Abstract
Knorringia sibirica is a typical species of Polygonaceae with high medicinal and ecological value. However, there are few available phylogenetic and genetic studies about the chloroplast genome of K. sibirica. In the current study, a comprehensive examination of the chloroplast genome of K. sibirica was performed. The K. sibirica chloroplast genome is 161,384 bp, circular with a typical quadripartite structure, and comprised of guanine + cytosine content of 37.63%. The genome consists of 131 genes, including 86 protein-coding genes, 37 tRNA genes, and 8 rRNA genes. Seventy simple sequence repeats were identified in the genome. When compared with three closely related species, the result revealed that the chloroplast genome of K. sibirica was conserved in genome composition and structure. The specific sites in the genome suitable for DNA barcodes were identified by calculation of the nucleotide diversity. Finally, a phylogenetic tree of 49 species in Polygonaceae was constructed using the whole chloroplast genome. The phylogenetic analysis suggested that Knorringia is an independent genus in the Polygonea tribe. This research could provide support for the utilization of genetic sources and the evolutionary study of K. sibirica.
1. Introduction
Knorringia sibirica is an annual herb belonging to the genus Knorringia of Polygonaceae and mainly distributed in Siberia, China, and other Himalayan regions [1]. Some species of Polygonaceae are widely distributed in China and are usually used in traditional Chinese medicine [2]. K. sibirica has been documented to possess numerous pharmacological applications and biological functions, including antioxidant and anti-aging properties. It has been utilized for the treatment of coughs, fatigue, weakness, diabetes mellitus, and various other health conditions. And the main medicinal ingredients of K. sibirica include polysaccharides, steroids, anthraquinone, alkaloids, cardiac glycosides, lignin, vitamins, various acids, and so on [3]. Apart from its excellent pharmacological effects, K. sibirica is also known for its strong tolerance to salinity and alkali. It can survive in wetlands, saline lands near river banks, and alkaline land. As one of the few important halophytes, it is often used as an important material for salt tolerance research and a potential genetic resource for genetic transformation [4]. However, the taxonomic history of Polygonaceae, especially K. sibirica, is convoluted and controversial, which has blocked the research and application of plants in Polygonaceae. Based on the tepal venation, cotyledon position, and other morphological features, Hong and Hedberg (1989) promoted the section Knorringia of Aconogonon to an independent genus [1]. Sanchez et al. (2009, 2011) and Schuster et al. (2011) used chloroplast genes and ribosomal internally transcribed spacers for the phylogenetic analysis of Polygonaceae, which supported the establishment of the genus Knorringia [5,6,7].
Chloroplasts are key metabolic centers that convert solar energy to carbohydrates, release oxygen, and are widely present in photoautotrophic organisms including land plants and certain algae [8,9]. In addition to photosynthesis, chloroplasts also play an important role in signaling and primary metabolism, such as adaption to abiotic stress and their defense against invading pathogens [10,11]. Despite the loss or transfer of some genes in the plastomes to the nuclear genome during coevolution, chloroplasts still maintain their own genetic system, which encodes crucial proteins and plays a vital role in plant viability [12,13,14,15]. Thus, in-depth research of chloroplast genes would be valuable for breeding and the identification and conservation of important traits [9]. Chloroplast genomes normally exhibit a closed circular structure ranging from 70,028 bp to 217,942 bp in length and have a conservative quartered form: a large single copy (LSC) region, a small single copy (SSC) region, and a pair of inverted repeat (IR) regions between them [16,17,18]. Plastomes are usually structurally stable, generally uniparental, haploid, and non-recombinant. Therefore, they provide a desirable model for plant taxonomic and phylogenetic studies [19,20]. High-throughput sequencing technology and third-generation DNA sequencing technologies facilitated rapid development in research on the chloroplast genome and enhanced our understanding of plant biology and phylogenetic classification [9,21]. By September 2023, the chloroplast genomes of more than 16,000 species were available in the chloroplast genome information resource (CGIR) [22]. The nomenclature of Polygonaceae has long been controversial [23]. K. sibirica, in particular, had been described in another genus and had several aliases [7]. In order to clarify the taxonomic status and make full use of K. sibirica, chloroplast genome sequencing and thorough analysis are indeed necessary. In this research, we successfully sequenced and assembled high-quality chloroplast genomes of K. sibirica (GenBank accession number: OR866441) and made comparisons to other species. This study will first offer a comprehensive analysis that supports the future development and utilization of K. sibirica.
2. Materials and Methods
2.1. DNA Extracting and Sequencing
The K. sibirica plant material used in the experiment was collected from Zaduo county, Qinghai province, China. Healthy fresh leaves, 2–3 g in weight, were used for DNA extraction. Leaves were snap-frozen with liquid nitrogen and stored at −80 °C before extracting chloroplast genome DNA. The total DNA was isolated using the cetyltrimethylammonium bromide method (CTAB) and its quality and quantity were measured by agarose gel electrophoresis and the Nanodrop method. The quantified DNA was used for library construction. A qualified 300 bp short-insert sequencing library (20 μL, 19.5 ng/μL) was constructed and sequenced with Illumina Nova-Seq (Wuhan Benagen Tech Solutions Company Limited, Wuhan, China). Sequencing was performed using a 2 × 150 paired-end configuration, and the raw sequencing reads were filtered for low-quality data to gain clean data using the filtering software (SOAPunke) (Version 2.1.0). The Nanopore sequencing was performed through a PromethION DNA sequencer (Oxford Nanopore Technologies, Oxford, UK). For the project, SOAPunke (Version: 2.1.0) was used as the filtering software. The filtering standards included removing reads with N base content exceeding 5%, removing reads with low quality (Q score ≤ 5) and a number of bases reaching 50%, and removing the adapter sequences contained in the reads [24].
