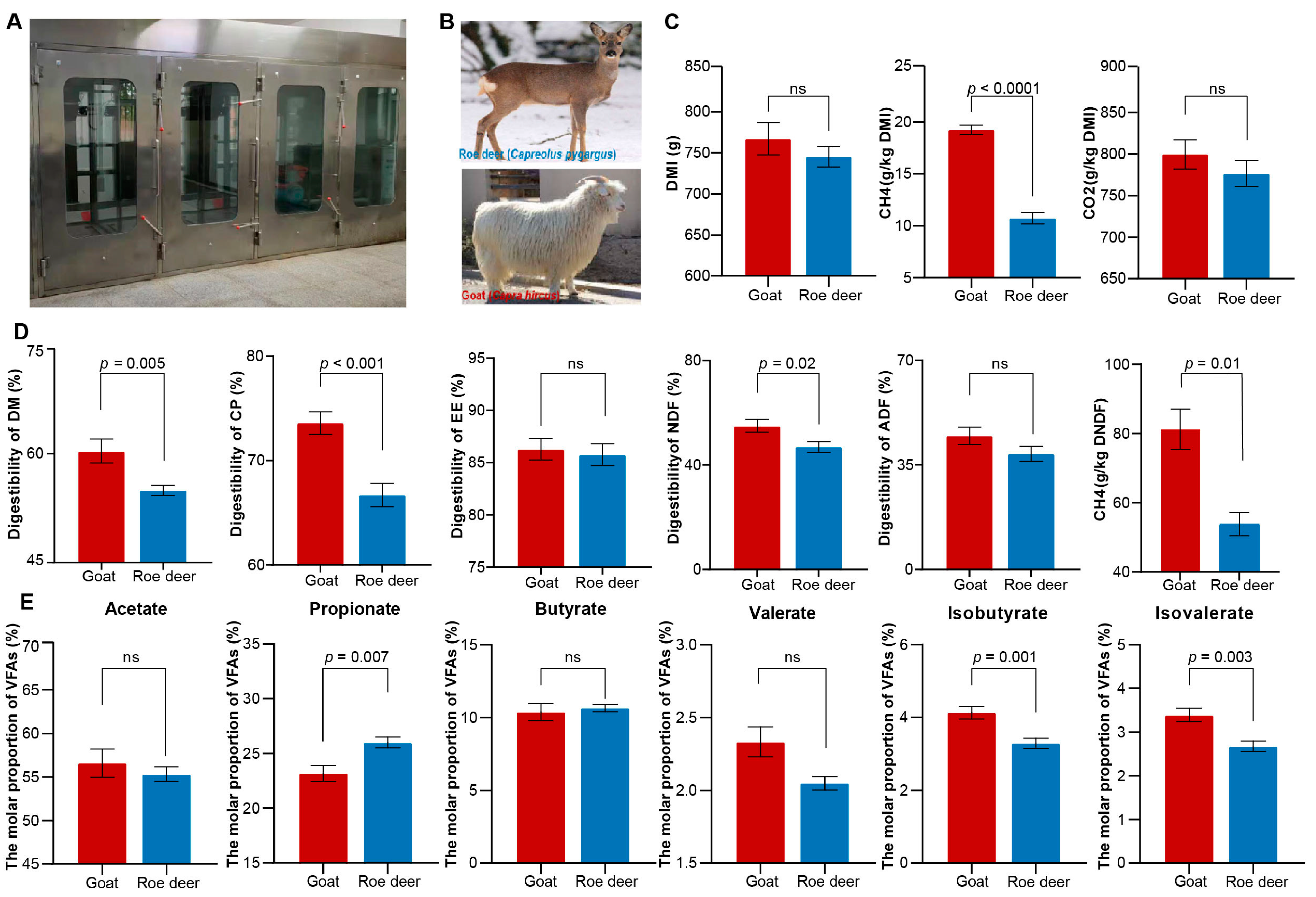
3.1. Variation in Gas Yields, Digestibility, and VFAs between Roe Deer and Goats
3.2. Bacterial Community Composition and Differences between Roe Deer and Goats
3.3. Methanogen Community Composition and Differences between Roe Deer and Goats
3.4. Co-occurrence Network among Bacteria, Methanogens, and Gas Yield
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Mizrahi, I.; Wallace, R.J.; Moraïs, S. The rumen microbiome: Balancing food security and environmental impacts. Nat. Rev. Microbiol. 2021, 19, 553–566. [Google Scholar] [CrossRef]
- Nikos, A.; Jelle, B. World Agriculture towards 2030/2050: The 2012 Revision; Agricultural Development Economics Division, Food and Agriculture Organization of the United Nations: Rome, Italy, 2012; pp. 1–47. [Google Scholar]
- Hill, J.; McSweeney, C.; Wright, A.D.G.; Bishop-Hurley, G.; Kalantar-zadeh, K. Measuring methane production from ruminants. Trends Biotech. 2016, 34, 26–35. [Google Scholar] [CrossRef]
- Gerber, P.J.; Steinfeld, H.; Henderson, B.; Mottet, A.; Opio, C.; Dijkman, J.; Alessandra, F.; Tempio, G. Tackling Climate Change through Livestock: A Global Assessment of Emissions and Mitigation Opportunities; Food and Agriculture Organization of the United Nations (FAO): Rome, Italy, 2013. [Google Scholar]
- Johnson, K.A.; Johnson, D.E. Methane emissions from cattle. J. Anim. Sci. 1995, 73, 2483–2492. [Google Scholar] [CrossRef]
- Morgavi, D.P.; Forano, E.; Martin, C.; Newbold, C.J. Microbial ecosystem and methanogenesis in ruminants. Animal 2010, 4, 1024–1036. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murray, R.M.; Bryant, A.M.; Leng, R.A. Rates of production of methane in the rumen and large intestine of sheep. Br. J. Nutr. 1976, 36, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- St-Pierre, B.; Wright, A.D.G. Diversity of gut methanogens in herbivorous animals. Animal 2013, 7, 49–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borrel, G.; Brugère, J.-F.; Gribaldo, S.; Schmitz, R.A.; Moissl-Eichinger, C. The host-associated archaeome. Nat. Rev. Microbiol. 2020, 18, 622–636. [Google Scholar] [CrossRef] [PubMed]
- Poulsen, M.; Schwab, C.; Jensen, B.B.; Engberg, R.M.; Spang, A.; Canibe, N.; Højberg, O.; Milinovich, G.; Fragner, L.; Schleper, C.; et al. Methylotrophic methanogenic Thermoplasmata implicated in reduced methane emissions from bovine rumen. Nat. Commun. 2013, 4, 1428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, W.; Moon, C.D.; Leahy, S.C.; Kang, D.; Froula, J.; Kittelmann, S.; Fan, C.; Deutsch, S.; Gagic, D.; Seedorf, H.; et al. Methane yield phenotypes linked to differential gene expression in the sheep rumen microbiome. Genome Res. 2014, 24, 1517–1525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamke, J.; Kittelmann, S.; Soni, P.; Li, Y.; Tavendale, M.; Ganesh, S.; Janssen, P.H.; Shi, W.; Shi, W.; Froula, J.; et al. Rumen metagenome and metatranscriptome analyses of low methane yield sheep reveals a Sharpea-enriched microbiome characterised by lactic acid formation and utilisation. Microbiome 2016, 4, 56. [Google Scholar] [CrossRef] [Green Version]
