
Ficus Genome Database: A Comprehensive Genomics and Transcriptomics Research Platform

Abstract
:1. Introduction
2. Construction and Content
2.1. Genome Data Source
2.2. Transcriptome Data Source
2.3. Genome Collinearity Analysis
2.4. Gene Expression Analysis
2.5. Noncoding RNA Analysis
2.6. Biochemical Pathways
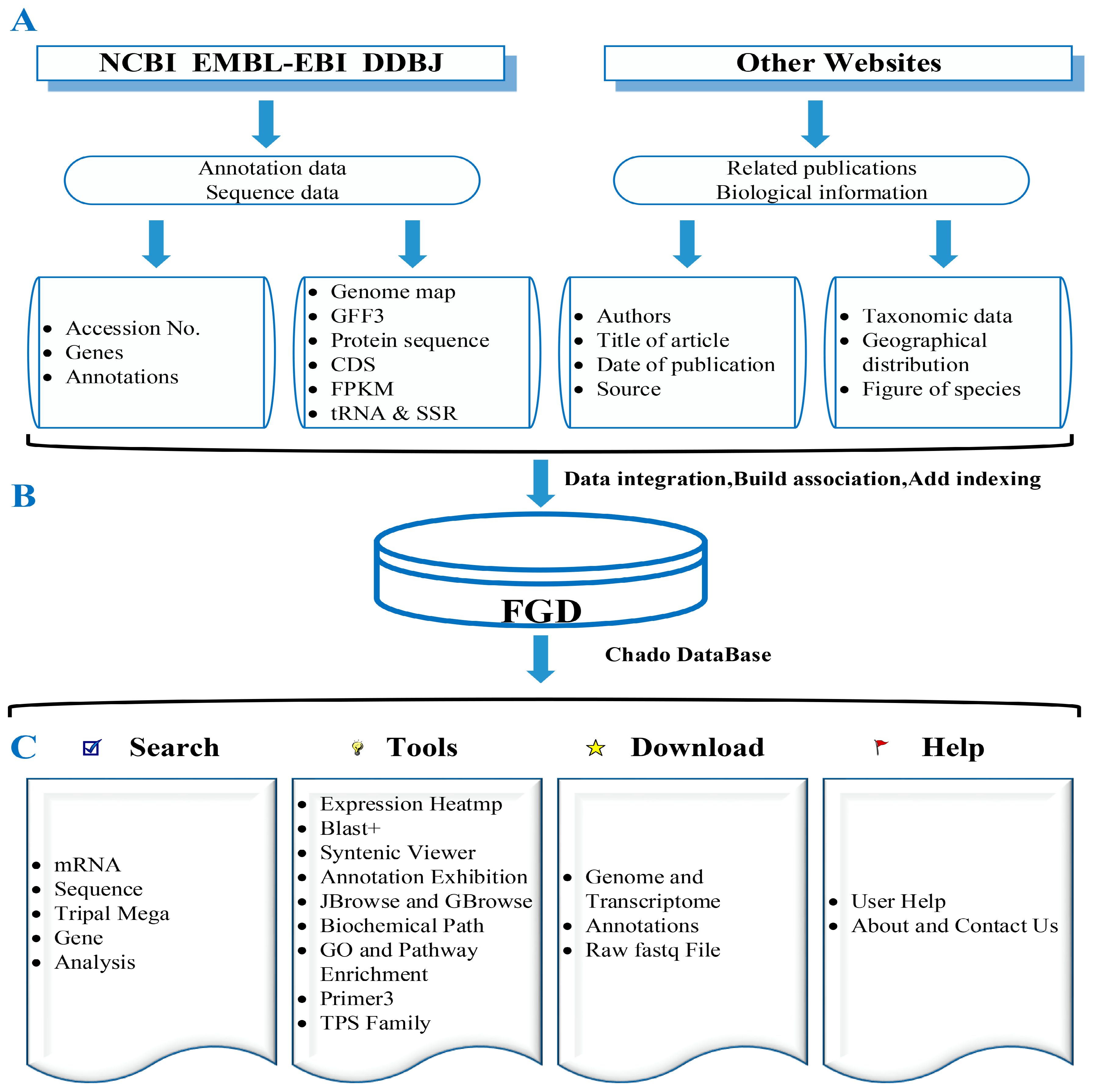
2.7. Database Construction
3. Utility and Discussion
3.1. A Brief Introduction to FGD
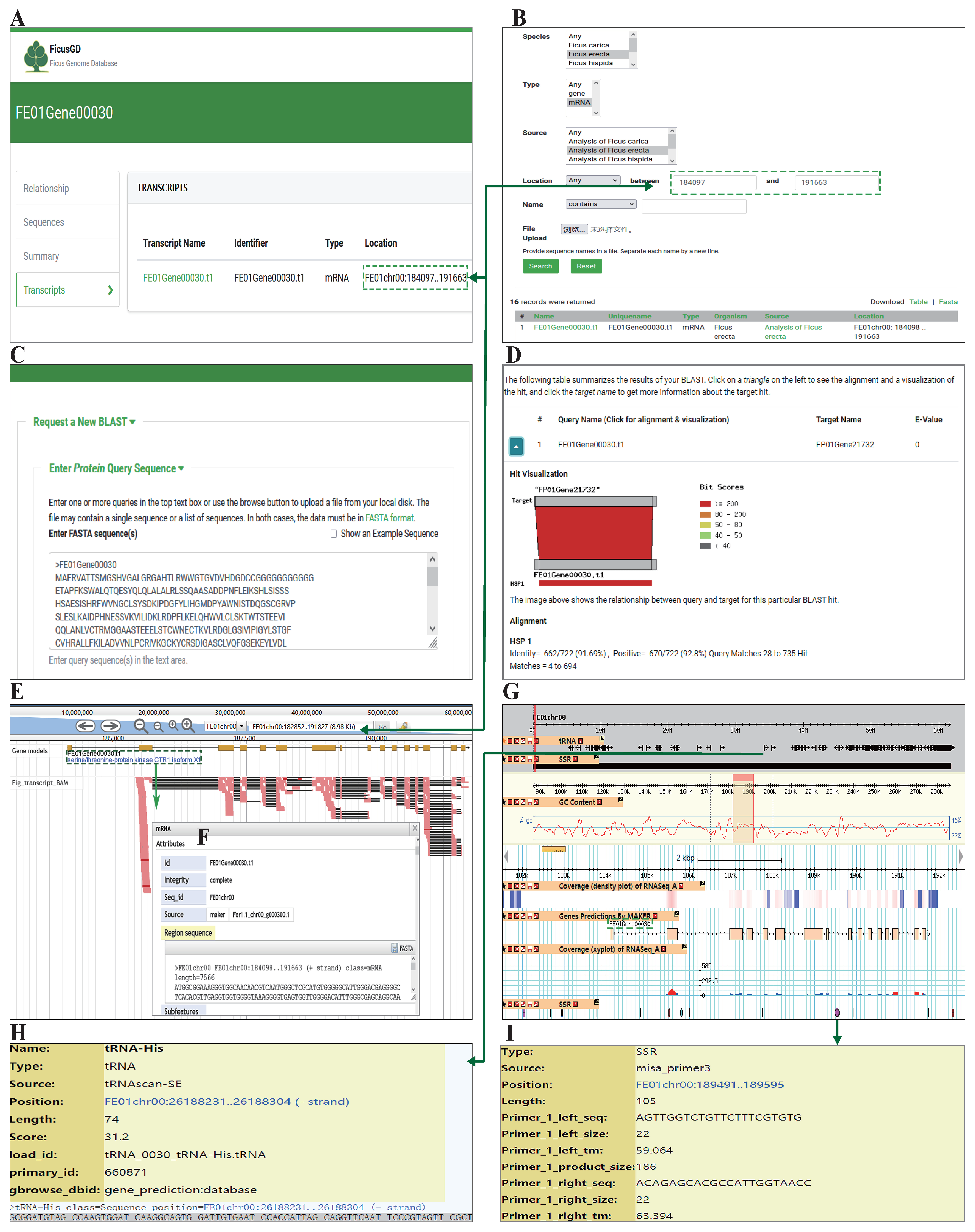
3.2. Search and Download
3.3. Homology Alignment and Searching
3.4. Genome Browsers
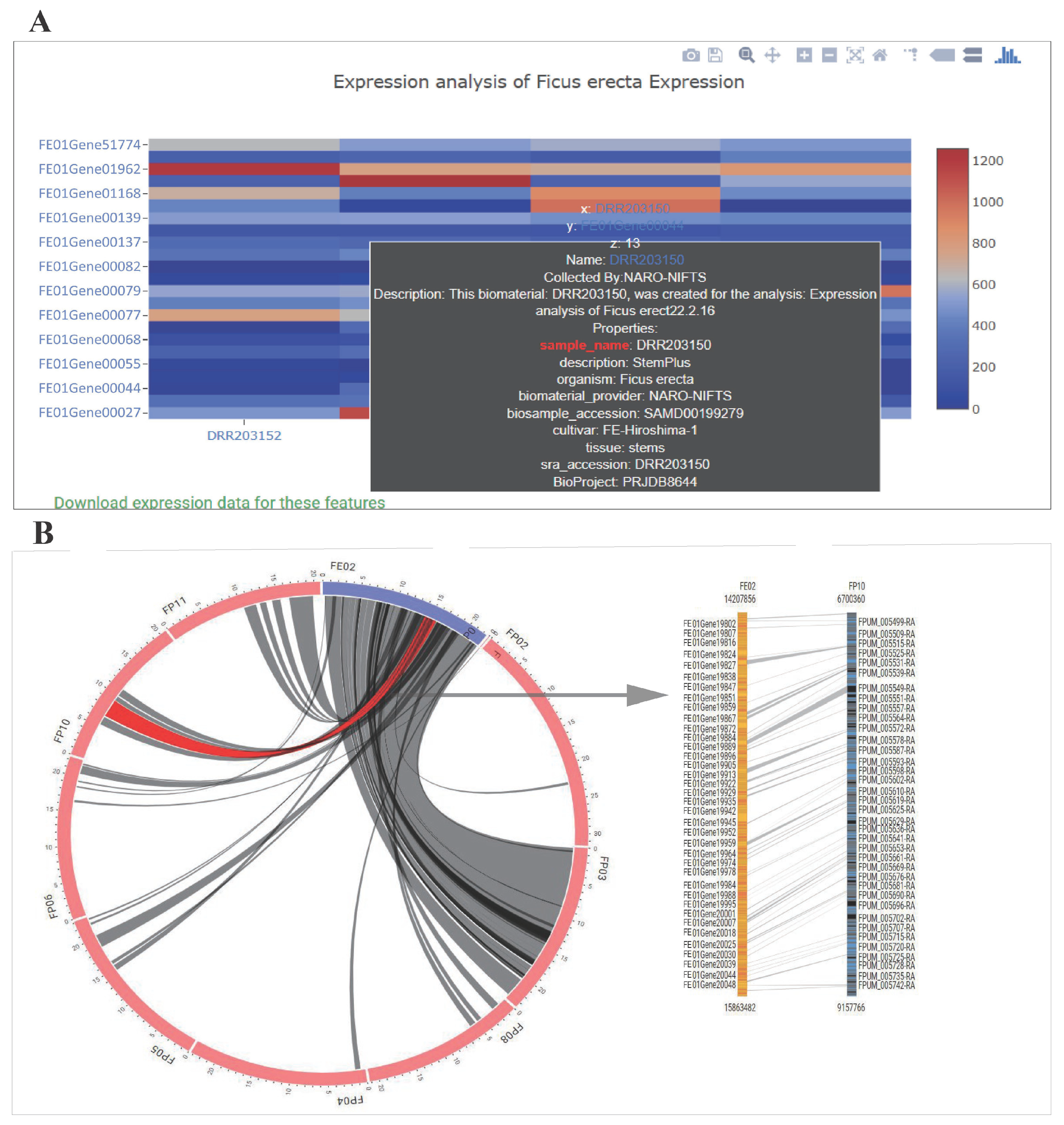
3.5. Expression Heatmap Exhibition
3.6. Synteny Viewer
3.7. Enrichment Analysis
3.8. Annotation Exhibition, FicusCyc, Primer3, and TPS Family
3.9. Download and Help
4. Conclusions and Future Directions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bain, A.; Tzeng, H.Y.; Wu, W.J.; Chou, L.S. Ficus (Moraceae) and fig wasps (Hymenoptera: Chalcidoidea) in Taiwan. Bot. Stud. 2015, 56, 11. [Google Scholar] [CrossRef] [PubMed]
- Barolo, M.I.; Ruiz Mostacero, N.; Lopez, S.N. Ficus carica L. (Moraceae): An ancient source of food and health. Food Chem. 2014, 164, 119–127. [Google Scholar] [CrossRef] [PubMed]
- Deepa, P.; Sowndhararajan, K.; Kim, S.; Park, S.J. A role of Ficus species in the management of diabetes mellitus: A review. J. Ethnopharmacol. 2018, 215, 210–232. [Google Scholar] [CrossRef] [PubMed]
- Mori, K.; Shirasawa, K.; Nogata, H.; Hirata, C.; Tashiro, K.; Habu, T.; Kim, S.; Himeno, S.; Kuhara, S.; Ikegami, H. Identification of RAN1 orthologue associated with sex determination through whole genome sequencing analysis in fig (Ficus carica L.). Sci. Rep. 2017, 7, 41124. [Google Scholar] [CrossRef] [PubMed]
- Mawa, S.; Husain, K.; Jantan, I. Ficus carica L. (Moraceae): Phytochemistry, Traditional Uses and Biological Activities. Evid. Based Complement. Altern. Med. 2013, 2013, 974256. [Google Scholar] [CrossRef] [PubMed]