2.2. Chloroplast Genome Assembly and Annotation
The length of the entire chloroplast genome was estimated using KmerGenie (v1.7051) [25]. The short reads were de novo assembled using the GetOrganelle toolkit (v1.7.7.0) with default parameters after quality controlling [26]. Subsequently, scaffolds with high coverage and length were pieced together into a more complete draft genome by BLASTN (v2.13.0) against the published whole chloroplast genome of K. sibirica (NC_082248.1). Annotation of the chloroplast genome includes the prediction of non-coding RNA and the structure and function of protein-coding genes. The gene annotation was implemented in CPGAVAS2 [27] and the sites of start and stop codons were calibrated by manual adjustment. To scan for the ribosome RNA, we used BLASTN (v2.13.0) to align rRNA sequences from accessible chloroplast genome to K. sibirica chloroplast genomein global coverage [28]. The tRNA was detected by tRNAscan-SE (v2.0.11) with pre-set parameters [29]. Then, the visual circle chloroplast genome map was generated by OGDRAW software (v1.1.1) [30]. Each annotation error was manually modified using CPGView and Apollo (v1.11.8) [31,32].
2.3. Comparative Genome Analysis
In order to further understand the structure and composition characteristics of the chloroplast genome of K. sibirica, we conducted a comparative genome analysis of the chloroplast genome of K. sibirica and its three closely related species, which included Fallopia aubertii (NC_068872.1), Muehlenbeckia australis (MG604297.1), and Polygonum cuspidatum (NC_057435.1). The whole chloroplast genomes of the four species were compared by MVISTA under the Shuffle-LAGAN model and the chloroplast genome of K. sibirica was used as a reference sequence [33]. IRScope was employed for the visual analysis of the junction between the four major regions in the chloroplast genomes of the four previously mentioned species [34]. The gene sequences were extracted from the four species and compared using MAFFT (v7.520) [35]. Then, the nucleotide diversity of each gene sequence between the four species was analyzed by MAFFT and R language (v4.2.3).
2.4. Analysis of Repeat Sequences and Codon Usage
To accurately annotate the repeat sequences in the chloroplast genome of K. sibirica, a combination of MISA (v2.1, https://webblast.ipk-gatersleben.de/misa/), TRF (v4.09, https://tandem.bu.edu/trf/trf.unix.help.html), and REPuter (https://bibiserv.cebitec.uni-bielefeld.de/reputer/) was used [36,37,38]. TRF was used to scan the tandem repeats, while REPuter was employed to estimate forward, palindromic, reverse, and complement repeats. MISA was utilized for exploring simple sequence repeats in the chloroplast genomes of K. sibirica and three related species. The parameters (SSR motif length - minimum number of repetitions) were set at 1-10, 2-5, 3-4, 4-3, 5-3, and 6-3.
Codon usage bias analysis was based on protein-coding sequences, which were extracted from the whole chloroplast genome by Phylosuite software (v1.1.16) [39]. CodonW (v1.4.4) was employed to calculate the relative synonymous codon usage in each CDS.
2.5. Phylogenetic Analyses
To clarify the taxonomic status of K. sibirica, a phylogenetic tree was constructed using the entire chloroplast genomes of K. sibirica and 48 other species in Polygonaceae, which were downloaded from the National Center for Biotechnology Information (NCBI). Symmeria paniculata was used as an outgroup. All of the tested sequences were aligned using MAFFT under standard parameters [35]. Primary alignments were cleaned by Gblocks (v0.91b) with default arguments [40]. For maximum likelihood (ML) phylogeny analysis, selection of the best DNA model and phylogenetic tree construction was performed by the IQ-TREE program [41]. For Bayesian inference (BI) analysis, the best-fit nucleotide substitution model was evaluated by PAUP (v4.0) and MrModeltest (v2.4) [42]. And BI phylogeny was deduced by MrBayes (v3.2.7) [43].
3. Results
3.1. Primary Features of K. sibirica Chloroplast Genome
Similar to most angiosperms, the chloroplast genome of K. sibirica exhibited a common closed circular structure with four parts and had a length of 161,384 bp with a LSC region of 87,085 bp in size, a SSC region 13,387 bp in size and two IR regions 30,456 bp in size, respectively. The GC content of the whole chloroplast genome was 37.63%, while the LSC, SSC, and IR regions showed 35.75%, 32.58%, and 41.43% GC contents, respectively (Table 1; Figure 1). A total of 131 genes were detected in the completely assembled chloroplast genome of K. sibirica, which included 86 protein-coding genes, 37 tRNA genes, and 8 rRNA genes (Table 2). Among them, seven protein-coding genes (ndhB, rps7, rps12, rpl23, rpl2, ycf1, ycf2), seven tRNA genes (trnA, trnI, trnI, trnL, trnN, trnR, trnV), and four rRNA genes (rrn4.5S, rrn5S, rrn16S, rrn23S) were contained in two IR regions. An intron is known as “a transcription unit containing regions which usually lost from the mature messenger”, which contributes to the diversity in protein isoforms [44,45]. There were 12 intron-containing genes in the chloroplast genome of K. sibirica. Only one gene (rps12) contained two introns, while the others (rps16, atpF, rpoC1, ycf3, clpP, petB, petD, rpl2, rpl16, ndhA, ndhB) embodied one intron.

Table 1.
Summary of the K. sibirica chloroplast genome.
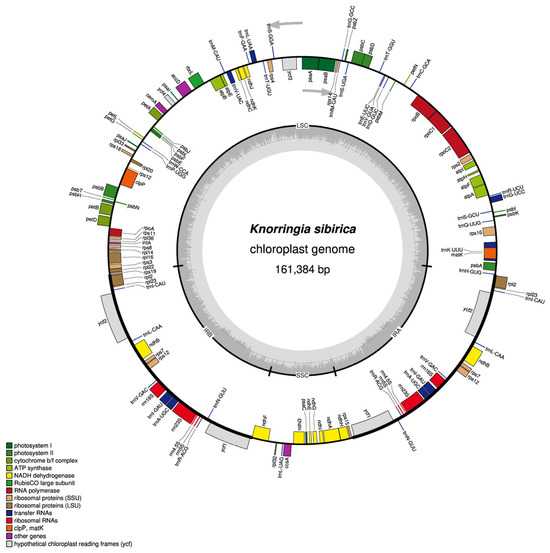
Figure 1.