- Wallace, R.J.; Sasson, G.; Garnsworthy, P.C.; Tapio, I.; Gregson, E.; Bani, P.; Huhtanen, P.; Bayat, A.R.; Strozzi, F.; Biscarini, F.; et al. A heritable subset of the core rumen microbiome dictates dairy cow productivity and emissions. Sci. Adv. 2019, 5, eaav8391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Difford, G.F.; Plichta, D.R.; Løvendahl, P.; Lassen, J.; Noel, S.J.; Højberg, O.; Wright, A.G.; Zhu, Z.; Kristensen, L.; Nielsen, H.B.; et al. Host genetics and the rumen microbiome jointly associate with methane emissions in dairy cows. PLoS Genet. 2018, 14, e1007580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Youngblut, N.D.; Reischer, G.H.; Dauser, S.; Maisch, S.; Walzer, C.; Stalder, G.; Farnleitner, A.H.; Ley, R.E. Vertebrate host phylogeny influences gut archaeal diversity. Nat. Microbiol. 2021, 6, 1443–1454. [Google Scholar] [CrossRef] [PubMed]
- Knight, T.; Ronimus, R.S.; Dey, D.; Tootill, C.; Naylor, G.; Evans, P.; Molano, G.; Smith, A.; Tavendale, M.; Pinares-Panino, C.; et al. Chloroform decreases rumen methanogenesis and methanogen populations without altering rumen function in cattle. Anim. Feed Sci. Tech. 2011, 166–167, 101–112. [Google Scholar] [CrossRef]
- Weimer, P.J.; Stevenson, D.M.; Mantovani, H.C.; Man, S.L.C. Host specificity of the ruminal bacterial community in the dairy cow following near-total exchange of ruminal contents1. J. Dairy Sci. 2010, 93, 5902–5912. [Google Scholar] [CrossRef]
- Pérez-Barbería, F.J. Scaling methane emissions in ruminants and global estimates in wild populations. Sci. Total Environ. 2017, 579, 1572–1580. [Google Scholar] [CrossRef]
- Crutzen, P.J.; Aselmann, I.; Seile, R.W. Methane production by domestic animals, wild ruminants, other herbivorous fauna, and humans. Tellus B 1986, 38B, 271–284. [Google Scholar] [CrossRef]
- Manafiazar, G.; Fitzsimmons, C.; Zhou, M.; Basarab, J.A.; Baron, V.S.; McKeown, L.; Guan, L.L. Association between fecal methanogen species with methane production and grazed forage intake of beef heifers classified for residual feed intake under drylot conditions. Animal 2021, 15, 100304. [Google Scholar] [CrossRef]
- AOAC. Official Methods of Analysis of the Association of Official’s Analytical Chemists; AOAC: Arlington, Virginia, 2003. [Google Scholar]
- Van Soest, P.J.; Robertson, J.B.; Lewis, B.A. Methods for Dietary Fiber, Neutral Detergent Fiber, and Nonstarch Polysaccharides in Relation to Animal Nutrition. J. Dairy Sci. 1991, 74, 3583–3597. [Google Scholar] [CrossRef]
- Liu, J.H.; Xu, T.T.; Zhu, W.Y.; Mao, S.Y. High-grain feeding alters caecal bacterial microbiota composition and fermentation and results in caecal mucosal injury in goats. Br. J. Nutr. 2014, 112, 416–427. [Google Scholar] [CrossRef] [Green Version]