- Cruaud, A.; Ronsted, N.; Chantarasuwan, B.; Chou, L.S.; Clement, W.L.; Couloux, A.; Cousins, B.; Genson, G.; Harrison, R.D.; Hanson, P.E.; et al. An extreme case of plant-insect codiversification: Figs and fig-pollinating wasps. Syst. Biol. 2012, 61, 1029–1047. [Google Scholar] [CrossRef] [PubMed]
- Hu, R.; Sun, P.; Yu, H.; Cheng, Y.; Wang, R.; Chen, X.; Kjellberg, F. Similitudes and differences between two closely related Ficus species in the synthesis by the ostiole of odors attracting their host-specific pollinators: A transcriptomic based investigation. Acta Oecologica 2020, 105, 103554. [Google Scholar] [CrossRef]
- Hossaert-McKey, M.; Soler, C.; Schatz, B.; Proffit, M. Floral scents: Their roles in nursery pollination mutualisms. Chemoecology 2010, 20, 75–88. [Google Scholar] [CrossRef]
- Proffit, M.; Johnson, S.D. Specificity of the signal emitted by figs to attract their pollinating wasps: Comparison of volatile organic compounds emitted by receptive syconia of Ficus sur and F. sycomorus in Southern Africa. S. Afr. J. Bot. 2009, 75, 771–777. [Google Scholar] [CrossRef]
- Souza, C.D.; Pereira, R.A.S.; Marinho, C.R.; Kjellberg, F.; Teixeira, S.P. Diversity of fig glands is associated with nursery mutualism in fig trees. Am. J. Bot. 2015, 102, 1564–1577. [Google Scholar] [CrossRef]
- Salehi, B.; Prakash Mishra, A.; Nigam, M.; Karazhan, N.; Shukla, I.; Kieltyka-Dadasiewicz, A.; Sawicka, B.; Glowacka, A.; Abu-Darwish, M.S.; Hussein Tarawneh, A.; et al. Ficus plants: State of the art from a phytochemical, pharmacological, and toxicological perspective. Phytother. Res. 2021, 35, 1187–1217. [Google Scholar] [CrossRef] [PubMed]
- Jung, S.; Lee, T.; Cheng, C.H.; Buble, K.; Zheng, P.; Yu, J.; Humann, J.; Ficklin, S.P.; Gasic, K.; Scott, K.; et al. 15 years of GDR: New data and functionality in the Genome Database for Rosaceae. Nucleic Acids Res. 2019, 47, D1137–D1145. [Google Scholar] [CrossRef] [PubMed]