Circular chloroplast genome map of K. sibirica. The squares on the outer ring represent genes and the color of squares indicates the function of each gene. The direction of gene transcription aligns with the gray arrow: genes inside the loop are transcribed clockwise, while those outside the loop are transcribed counterclockwise. The inner ring shows the location of LSC, SSC, and IRs regions. The ratio of dark to light gray regions inside the inner ring represents the contents of guanine + cytosine (GC) and adenine + thymine (AT), respectively.
Table 2.
List of annotated genes of the chloroplast genome of K. sibirica.
3.2. Codon Usage Bias
Synonymous codon usage bias refers to the difference in the frequency of use of synonymous codons for individual amino acids. The major forces affecting codon usage bias include mutation, selection, and genetic drift [46,47]. Relative synonymous codon usage (RSCU) quantifies the extent of the codon usage bias and drives a deeper understanding of species evolution. The codon usage bias was calculated based on 79 protein-coding gene sequences. There were 22,560 codons detected in all protein-coding genes (Table S1). The codons for leucine (2427, 10.76%) was the most frequently occurring in the chloroplast genome of K. sibirica, followed by isoleucine (1897, 8.41%). The least abundant codons encoded cysteine (246, 1.09%). Judging from the RSCU value, twenty-nine codons had a high preference for use (RSCU > 1), of which only one codon (UUG) bases not ending in A/U. This indicated that the protein-coding genes exhibited codon usage bias and prioritized the use of A/U (Figure 2, Table S1).
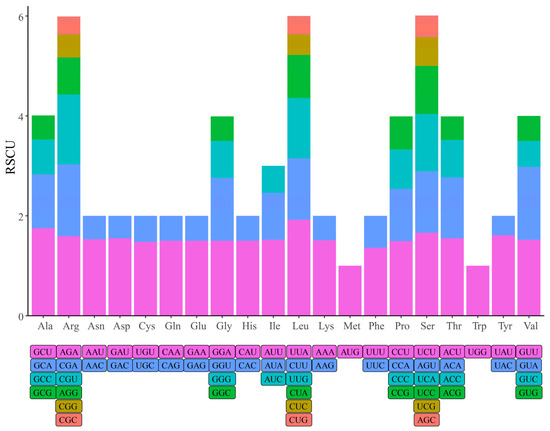
Figure 2.
Relative synonymous codon usage (RSCU) of 20 amino acids in all protein-coding genes of the K. sibirica chloroplast genome.
3.3. Repeat Sequence Analysis
Simple sequence repeats (SSRs), DNA segments with a tandem repeat sequence of 1–6 bases as the basic unit, also described as microsatellites, are widely used as molecular markers for the study of genetic variation [48,49,50]. In the chloroplast genome of K. sibirica, a total of 70 SSRs were found, which contained 30 distinct forms. These included 46 monomeric, 10 dimeric, 5 trimeric, 8 tetrameric, and 1 pentameric SSRs (Figure 3A; Table S2). Similar to other reports of chloroplast genomes, most types of the SSRs (n = 22, 71%) in the K. sibirica chloroplast genome simply consisted of A or T, and the content of GC of the SSRs was not more than 50%, mirroring the overall AT richness of the whole chloroplast sequence. The majority of the SSRs were located in the LSC region (n = 48, 68.57%), with 12 (17.14%) and 10 (14.29%) SSRs in the IR and SSC regions, respectively (Figure 3B). For the related species, we identified 57, 79, and 66 SSRs in the chloroplast genomes of F. aubertii, M. australis, and P. cuspidatum, respectively (Table S3). They shared the same base preference as K. sibirica but differed in type and quantity. Furthermore, we also detected 39 dispersed repeat sequences and 42 tandem repeat sequences in the chloroplast genome (Figure 3C; Tables S4 and S5). The dispersed repeat sequences included 17 palindromic repeats, 20 forward repeats, 2 reverse repeats, and 0 complementary repeats. Tandem repeat sequences ranged from 24 to 166 bp and were only distributed in the LSC and IR regions.
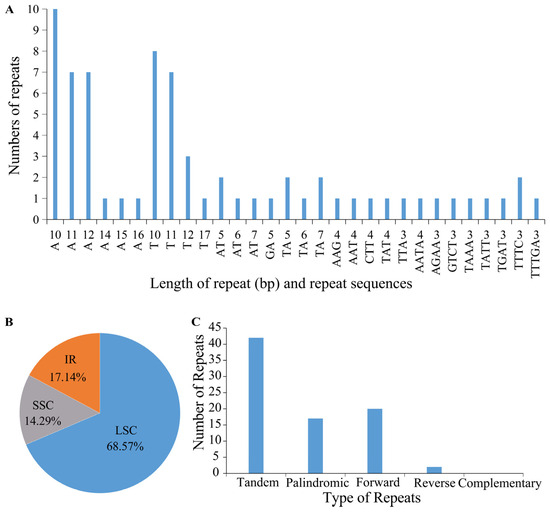
Figure 3.
Repeat sequences and SSR analysis of the K. sibirica chloroplast genome. (A) The number and length of each SSR. (B) Distribution of SSRs in the LSC, SSC, and IR regions of the K. sibirica chloroplast genome. (C) The type and number of repeat sequences other than SSRs.
3.4. Comparative Genome Analyses
The expansion and contraction in IR region boundaries are common in species evolution and are recognized as one of the causes of the variation in chloroplast genome size [34,51]. To investigate the expansion and contraction of the K. sibirica chloroplast genome, we detected the location of the junction between LSC and IRb (JLB), SSC and IRb (JSB), SSC and IRa (JSA), and LSC and IRa (JLA) (Figure 4). The JLB of four species intersected in the rps19 gene, of which the length extending to the IRb region was 41–108 bp. The junction line of LSC and IRa of the four species was located at the gene trnH or between trnH and rpl2. In the chloroplast genomes of K. sibirica, F. aubertii, and P. cuspidatum, the JSB was found within the ndhF gene, and the JSA was located 49–60 bp away from the rps15 gene. However, in the chloroplast genome of M. australis, the JSA was situated within the ndhF gene, and the JSB was 61 bp away from the rps15 gene.