- The Human Microbiome Project Consortium. Structure, function and diversity of the healthy human microbiome. Nature 2012, 486, 207–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klindworth, A.; Pruesse, E.; Schweer, T.; Peplies, J.; Quast, C.; Horn, M.; Glockner, F.O. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. 2012, 41, e1. [Google Scholar] [CrossRef] [PubMed]
- Magoč, T.; Salzberg, S.L. FLASH: Fast length adjustment of short reads to improve genome assemblies. Bioinformatics 2011, 27, 2957–2963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Peña, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [Green Version]
- Edgar, R.C. UPARSE: Highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 2013, 10, 996–998. [Google Scholar] [CrossRef]
- Edgar, R.C.; Haas, B.J.; Clemente, J.C.; Quince, C.; Knight, R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 2011, 27, 2194–2200. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Garrity, G.M.; Tiedje, J.M.; Cole, J.R. Naïve bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 2007, 73, 5261–5267. [Google Scholar] [CrossRef] [Green Version]
- Langille, M.G.I.; Zaneveld, J.; Caporaso, J.G.; McDonald, D.; Knights, D.; Reyes, J.A.; Clemente, J.C.; Burkepile, D.E.; Vega Thurber, R.L.; Knight, R.; et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotech. 2013, 31, 814–821. [Google Scholar] [CrossRef]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Scardoni, G.; Petterlini, M.; Laudanna, C. Analyzing biological network parameters with CentiScaPe. Bioinformatics 2009, 25, 2857–2859. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bader, G.D.; Hogue, C.W.V. An automated method for finding molecular complexes in large protein interaction networks. BMC Bioinform. 2003, 4, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hofmann, R.R. Evolutionary steps of ecophysiological adaptation and diversification of ruminants: A comparative view of their digestive system. Oecologia 1989, 78, 443–457. [Google Scholar] [CrossRef] [PubMed]
- Na, Y.Y.; Li, D.H.; Choi, Y.J.; Kim, K.H.; Lee, S.R. Effects of feeding level on nutrient digestibility and enteric methane production in growing goats (Capra hircus hircus) and Sika deer (Cervus nippon hortulorum). Asian Austral. J. Anim. Sci. 2018, 31, 1238–1243. [Google Scholar] [CrossRef]
- Swainson, N.M.; Hoskin, S.O.; Clark, H.; Pinares-Patino, C.S.; Brookes, I.M. Comparative methane emissions from cattle, red deer and sheep. Proceed. N. Zeal. Soc. Anim. Prod. 2008, 68, 59–62. [Google Scholar]
- Olijhoek, D.W.; Løvendahl, P.; Lassen, J.; Hellwing, A.L.F.; Höglund, J.K.; Weisbjerg, M.R.; Noel, S.J.; McLean, F.; Højberg, O.; Lund, P. Methane production, rumen fermentation, and diet digestibility of Holstein and Jersey dairy cows being divergent in residual feed intake and fed at 2 forage-to-concentrate ratios. J. Dairy Sci. 2018, 101, 9926–9940. [Google Scholar] [CrossRef] [Green Version]