- Humann, J.L.; Cheng, C.H.; Lee, T.; Buble, K.; Jung, S.; Yu, J.; Zheng, P.; Hough, H.; Crabb, J.; Frank, M.; et al. Using the Genome Database for Vaccinium for genetics, genomics, and breeding research. In Proceedings of the XII International Vaccinium Symposium 1357, Debert, NS, Canada, 30 August–1 September 2021; ISHS: Bierbeek, Belgium, 2023; pp. 115–122. [Google Scholar]
- Yu, J.; Jung, S.; Cheng, C.H.; Ficklin, S.P.; Lee, T.; Zheng, P.; Jones, D.; Percy, R.G.; Main, D. CottonGen: A genomics, genetics and breeding database for cotton research. Nucleic Acids Res. 2014, 42, D1229–D1236. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Wu, S.; Sun, H.; Wang, X.; Tang, X.; Guo, S.; Zhang, Z.; Huang, S.; Xu, Y.; Weng, Y.; et al. CuGenDBv2: An updated database for cucurbit genomics. Nucleic Acids Res. 2023, 51, D1457–D1464. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Jung, S.; Cheng, C.-H.; Lee, T.; Zheng, P.; Buble, K.; Crabb, J.; Humann, J.; Hough, H.; Jones, D.; et al. CottonGen: The Community Database for Cotton Genomics, Genetics, and Breeding Research. Plants 2021, 10, 2805. [Google Scholar] [CrossRef]
- Chen, B.; Yu, T.; Xie, S.; Du, K.; Liang, X.; Lan, Y.; Sun, C.; Lu, X.; Shao, Y. Comparative shotgun metagenomic data of the silkworm Bombyx mori gut microbiome. Sci. Data 2018, 5, 180285. [Google Scholar] [CrossRef]
- Joshi, P.; Banerjee, S.; Hu, X.; Khade, P.M.; Friedberg, I. GOThresher: A program to remove annotation biases from protein function annotation datasets. Bioinformatics 2023, 39, btad048. [Google Scholar] [CrossRef]
- Conesa, A.; Gotz, S. Blast2GO: A comprehensive suite for functional analysis in plant genomics. Int. J. Plant Genom. 2008, 2008, 619832. [Google Scholar] [CrossRef]
- Feng, Y.; Zou, S.; Chen, H.; Yu, Y.; Ruan, Z. BacWGSTdb 2.0: A one-stop repository for bacterial whole-genome sequence typing and source tracking. Nucleic Acids Res. 2021, 49, D644–D650. [Google Scholar] [CrossRef]