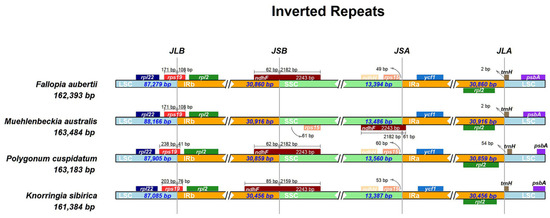
Figure 4.
Comparison of the borders of the LSC, SSC, IRa (equal to IRA), and IRb (equal to IRB) regions between K. sibirica, F. aubertii, M. australis, and P. cuspidatum.
To explore the characteristics of the K. sibirica chloroplast genome, it was compared with F. aubertii, M. australis, and P. cuspidatum by the MVISTA program (Figure 5). Overall, there was low sequence divergence between the four species, except for a gene (petD) and a few conserved non-coding regions. The gene petD of K. sibirica exhibited significant similarity to M. australis, while it diverged from F. aubertii and P. cuspidatum, with a similarity score below 50%. The highly divergent non-coding sequences between the four species include rps16-trnQ, trnS-trnG, trnC-petN, ndhF-rpl32, rpl32-trnL, and the intron of rpl16, which were possible to develop into molecular markers. The regions with high divergence were mainly located in the LSC and SSC regions. Consistent with previous studies, the IR regions contained low substitution for their duplicative nature [52]. Furthermore, the polymorphism analysis of conserved domains was used to detect the low- and high-variable sites in the chloroplast genome of the four species (Figure 6). The average polymorphism value of genes was 0.0167. And LSC and SSC regions were more diverse than IR regions. In particular, the genes matK, ndhF, and rps15 showed high diversity among the four species, which had the potential to develop molecular markers.

Figure 5.
Comparison of the chloroplast genomes of K. sibirica, F. aubertii, M. australis, and P. cuspidatum performed by MVISTA. The x-axis and y-axis represent the coordinate and sequence similarity. The arrows above the alignment imply the direction of gene transcription.
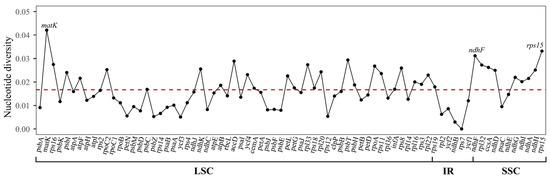
Figure 6.
Nucleotide diversity of conserved gene regions and inter-gene regions between K. sibirica, F. aubertii, M. australis, and P. cuspidatum. The dotted line represents for the average value (n = 0.0167).
3.5. Phylogenetic Relationships Analysis
Compared to the nuclear genome and mitochondrial genome, the chloroplast genome is more conserved and maintains low heteroplasmy and recombination, making the chloroplast genome more convenient for phylogenetic analysis [53]. To determine the phylogenetic position of K. sibirica in Polygonaceae, the whole chloroplast genomes of some plants in Polygonaceae were chosen for constructing a phylogenetic tree and Symmeria paniculata was used as an outgroup. The trees were constructed by maximum likelihood (ML) method with a TVM+F+I+I+R3 model and Bayesian inference (BI) method with a GTR+I+G model. The analysis results of both methods were largely identical in topology and had highly supported values. Figure 7 shows the phylogenetic tree topology from the ML method of the whole chloroplast genome, containing the ML bootstrap and BI posterior probability. The phylogenetic tree revealed that Muehlenbeckia, Fallopia, Reynoutria, Polygonum, and Atraphaxis were grouped into a clade. K. sibirica was listed as a sister to this clade, consistent with the result of Sanchez’s research [5].
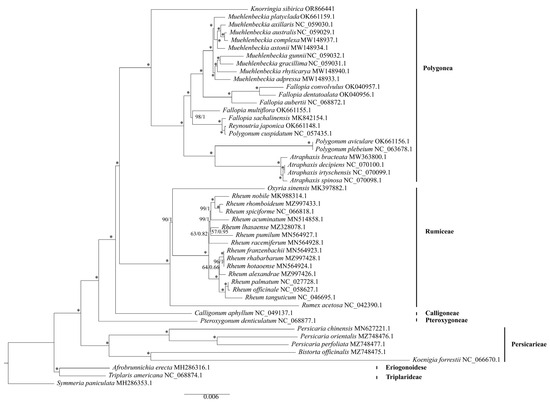
Figure 7.
Phylogenetic tree contracted from the whole chloroplast genome of 49 species in Polygonaceae. Each node is marked with ML bootstrap supports and Bayesian posterior probability values. Unless otherwise noted, an asterisk (*) indicates the branch with the maximum index value.
4. Discussion
In this study, the chloroplast genome of K. sibirica was de novo assembled and annotated, and it was the first report of a comprehensive analysis of K. sibirica chloroplast genetic information. The comparative analysis with other Polygonaceae plants suggested that the K. sibirica chloroplast genome was conserved in size, gene composition, and structure. In the analyses of the junction between the four regions in the chloroplast genome, no significant expansion or contraction was observed in the IRs regions of the K. sibirica chloroplast genome. And K. sibirica exhibited a similar structure to related species, except for M. australis, the SSC region of which was inverted compared to the other three species (Figure 4). Like most species in Polygonaceae, K. sibirica has a chloroplast genome containing 113 unique genes, with 18 of them having two copies. Meanwhile, some species exhibit partial gene loss or pseudogenization, such as the absence of rpl23 in the plastomes of P. aviculare, P. bistorta, P. orientale, and P. perfoliatum [2,54]. And similar to other plants, the GC content in the IRs regions of the chloroplast genome of K. sibirica is higher than that of the LSC and SSC regions (Table 1). The elevated GC content in the IRs regions is primarily due to the presence of rRNA and tRNA genes [2].