- Goopy, J.P.; Donaldson, A.; Hegarty, R.; Vercoe, P.E.; Haynes, F.; Barnett, M.; Oddy, V.H. Low-methane yield sheep have smaller rumens and shorter rumen retention time. Br. J. Nutr. 2014, 111, 578–585. [Google Scholar] [CrossRef] [Green Version]
- Tavendale, M.H.; Meagher, L.P.; Pacheco, D.; Walker, N.; Attwood, G.T.; Sivakumaran, S. Methane production from in vitro rumen incubations with Lotus pedunculatus and Medicago sativa, and effects of extractable condensed tannin fractions on methanogenesis. Anim. Feed Sci. Tech. 2005, 123, 403–419. [Google Scholar] [CrossRef]
- Li, Z.P.; Henderson, G.; Yang, Y.H.; Li, G.Y. Diversity of formyltetrahydrofolate synthetase genes in the rumens of roe deer (Capreolus pygargus) and sika deer (Cervus nippon) fed different diets. Can. J. Microbiol. 2017, 63, 11–19. [Google Scholar] [CrossRef] [Green Version]
- Moss, A.R.; Jouany, J.P.; Newbold, J. Methane production by ruminants: Its contribution to global warming. Annal. Zootech. 2000, 49, 231–253. [Google Scholar] [CrossRef] [Green Version]
- Kittelmann, S.; Pinares-Patiño, C.S.; Seedorf, H.; Kirk, M.R.; Ganesh, S.; McEwan, J.C.; Janssen, P.H. Two different bacterial community types are linked with the low-methane emission trait in sheep. PLoS ONE 2014, 9, e103171. [Google Scholar] [CrossRef] [PubMed]
- Danielsson, R.; Dicksved, J.; Sun, L.; Gonda, H.; Müller, B.; Schnürer, A.; Bertilsson, J. Methane Production in Dairy Cows Correlates with Rumen Methanogenic and Bacterial Community Structure. Front. Microbiol. 2017, 8, 226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, F.; Jin, W.; Si, H.Z.; Yuan, Y.; Tao, Y.; Liu, J.; Wang, X.; Yang, C.; Li, Q.; Yan, X.; et al. An integrated gene catalog and over 10,000 metagenome-assembled genomes from the gastrointestinal microbiome of ruminants. Microbiome 2021, 9, 137. [Google Scholar] [CrossRef] [PubMed]
- Narabe, C.; Kamiyama, S.; Saito, M.; Boonsaen, P.; Khongpradit, A.; Sawanon, S.; Suzuki, Y.; Koike, S.; Kobayashi, Y. Cashew nut shell liquid potentially mitigates methane emission from the feces of Thai native ruminant livestock by modifying fecal microbiota. Anim. Sci. J. 2021, 92, e13614. [Google Scholar] [CrossRef]
- Cunha, C.S.; Veloso, C.M.; Marcondes, M.I.; Mantovani, H.C.; Tomich, T.R.; Pereira, L.G.R.; Ferreira, M.F.L.; Dill-McFarland, K.A.; Suen, G. Assessing the impact of rumen microbial communities on methane emissions and production traits in Holstein cows in a tropical climate. Syst. Appl. Microbiol. 2017, 40, 492–499. [Google Scholar] [CrossRef] [PubMed]
- Lyons, T.; Bielak, A.; Doyle, E.; Kuhla, B. Variations in methane yield and microbial community profiles in the rumen of dairy cows as they pass through stages of first lactation. J. Dairy Sci. 2018, 101, 5102–5114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pope, P.B.; Smith, W.; Denman, S.E.; Tringe, S.G.; Barry, K.; Hugenholtz, P.; McSweeney, C.S.; McHardy, A.C.; Morrison, M. Isolation of Succinivibrionaceae implicated in low methane emissions from Tammar wallabies. Science 2011, 333, 646–648. [Google Scholar] [CrossRef]
