
Interspecies Conservation of Gene Expression Patterns in Brassica Reproductive Organs Unveiled by Comparative Transcriptomics






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Transcriptome and Reference Genome Data Collection
2.2. RNA-Seq Data Processing
2.3. Organ Specific Genes (SPM Genes) Identification
2.4. Calculation of Jaccard Coefficients (JCCs) Between Organs of Different Genomes and Subgenomes
2.5. Enrichment Analysis of Gene Regulatory Pathways and TFs
2.6. Phylogenetic Analysis of TF Families
2.7. Image Plotting
3. Results
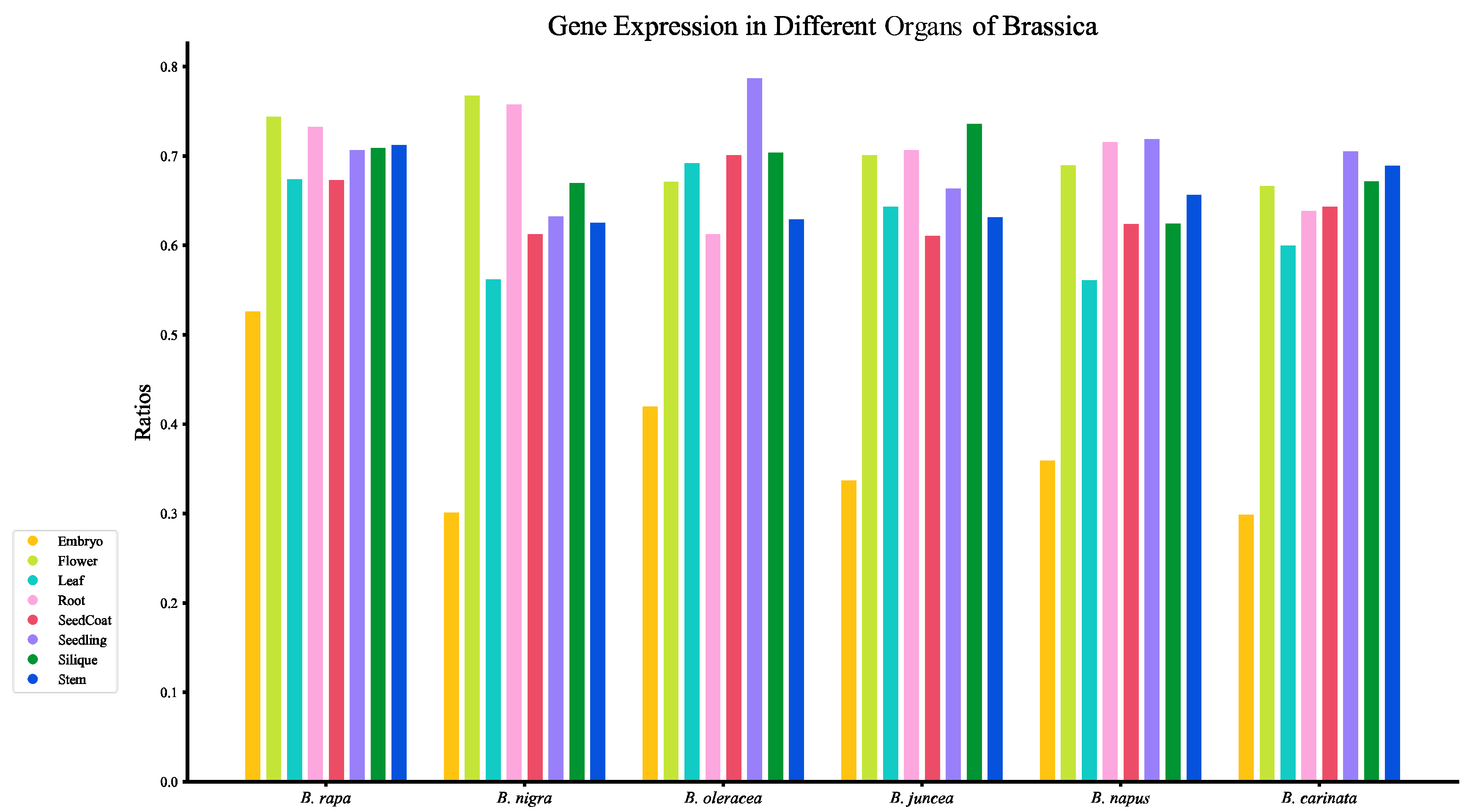
3.1. Multi-Organ Gene Expression Atlas of 6 Brassica Species
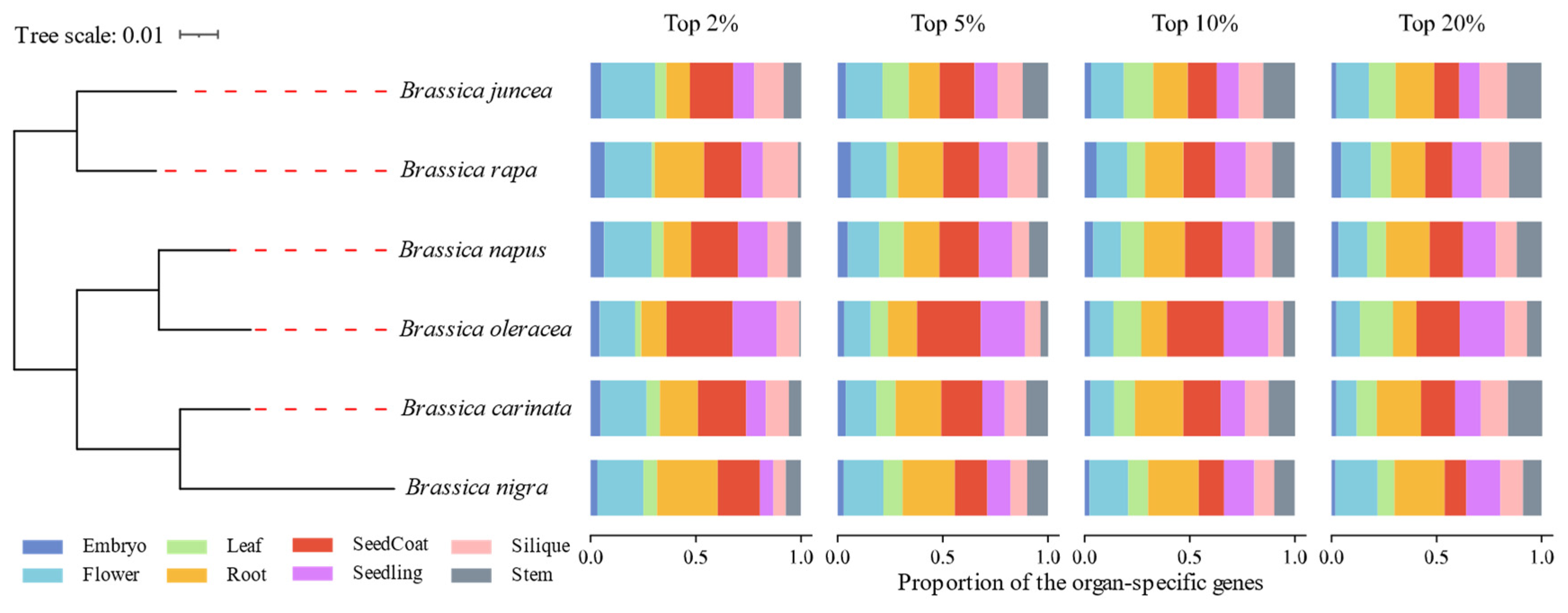
3.2. Conserved SPM Genes Allocation Patterns
3.3. Prominent GEC in the Reproductive Organs of Brassica
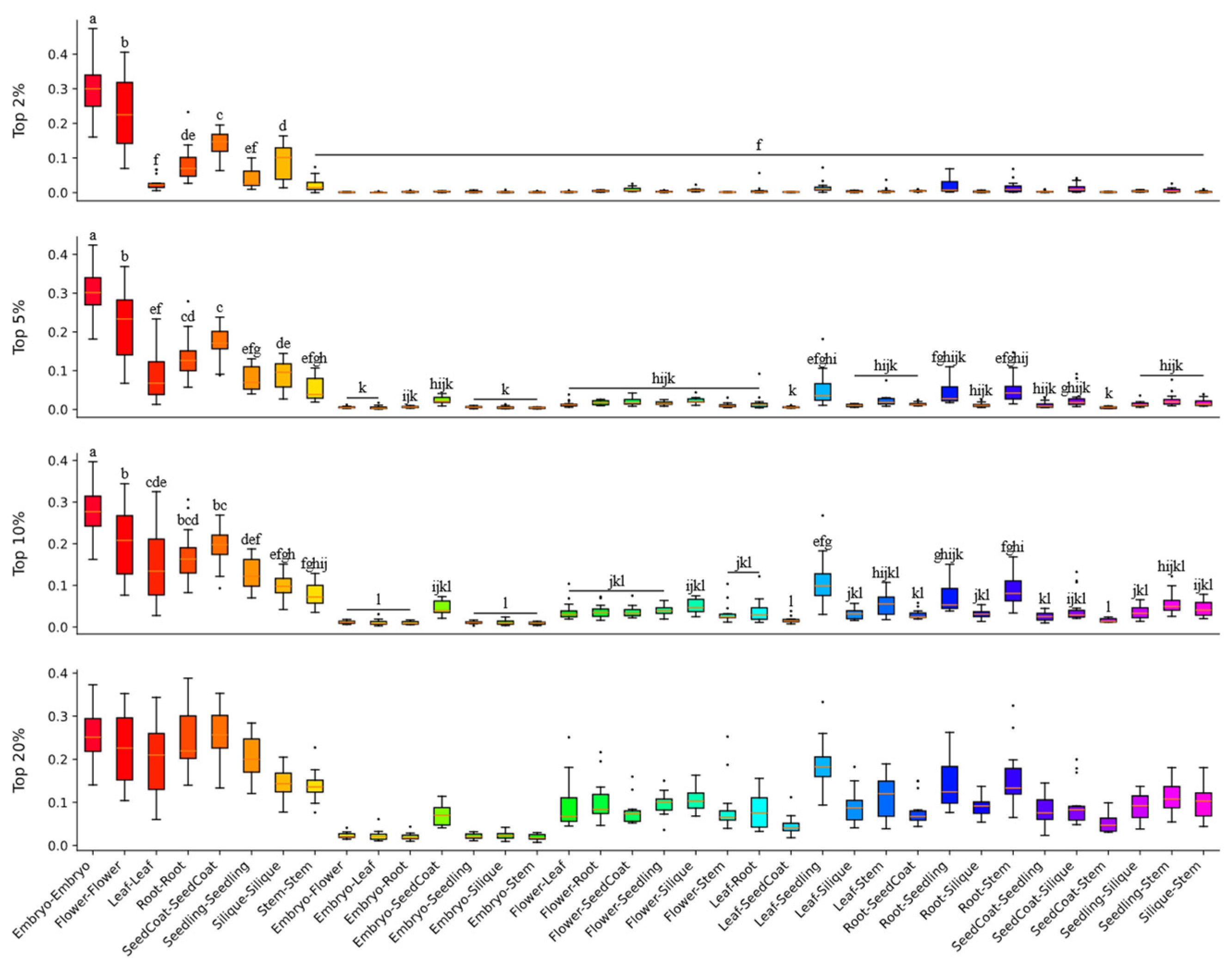