SSRs, which are present throughout the genome of eukaryotes, are highly informative and codominant multi-allele genetic markers. They are extensively used in research related to genetic mapping, population structure, and evolutionary processes [55]. The SSRs in chloroplast genomes are always used in genetic studies for their extreme variability and they provide a new opportunity for studies of species origin and evolution [56,57]. We identified the SSRs in the chloroplast genomes of K. sibirica and its related species by MISA, and 57–79 SSRs were found in the four species. In terms of the types, most SSRs consisted of A or T bases in the four species (Table S3). The results obtained in the present study were consistent with the previous opinion that SSRs in the chloroplast genome are predominantly composed of polyA and polyT mononucleotide repeat sequences. This observation may be attributed to the higher likelihood of A-T transformations occurring compared to G-C transformations [58]. And the SSRs in the chloroplast genome of K. sibirica were observed to be randomly distributed and distinct from those of the related species, making them suitable for molecular marker development.
Species identification is an essential part of biological research. There is indeed a vast number of species in the world, approximately 100 million [59]. Traditional taxonomic practices could be time-consuming and have significant limitations in accuracy, comprehensiveness, and objectivity. To address this issue, DNA barcodes were developed for biological identification [60,61]. The chloroplast genome has a higher interspecific and lower intraspecific divergence, making it suitable for DNA barcodes [62]. Similarly, there are many kilobase conserved sequences in the whole chloroplast genomes, which contain a tremendous amount of genetic information and make them more sensitive than traditional DNA barcodes [63,64,65]. Scientists have recently suggested utilizing the complete plastid genome sequence to improve phylogenetic resolution, particularly for species at a low taxonomic level [64,66]. Therefore, we constructed a phylogenetic tree using all of the species of Polygonaceae with accessible chloroplast genomes. Our results supported Knorringia as a separate genus and a sister clade to other clades including Atraphaxis, Polygonum, Fallopia, Reynoutria, and Muehlenbeckia. The overall topology of the phylogenetic tree was highly consistent with previous studies [5].
Single-nucleotide polymorphisms (SNPs) are the basis for the development of barcodes. The widespread use of chloroplast genome SNPs has greatly aided in species identification and evolutionary analysis [67,68]. DNA sequences with high mutation rates within a given taxonomic group can be used as ‘specific barcodes’, which can accurately identify specific taxa at a low cost [62]. In this study, we analyzed the nucleotide diversity of conserved regions of each gene and inter-gene sequence between K. sibirica and three related species (Figure 5 and Figure 6). The gene with the highest nucleotide diversity value was MatK, a traditional barcode highly desirable for species identification in plants [69]. The non-coding sequences ndhJ-ndhK, petL-petG, petG-UGG-psaJ, rps18-rpl20, psbE-petL, and trnH-GUG-psbA were quite diverse among the four species. Consistent with previous studies, trnH-GUG-psbA was highly variable in different species. Furthermore, both sides of trnH-GUG-psbA contained a relatively conserved sequence that can be utilized for designing primers for plant classification [70]. Other than that, all the above regions could be utilized as specific barcodes in the phylogenetic study of Polygonaceae and validated in future studies.
5. Conclusions
Overall, this research has assembled and annotated the complete chloroplast genome of K. sibirica. The chloroplast genome revealed a typical tetrameric structure of 161,384 bp in length, comprising 86 protein-coding genes, 37 tRNAs, and 8 rRNAs. The structure of the chloroplast genome of K. sibirica showed a high similarity with related species, although there was an inverted SSC region in M. australis. Through genome comparison analysis, both highly conserved and variable gene and inter-gene sequences among Polygonaceae species were observed, which could assist in species identification and further phylogenetic studies. The present study provides valuable resources for taxonomic and genetic research in K. sibirica.
Supplementary Materials
The following supporting information can be downloaded at https://www.mdpi.com/article/10.3390/horticulturae10030268/s1, Table S1: Usage counter and relative synonymous codon usage of each codon in protein-coding genes of K. sibirica chloroplast genome; Table S2: Size, type, and location of SSRs in K. sibirica chloroplast genome; Table S3: The number of each type of SSRs in the chloroplast genomes of K. sibirica, F. aubertii, M. australis, and P. cuspidatum; Table S4: Size, type, and location of dispersed repeats in K. sibirica chloroplast genome; Table S5: Tandem repeat sequences in K. sibirica chloroplast genome.
Author Contributions
Conceptualization, C.L.; formal analysis, K.Q.; investigation, Z.C., X.L. and K.Q.; resources, C.L.; data curation, K.Q.; writing—original draft preparation, K.Q.; writing—review and editing, Z.C. and K.M.; visualization, K.Q.; supervision, C.L. and Z.C.; project administration, C.L.; funding acquisition, C.L. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Major Science and Technology Project of Qinghai Province (2021-SF-A4); Chinese Academy of Sciences–People’s Government of Qinghai Province on Sanjiangyuan National Park (LHZX-2022-01); Gansu Province Grassland Monitoring and Evaluation Technology Support Project of Gansu Province Forestry and Grassland Administration (2021794).
Data Availability Statement
The data presented in this study are openly available in NCBI (GenBank accession number: OR866441).