- Wallace, R.J.; Rooke, J.A.; McKain, N.; Duthie, C.A.; Hyslop, J.J.; Ross, D.W.; Waterhouse, A.; Watson, M.; Roehe, R. The rumen microbial metagenome associated with high methane production in cattle. BMC Genom. 2015, 16, 839. [Google Scholar] [CrossRef] [Green Version]
- Stackebrandt, E.; Hespell, R. The family succinivibrionaceae. In The Prokaryotes; Dworkin, M., Falkow, S., Rosenberg, E., Schleifer, K.-H., Stackebrandt, E., Eds.; Springer: New York, NY, USA, 2006; pp. 419–429. [Google Scholar]
- Russell, J.B.; Muck, R.E.; Weimer, P.J. Quantitative analysis of cellulose degradation and growth of cellulolytic bacteria in the rumen. FEMS Microbiol. Ecol. 2009, 67, 183–197. [Google Scholar] [CrossRef]
- Holman, D.B.; Gzyl, K.E. A meta-analysis of the bovine gastrointestinal tract microbiota. FEMS Microbiol. Ecol. 2019, 95, fiz072. [Google Scholar] [CrossRef]
- Stevenson, D.M.; Weimer, P.J. Dominance of Prevotella and low abundance of classical ruminal bacterial species in the bovine rumen revealed by relative quantification real-time PCR. Appl. Microbiol. Biotech. 2007, 75, 165–174. [Google Scholar] [CrossRef]
- Moissl-Eichinger, C.; Pausan, M.; Taffner, J.; Berg, G.; Bang, C.; Schmitz, R.A. Archaea are interactive components of complex microbiomes. Trends Microbiol. 2018, 26, 70–85. [Google Scholar] [CrossRef]
- Li, Z.P.; Wang, X.X.; Zhang, T.; Si, H.Z.; Xu, C.; Wright, A.D.G.; Li, G. Heterogeneous development of methanogens and the correlation with bacteria in the rumen and cecum of sika deer (Cervus nippon) during early life suggest different ecology relevance. BMC Microbiol. 2019, 19, 129. [Google Scholar] [CrossRef] [PubMed]
- Raymann, K.; Moeller, A.H.; Goodman, A.L.; Ochman, H.; Tringe, S.G. Unexplored archaeal diversity in the great ape gut microbiome. mSphere 2017, 2, e00026-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bapteste, É.; Brochier, C.; Boucher, Y. Higher-level classification of the Archaea: Evolution of methanogenesis and methanogens. Archaea 2005, 1, 353–363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Browne, P.; Tamaki, H.; Kyrpides, N.; Woyke, T.; Goodwin, L.; Imachi, H.; Bräuer, H.; Yavitt, J.B.; Liu, W.T.; Zinder, S.; et al. Genomic composition and dynamics among Methanomicrobiales predict adaptation to contrasting environments. ISME J. 2017, 11, 87–99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hook, S.E.; Wright, A.D.G.; McBride, B.W. Methanogens: Methane producers of the rumen and mitigation strategies. Archaea 2010, 2010, 945785. [Google Scholar] [CrossRef] [Green Version]
- King, E.E.; Smith, R.P.; St-Pierre, B.; Wright, A.D.G. Differences in the rumen methanogen populations of lactating Jersey and Holstein dairy cows under the same diet regimen. Appl. Environ. Microbiol. 2011, 77, 5682–5687. [Google Scholar] [CrossRef] [Green Version]