- Sun, P.; Chen, X.; Chantarasuwan, B.; Zhu, X.; Deng, X.; Bao, Y.; Yu, H. Composition Diversity and Expression Specificity of the TPS Gene Family among 24 Ficus Species. Diversity 2022, 14, 721. [Google Scholar] [CrossRef]
- Liyanage, N.M.N.; Chandrasekara, B.; Bandaranayake, P.C.G. A CTAB protocol for obtaining high-quality total RNA from cinnamon (Cinnamomum zeylanicum Blume). 3 Biotech 2021, 11, 201. [Google Scholar] [CrossRef] [PubMed]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [PubMed]
- Haas, B.J.; Papanicolaou, A.; Yassour, M.; Grabherr, M.; Blood, P.D.; Bowden, J.; Couger, M.B.; Eccles, D.; Li, B.; Lieber, M.; et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 2013, 8, 1494–1512. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Tang, H.; Debarry, J.D.; Tan, X.; Li, J.; Wang, X.; Lee, T.H.; Jin, H.; Marler, B.; Guo, H.; et al. MCScanX: A toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 2012, 40, e49. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef]
- You, Q.; Xu, W.; Zhang, K.; Zhang, L.; Yi, X.; Yao, D.; Wang, C.; Zhang, X.; Zhao, X.; Provart, N.J.; et al. ccNET: Database of co-expression networks with functional modules for diploid and polyploid Gossypium. Nucleic Acids Res. 2017, 45, D1090–D1099. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.; Pirrung, M.; McCue, L.A. FQC Dashboard: Integrates FastQC results into a web-based, interactive, and extensible FASTQ quality control tool. Bioinformatics 2017, 33, 3137–3139. [Google Scholar] [CrossRef] [PubMed]
- Sewe, S.O.; Silva, G.; Sicat, P.; Seal, S.E.; Visendi, P. Trimming and Validation of Illumina Short Reads Using Trimmomatic, Trinity Assembly, and Assessment of RNA-Seq Data. Methods Mol. Biol. 2022, 2443, 211–232. [Google Scholar] [CrossRef]
- Pertea, M.; Kim, D.; Pertea, G.M.; Leek, J.T.; Salzberg, S.L. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat. Protoc. 2016, 11, 1650–1667. [Google Scholar] [CrossRef]
- Chan, P.P.; Lin, B.Y.; Mak, A.J.; Lowe, T.M. tRNAscan-SE 2.0: Improved detection and functional classification of transfer RNA genes. Nucleic Acids Res. 2021, 49, 9077–9096. [Google Scholar] [CrossRef]