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Hong, S.P. Knorringia (= Aconogonon sect. Knorringia), a new genus in the Polygonaceae. Nord. J. Bot. 1989, 9, 343–357. [Google Scholar] [CrossRef]
- Guo, S.; Liao, X.; Chen, S.; Liao, B.; Guo, Y.; Cheng, R.; Xiao, S.; Hu, H.; Chen, J.; Pei, J.; et al. A Comparative Analysis of the Chloroplast Genomes of Four Polygonum Medicinal Plants. Front. Genet. 2022, 13, 765434. [Google Scholar] [CrossRef] [PubMed]
- Cui, X.; Wang, S.; Cao, H.; Guo, H.; Li, Y.; Xu, F.; Zheng, M.; Xi, X.; Han, C. A Review: The Bioactivities and Pharmacological Applications of Polygonatum sibiricum polysaccharides. Molecules 2018, 23, 1170. [Google Scholar] [CrossRef] [PubMed]
- Qu, C.; Xu, Z.; Liu, G.; Liu, C.; Li, Y.; Wei, Z.; Liu, G. Differential Expression of Copper-Zinc Superoxide Dismutase Gene of Polygonum sibiricum Leaves, Stems and Underground Stems, Subjected to High-Salt Stress. Int. J. Mol. Sci. 2010, 11, 5235–5246. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, A.; Schuster, T.M.; Kron, K.A. A large-scale phylogeny of Polygonaceae based on molecular data. Int. J. Plant Sci. 2009, 170, 1044–1055. [Google Scholar] [CrossRef]
- Schuster, T.M.; Reveal, J.L.; Kron, K.A. Phylogeny of Polygoneae (Polygonaceae: Polygonoideae). Taxon 2011, 60, 1653–1666. [Google Scholar] [CrossRef]
- Sanchez, A.; Schuster, T.M.; Burke, J.M.; Kron, K.A. Taxonomy of Polygonoideae (Polygonaceae): A new tribal classification. Taxon 2011, 60, 151–160. [Google Scholar] [CrossRef]
- Asaf, S.; Khan, A.L.; Khan, M.A.; Imran, Q.M.; Kang, S.-M.; Al-Hosni, K.; Jeong, E.J.; Lee, K.E.; Lee, I.-J. Comparative analysis of complete plastid genomes from wild soybean (Glycine soja) and nine other Glycine species. PLoS ONE 2017, 12, e182281. [Google Scholar] [CrossRef] [PubMed]
- Daniell, H.; Lin, C.-S.; Yu, M.; Chang, W.-J. Chloroplast genomes: Diversity, evolution, and applications in genetic engineering. Genome Biol. 2016, 17, 134. [Google Scholar] [CrossRef]
- Bose, J.; Munns, R.; Shabala, S.; Gilliham, M.; Pogson, B.; Tyerman, S.D. Chloroplast function and ion regulation in plants growing on saline soils: Lessons from halophytes. J. Exp. Bot. 2017, 68, 3129–3143. [Google Scholar] [CrossRef]
- Littlejohn, G.R.; Breen, S.; Smirnoff, N.; Grant, M. Chloroplast immunity illuminated. New Phytol. 2021, 229, 3088–3107. [Google Scholar] [CrossRef]
- Martin, W.; Rujan, T.; Richly, E.; Hansen, A.; Cornelsen, S.; Lins, T.; Leister, D.; Stoebe, B.; Hasegawa, M.; Penny, D. Evolutionary analysis of Arabidopsis, cyanobacterial, and chloroplast genomes reveals plastid phylogeny and thousands of cyanobacterial genes in the nucleus. Proc. Natl. Acad. Sci. USA 2002, 99, 12246–12251. [Google Scholar] [CrossRef] [PubMed]
- Green, B.R. Chloroplast genomes of photosynthetic eukaryotes. Plant J. 2011, 66, 34–44. [Google Scholar] [CrossRef] [PubMed]
- Allen, J.F.; de Paula, W.B.M.; Puthiyaveetil, S.; Nield, J. A structural phylogenetic map for chloroplast photosynthesis. Trends Plant Sci. 2011, 16, 645–655. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, A.; Li, X.; Lu, C. The Role of Chloroplast Gene Expression in Plant Responses to Environmental Stress. Int. J. Mol. Sci. 2020, 21, 6082. [Google Scholar] [CrossRef] [PubMed]
- Asaf, S.; Khan, A.L.; Khan, M.A.; Waqas, M.; Kang, S.-M.; Yun, B.-W.; Lee, I.-J. Chloroplast genomes of Arabidopsis halleri ssp. gemmifera and Arabidopsis lyrata ssp. petraea: Structures and comparative analysis. Sci. Rep. 2017, 7, 7556. [Google Scholar] [CrossRef]
- Kolodner, R.; Tewari, K.K. Inverted repeats in chloroplast DNA from higher plants. Proc. Natl. Acad. Sci. USA 1979, 76, 41–45. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Wang, J.; Feng, L.; Pang, H.; Qi, L.; Li, J.; Sun, Y.; Qiao, W.; Zhang, L.; Cheng, Y.; et al. Inferring the evolutionary mechanism of the chloroplast genome size by comparing whole-chloroplast genome sequences in seed plants. Sci. Rep. 2017, 7, 1555. [Google Scholar]
- Hollingsworth, P.M. Refining the DNA barcode for land plants. Proc. Natl. Acad. Sci. USA 2011, 108, 19451–19452. [Google Scholar] [CrossRef]
- Small, R.L.; Cronn, R.C.; Wendel, J.F. Use of nuclear genes for phylogeny reconstruction in plants. Aust. Syst. Bot. 2004, 17, 145–170. [Google Scholar] [CrossRef]
- van Dijk, E.L.; Auger, H.; Jaszczyszyn, Y.; Thermes, C. Ten years of next-generation sequencing technology. Trends Genet. 2014, 30, 418–426. [Google Scholar] [CrossRef]
- Hua, Z.; Tian, D.; Jiang, C.; Song, S.; Chen, Z.; Zhao, Y.; Jin, Y.; Huang, L.; Zhang, Z.; Yuan, Y. Towards comprehensive integration and curation of chloroplast genomes. Plant Biotechnol. J. 2022, 20, 2239–2241. [Google Scholar] [CrossRef]