- Danielsson, R.; Schnurer, A.; Arthurson, V.; Bertilsson, J. Methanogenic population and CH4 production in swedish dairy cows fed different levels of forage. Appl. Environ. Microbiol. 2012, 78, 6172–6179. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.; Guo, T.J.; Chen, Y.X.; Meng, Q.H.; Zhu, C.X.; Huang, H.K. Physicochemical characteristics of stored cattle manure affect methane emissions by inducing divergence of methanogens that have different interactions with bacteria. Agri. Ecosys. Environ. 2018, 253, 38–47. [Google Scholar] [CrossRef]
- Li, Z.P.; Wang, X.X.; Alberdi, A.; Deng, J.B.; Zhong, Z.Y.; Si, H.Z.; Zheng, C.; Zhou, H.; Wang, J.; Yang, Y.; et al. Comparative microbiome analysis reveals the ecological relationships between rumen methanogens, acetogens, and their hosts. Front. Microbiol. 2020, 11, 1311. [Google Scholar] [CrossRef]
- Kumpitsch, C.; Fischmeister, F.P.S.; Mahnert, A.; Lackner, S.; Wilding, M.; Sturm, C.; Springer, A.; Madl, T.; Madl, T.; Högenauer, C.; et al. Reduced B12 uptake and increased gastrointestinal formate are associated with archaeome-mediated breath methane emission in humans. Microbiome 2021, 9, 193. [Google Scholar] [CrossRef] [PubMed]
- Greening, C.; Geier, R.; Wang, C.; Woods, L.C.; Morales, S.E.; McDonald, M.J.; Rushton-Green, R.; Morgan, X.C.; Koike, S.; Leahy, S.C.; et al. Diverse hydrogen production and consumption pathways influence methane production in ruminants. ISME J. 2019, 13, 2617–2632. [Google Scholar] [CrossRef] [PubMed]
- Ramayo-Caldas, Y.; Zingaretti, L.; Popova, M.; Estellé, J.; Bernard, A.; Pons, N.; Bellot, P.; Mach, N.; Rau, A.; Roume, H.; et al. Identification of rumen microbial biomarkers linked to methane emission in Holstein dairy cows. J. Anim. Breed. Genet. 2020, 137, 49–59. [Google Scholar] [CrossRef] [Green Version]
- Shabat, S.K.B.; Sasson, G.; Doron-Faigenboim, A.; Durman, T.; Yaacoby, S.; Berg Miller, M.E.; White, B.A.; Shterzer, N.; Mizrahi, I. Specific microbiome-dependent mechanisms underlie the energy harvest efficiency of ruminants. ISME J. 2016, 10, 2958–2972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maman, L.G.; Palizban, F.; Atanaki, F.F.; Ghiasi, N.E.; Ariaeenejad, S.; Ghaffari, M.R.; Salekdeh, G.H.; Kavousi, K. Co-abundance analysis reveals hidden players associated with high methane yield phenotype in sheep rumen microbiome. Sci. Rep. 2020, 10, 4995. [Google Scholar] [CrossRef] [Green Version]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Han, Y.; Li, S.; Mu, R.; Zhao, F.; Yan, X.; Si, H.; Li, Z. Roe Deer Produce Less Methane and Harbor Distinct Gut Microbiota. Fermentation 2023, 9, 186. https://doi.org/10.3390/fermentation9020186
Han Y, Li S, Mu R, Zhao F, Yan X, Si H, Li Z. Roe Deer Produce Less Methane and Harbor Distinct Gut Microbiota. Fermentation. 2023; 9(2):186. https://doi.org/10.3390/fermentation9020186
Chicago/Turabian StyleHan, Yu, Songze Li, Ruina Mu, Fei Zhao, Xiaogang Yan, Huazhe Si, and Zhipeng Li. 2023. "Roe Deer Produce Less Methane and Harbor Distinct Gut Microbiota" Fermentation 9, no. 2: 186. https://doi.org/10.3390/fermentation9020186