- Tang, H.; Saina, J.K.; Long, Z.C.; Chen, J.; Dai, C. De novo transcriptome assembly using Illumina sequencing and development of EST-SSR markers in a monoecious herb Sagittaria trifolia Linn. PeerJ 2022, 10, e14268. [Google Scholar] [CrossRef] [PubMed]
- Beier, S.; Thiel, T.; Munch, T.; Scholz, U.; Mascher, M. MISA-web: A web server for microsatellite prediction. Bioinformatics 2017, 33, 2583–2585. [Google Scholar] [CrossRef]
- Caspi, R.; Altman, T.; Billington, R.; Dreher, K.; Foerster, H.; Fulcher, C.A.; Holland, T.A.; Keseler, I.M.; Kothari, A.; Kubo, A.; et al. The MetaCyc database of metabolic pathways and enzymes and the BioCyc collection of Pathway/Genome Databases. Nucleic Acids Res. 2014, 42, D459–D471. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Papanicolaou, A.; Heckel, D.G. The GMOD Drupal Bioinformatic Server Framework. Bioinformatics 2010, 26, 3119–3124. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Henry, N.; Almsaeed, A.; Zhou, X.; Wegrzyn, J.; Ficklin, S.; Staton, M. New extension software modules to enhance searching and display of transcriptome data in Tripal databases. Database 2017, 2017, bax052. [Google Scholar] [CrossRef]
- Yue, J.; Liu, J.; Tang, W.; Wu, Y.Q.; Tang, X.; Li, W.; Yang, Y.; Wang, L.; Huang, S.; Fang, C.; et al. Kiwifruit Genome Database (KGD): A comprehensive resource for kiwifruit genomics. Hortic. Res. 2020, 7, 117. [Google Scholar] [CrossRef]
- Elsik, C.G.; Tayal, A.; Diesh, C.M.; Unni, D.R.; Emery, M.L.; Nguyen, H.N.; Hagen, D.E. Hymenoptera Genome Database: Integrating genome annotations in HymenopteraMine. Nucleic Acids Res. 2016, 44, D793–D800. [Google Scholar] [CrossRef] [PubMed]
- Buble, K.; Jung, S.; Humann, J.L.; Yu, J.; Cheng, C.H.; Lee, T.; Ficklin, S.P.; Hough, H.; Condon, B.; Staton, M.E.; et al. Tripal MapViewer: A tool for interactive visualization and comparison of genetic maps. Database 2019, 2019, baz100. [Google Scholar] [CrossRef]