- Zhang, H.J.; Zhang, X.; Sun, Y.X.; Landis, J.B.; Li, L.J.; Hu, G.W.; Sun, J.; Tiamiyu, B.B.; Kuang, T.H.; Deng, T.; et al. Plastome phylogenomics and biogeography of the subfam. Polygonoideae (Polygonaceae). Front. Plant Sci. 2022, 13, 893201. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Chen, Y.; Shi, C.; Huang, Z.; Zhang, Y.; Li, S.; Li, Y.; Ye, J.; Yu, C.; Li, Z.; et al. SOAPnuke: A MapReduce acceleration-supported software for integrated quality control and preprocessing of high-throughput sequencing data. Gigascience 2017, 7, gix120. [Google Scholar] [CrossRef] [PubMed]
- Chikhi, R.; Medvedev, P. Informed and automated k-mer size selection for genome assembly. Bioinformatics 2014, 30, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Jin, J.; Yu, W.; Yang, J.; Song, Y.; dePamphilis, C.W.; Yi, T.; Li, D. GetOrganelle: A fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome Biol. 2020, 21, 241. [Google Scholar] [CrossRef] [PubMed]
- Shi, L.; Chen, H.; Jiang, M.; Wang, L.; Wu, X.; Huang, L.; Liu, C. CPGAVAS2, an integrated plastome sequence annotator and analyzer. Nucleic Acids Res. 2019, 47, W65–W73. [Google Scholar] [CrossRef]
- Chen, Y.; Ye, W.; Zhang, Y.; Xu, Y. High speed BLASTN: An accelerated MegaBLAST search tool. Nucleic Acids Res. 2015, 43, 7762–7768. [Google Scholar] [CrossRef] [PubMed]
- Lowe, T.M.; Eddy, S.R. tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997, 25, 955–964. [Google Scholar] [CrossRef] [PubMed]
- Lohse, M.; Drechsel, O.; Bock, R. OrganellarGenomeDRAW (OGDRAW): A tool for the easy generation of high-quality custom graphical maps of plastid and mitochondrial genomes. Curr. Genet. 2007, 52, 267–274. [Google Scholar] [CrossRef]
- Liu, S.; Ni, Y.; Li, J.; Zhang, X.; Yang, H.; Chen, H.; Liu, C. CPGView: A package for visualizing detailed chloroplast genome structures. Mol. Ecol. Resour. 2023, 23, 694–704. [Google Scholar] [CrossRef]
- Lewis, S.E.; Searle, S.M.J.; Harris, N.; Gibson, M.; Lyer, V.; Richter, J.; Wiel, C.; Bayraktaroglu, L.; Birney, E.; Crosby, M.A.; et al. Apollo: A sequence annotation editor. Genome Biol. 2002, 3, RESEARCH0082. [Google Scholar] [CrossRef]
- Frazer, K.A.; Pachter, L.; Poliakov, A.; Rubin, E.M.; Dubchak, I. VISTA: Computational tools for comparative genomics. Nucleic Acids Res. 2004, 32, W273–W279. [Google Scholar] [CrossRef]
- Amiryousefi, A.; Hyvonen, J.; Poczai, P. IRscope: An online program to visualize the junction sites of chloroplast genomes. Bioinformatics 2018, 34, 3030–3031. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Beier, S.; Thiel, T.; Muench, T.; Scholz, U.; Mascher, M. MISA-web: A web server for microsatellite prediction. Bioinformatics 2017, 33, 2583–2585. [Google Scholar] [CrossRef] [PubMed]
- Benson, G. Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res. 1999, 27, 573–580. [Google Scholar] [CrossRef]
- Kurtz, S.; Choudhuri, J.V.; Ohlebusch, E.; Schleiermacher, C.; Stoye, J.; Giegerich, R. REPuter: The manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 2001, 29, 4633–4642. [Google Scholar] [CrossRef]
- Zhang, D.; Gao, F.; Jakovlic, I.; Zou, H.; Zhang, J.; Li, W.X.; Wang, G.T. PhyloSuite: An integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol. Ecol. Resour. 2020, 20, 348–355. [Google Scholar] [CrossRef] [PubMed]
- Talavera, G.; Castresana, J. Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst. Biol. 2007, 56, 564–577. [Google Scholar] [CrossRef] [PubMed]
- Lam-Tung, N.; Schmidt, H.A.; von Haeseler, A.; Bui Quang, M. IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar]
- Pereira, T.L.; Santos, U.; Schaefer, C.E.; Souza, G.O.; Paiva, S.R.; Malabarba, L.R.; Schmidt, E.E.; Dergam, J.A. Dispersal and vicariance of Hoplias malabaricus (Bloch, 1794) (Teleostei, Erythrinidae) populations of the Brazilian continental margin. J. Biogeogr. 2013, 40, 905–914. [Google Scholar] [CrossRef]
- Ronquist, F.; Huelsenbeck, J.P. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 2003, 19, 1572–1574. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, W. Why genes in pieces? Nature 1978, 271, 501. [Google Scholar] [CrossRef] [PubMed]
- Min, X.J.; Powell, B.; Braessler, J.; Meinken, J.; Yu, F.; Sablok, G. Genome-wide cataloging and analysis of alternatively spliced genes in cereal crops. BMC Genom. 2015, 16, 721. [Google Scholar] [CrossRef]
- Brandis, G.; Hughes, D. The Selective Advantage of Synonymous Codon Usage Bias in Salmonella. PLoS Genet. 2016, 12, e1005926. [Google Scholar] [CrossRef]
- Bulmer, M. The selection-mutation-drift theory of synonymous codon usage. Genetics 1991, 129, 897–907. [Google Scholar] [CrossRef]
- Kpatenon, M.J.; Salako, K.V.; Santoni, S.; Zekraoui, L.; Latreille, M.; Tollon-Cordet, C.; Mariac, C.; Jaligot, E.; Beule, T.; Adeoti, K. Transferability, development of simple sequence repeat (SSR) markers and application to the analysis of genetic diversity and population structure of the African fan palm (Borassus aethiopum Mart.) in Benin. BMC Genet. 2020, 21, 145. [Google Scholar] [CrossRef]