- Jung, S.; Cheng, C.H.; Buble, K.; Lee, T.; Humann, J.; Yu, J.; Crabb, J.; Hough, H.; Main, D. Tripal MegaSearch: A tool for interactive and customizable query and download of big data. Database 2021, 2021, baab023. [Google Scholar] [CrossRef]
- Youens-Clark, K.; Faga, B.; Yap, I.V.; Stein, L.; Ware, D. CMap 1.01: A comparative mapping application for the Internet. Bioinformatics 2009, 25, 3040–3042. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Zhang, L.; Zhou, Y.; Tu, M.; Wu, Z.; Gui, D.; Ma, Y.; Wang, J.; Zhang, C. The Rhododendron Plant Genome Database (RPGD): A comprehensive online omics database for Rhododendron. BMC Genom. 2021, 22, 376. [Google Scholar] [CrossRef] [PubMed]
- Sanderson, L.A.; Ficklin, S.P.; Cheng, C.H.; Jung, S.; Feltus, F.A.; Bett, K.E.; Main, D. Tripal v1.1: A standards-based toolkit for construction of online genetic and genomic databases. Database 2013, 2013, bat075. [Google Scholar] [CrossRef] [PubMed]
- Gui, S.; Yang, L.; Li, J.; Luo, J.; Xu, X.; Yuan, J.; Chen, L.; Li, W.; Yang, X.; Wu, S.; et al. ZEAMAP, a Comprehensive Database Adapted to the Maize Multi-Omics Era. iScience 2020, 23, 101241. [Google Scholar] [CrossRef]
- Memczak, S.; Jens, M.; Elefsinioti, A.; Torti, F.; Krueger, J.; Rybak, A.; Maier, L.; Mackowiak, S.D.; Gregersen, L.H.; Munschauer, M.; et al. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature 2013, 495, 333–338. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Data Type | Entries No. | Details |
|---|---|---|
| Species | 26 | Origin, genome group, germplasm, sequences and libraries, specific species pages with hyperlinks to various data and tools. |
| Genome | 5 | Whole genome assemblies and annotations from five Ficus *. |
| Transcriptome | 24 | Ositolar bracts transcriptome assemblies from 24 Ficus receptive figs **. |
| Unigenes | 702,402 | All assembled unigenes from 24 Ficus transcriptome. |
| Gene and mRNA | 171,752 genes and 171,867 mRNAs | Genes and mRNAs from five Ficus * whole genome assemblies and parsed from NCBI nucleotide sequences. |
| Annotations | 5 Nr and Swissprot, 1 Interpro | Annotated to Nr and Swissprot for five Ficus *, Interpro Annotation for F. erecta var. erecta, imported into the Chado database. |
| SRA project | 3 | PRJNA623468, PRJNA397979, PRJDB8644 Project of SRA database. |
| Syntenic blocks | 425,439 | 425,439 homologous gene pairs of five Ficus * genomes. |
| tRNA | 3526 | tRNA for five Ficus * species. |
| SSRs | 563,787 | Simple Sequence Repeats (SSRs) for five Ficus * species. |
| Species | Genome Version | Assembled Size (Mb) | Ploidy | Scaffold N50 | Busco V5 (%) | Gene No. | mRNA No. | Protein No. | SRA Project |
|---|---|---|---|---|---|---|---|---|---|
| F. carica L. | V1 | 247.1 | 2n = 2x = 26 | 166.1 kb | 95.1 | 36,107 | 36,115 | 36,115 | PRJNA623468 PRJNA397979 |
| F. erecta var. erecta | V201909 | 595.8 | 2n = 2x = 26 | / | 94.6 | 51,801 | 51,826 | 51,826 | PRJDB8644 |
| F. hispida L.f. | V202209 | 369.8 | 2n = 2x = 28 | 24.1 Mb | 97.2 | 27,207 | 27,247 | 27,247 | / |
| F. microcarpa L.f. | V202209 | 426.6 | 2n = 2x = 26 | 29.3 Mb | 94.8 | 28,453 | 28,478 | 28,478 | / |
| F. pumila var. pumila | V202105 | 315.7 | 2n = 2x = 26 | 2.3 Mb | 96.6 | 28,184 | 28,201 | 28,201 | / |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, P.; Yang, L.; Yu, H.; Chen, L.; Bao, Y. Ficus Genome Database: A Comprehensive Genomics and Transcriptomics Research Platform. Horticulturae 2024, 10, 613. https://doi.org/10.3390/horticulturae10060613
Sun P, Yang L, Yu H, Chen L, Bao Y. Ficus Genome Database: A Comprehensive Genomics and Transcriptomics Research Platform. Horticulturae. 2024; 10(6):613. https://doi.org/10.3390/horticulturae10060613
Chicago/Turabian StyleSun, Peng, Lei Yang, Hui Yu, Lianfu Chen, and Ying Bao. 2024. "Ficus Genome Database: A Comprehensive Genomics and Transcriptomics Research Platform" Horticulturae 10, no. 6: 613. https://doi.org/10.3390/horticulturae10060613