- Powell, W.; Morgante, M.; McDevitt, R.; Vendramin, G.G.; Rafalski, J.A. Polymorphic simple sequence repeat regions in chloroplast genomes: Applications to the population genetics of pines. Proc. Natl. Acad. Sci. USA 1995, 92, 7759–7763. [Google Scholar] [CrossRef]
- Bhattarai, G.; Shi, A.; Kandel, D.R.; Solis-Gracia, N.; da Silva, J.A.; Avila, C.A. Genome-wide simple sequence repeats (SSR) markers discovered from whole-genome sequence comparisons of multiple spinach accessions. Sci. Rep. 2021, 11, 9999. [Google Scholar] [CrossRef]
- Goulding, S.E.; Olmstead, R.G.; Morden, C.W.; Wolfe, K.H. Ebb and flow of the chloroplast inverted repeat. Mol. Gen. Genet. 1996, 252, 195–206. [Google Scholar] [CrossRef]
- Zhu, A.; Guo, W.; Gupta, S.; Fan, W.; Mower, J.P. Evolutionary dynamics of the plastid inverted repeat: The effects of expansion, contraction, and loss on substitution rates. New Phytol. 2016, 209, 1747–1756. [Google Scholar] [CrossRef]
- Provan, J.; Powell, W.; Hollingsworth, P.M. Chloroplast microsatellites: New tools for studies in plant ecology and evolution. Trends Ecol. Evol. 2001, 16, 142–147. [Google Scholar] [CrossRef] [PubMed]
- Cao, D.L.; Zhang, X.J.; Qu, X.J.; Fan, S.J. Plastid phylogenomics sheds light on divergence time and ecological adaptations of the tribe Persicarieae (Polygonaceae). Front. Plant Sci. 2022, 13, 1046253. [Google Scholar] [CrossRef] [PubMed]
- Carneiro Vieira, M.L.; Santini, L.; Diniz, A.L.; Munhoz, C.d.F. Microsatellite markers: What they mean and why they are so useful. Genet. Mol. Biol. 2016, 39, 312–328. [Google Scholar] [CrossRef] [PubMed]
- Nishikawa, T.; Vaughan, D.A.; Kadowaki, K. Phylogenetic analysis of Oryza species, based on simple sequence repeats and their flanking nucleotide sequences from the mitochondrial and chloroplast genomes. Theor. Appl. Genet. 2005, 110, 696–705. [Google Scholar] [CrossRef] [PubMed]
- Ebert, D.; Peakall, R. Chloroplast simple sequence repeats (cpSSRs): Technical resources and recommendations for expanding cpSSR discovery and applications to a wide array of plant species. Mol. Ecol. Resour. 2009, 9, 673–690. [Google Scholar] [CrossRef] [PubMed]
- Xie, D.-F.; Yu, Y.; Deng, Y.-Q.; Li, J.; Liu, H.-Y.; Zhou, S.-D.; He, X.-J. Comparative Analysis of the Chloroplast Genomes of the Chinese Endemic Genus Urophysa and Their Contribution to Chloroplast Phylogeny and Adaptive Evolution. Int. J. Mol. Sci. 2018, 19, 1847. [Google Scholar] [CrossRef] [PubMed]
- Costello, M.J.; May, R.M.; Stork, N.E. Can We Name Earth’s Species before They Go Extinct? Science 2013, 339, 413–416. [Google Scholar] [CrossRef]
- Chase, M.W.; Fay, M.F. Barcoding of Plants and Fungi. Science 2009, 325, 682–683. [Google Scholar] [CrossRef]
- Hebert, P.D.N.; Cywinska, A.; Ball, S.L.; DeWaard, J.R. Biological identifications through DNA barcodes. Proc. R. Soc. B-Biol. Sci. 2003, 270, 313–321. [Google Scholar] [CrossRef]
- Li, X.; Yang, Y.; Henry, R.J.; Rossetto, M.; Wang, Y.; Chen, S. Plant DNA barcoding: From gene to genome. Biol. Rev. 2015, 90, 157–166. [Google Scholar] [CrossRef] [PubMed]
- Kane, N.; Sveinsson, S.; Dempewolf, H.; Yang, J.Y.; Zhang, D.; Engels, J.M.M.; Cronk, Q. Ultra-barcoding in cacao (Theobroma spp.; Malvaceae) using whole chloroplast genomes and nuclear ribosomal DNA. Am. J. Bot. 2012, 99, 320–329. [Google Scholar] [CrossRef] [PubMed]
- Nock, C.J.; Waters, D.L.E.; Edwards, M.A.; Bowen, S.G.; Rice, N.; Cordeiro, G.M.; Henry, R.J. Chloroplast genome sequences from total DNA for plant identification. Plant Biotechnol. J. 2011, 9, 328–333. [Google Scholar] [CrossRef] [PubMed]
- Steele, P.R.; Pires, J.C. Biodiversity assessment: State-of-the-aart techniques in phylogenomics and species identification. Am. J. Bot. 2011, 98, 415–425. [Google Scholar] [CrossRef] [PubMed]
- Parks, M.; Cronn, R.; Liston, A. Increasing phylogenetic resolution at low taxonomic levels using massively parallel sequencing of chloroplast genomes. BMC Biol. 2009, 7, 84. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Fang, Z.; Wang, Q.; Liu, Y.; Yang, L.; Zhuang, M.; Sun, P. Chloroplast Subspecies-Specific SNP Detection and Its Maternal Inheritance in Brassica oleracea L. by Using a dCAPS Marker. J. Hered. 2012, 103, 606–611. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Cui, H.; Ding, Z.; Zhu, Q.; Wu, Y.; Gao, P. Population structure and genetic diversity of watermelon (Citrullus lanatus) based on SNP of chloroplast genome. 3 Biotech 2020, 10, 374. [Google Scholar] [CrossRef] [PubMed]
- Hollingsworth, P.M.; Forrest, L.L.; Spouge, J.L.; Hajibabaei, M.; Ratnasingham, S.; van der Bank, M.; Chase, M.W.; Cowan, R.S.; Erickson, D.L.; Fazekas, A.J.; et al. A DNA barcode for land plants. Proc. Natl. Acad. Sci. USA 2009, 106, 12794–12797. [Google Scholar]
- Schilling, E.E.; Small, R.L. The tortoise and the hare II: Relative utility of 21 noncoding chloroplast DNA sequences for phylogenetic analysis. Am. J. Bot. 2005, 92, 142–166. [Google Scholar]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).