3.3.1. Significant GEC in Reproductive Organs Across Brassica Genomes
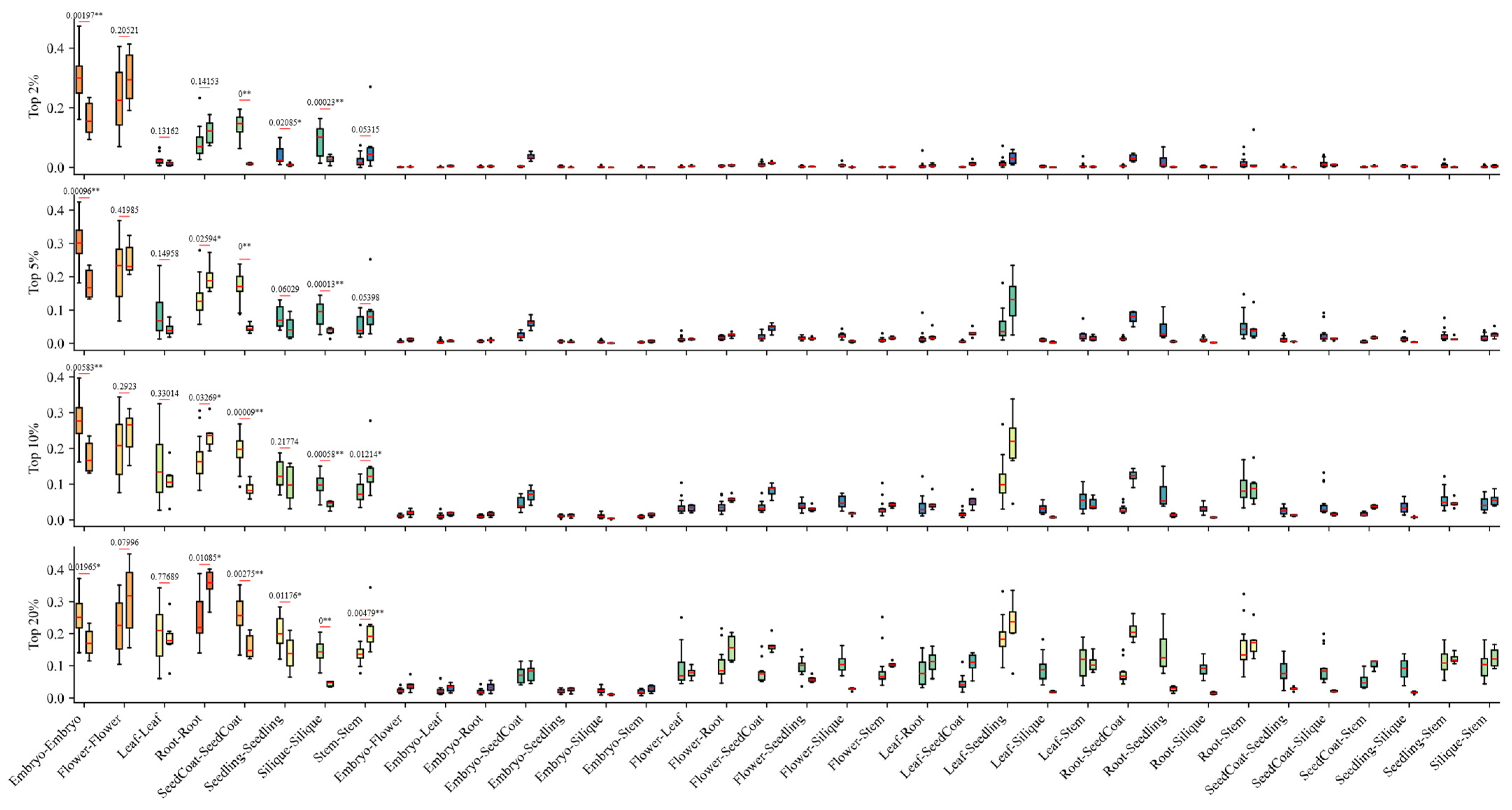
3.3.2. Significant GEC in Reproductive Organs Across Brassica Subgenomes
3.4. Significant GEC in Reproductive Organs Between Brassica and A. thaliana
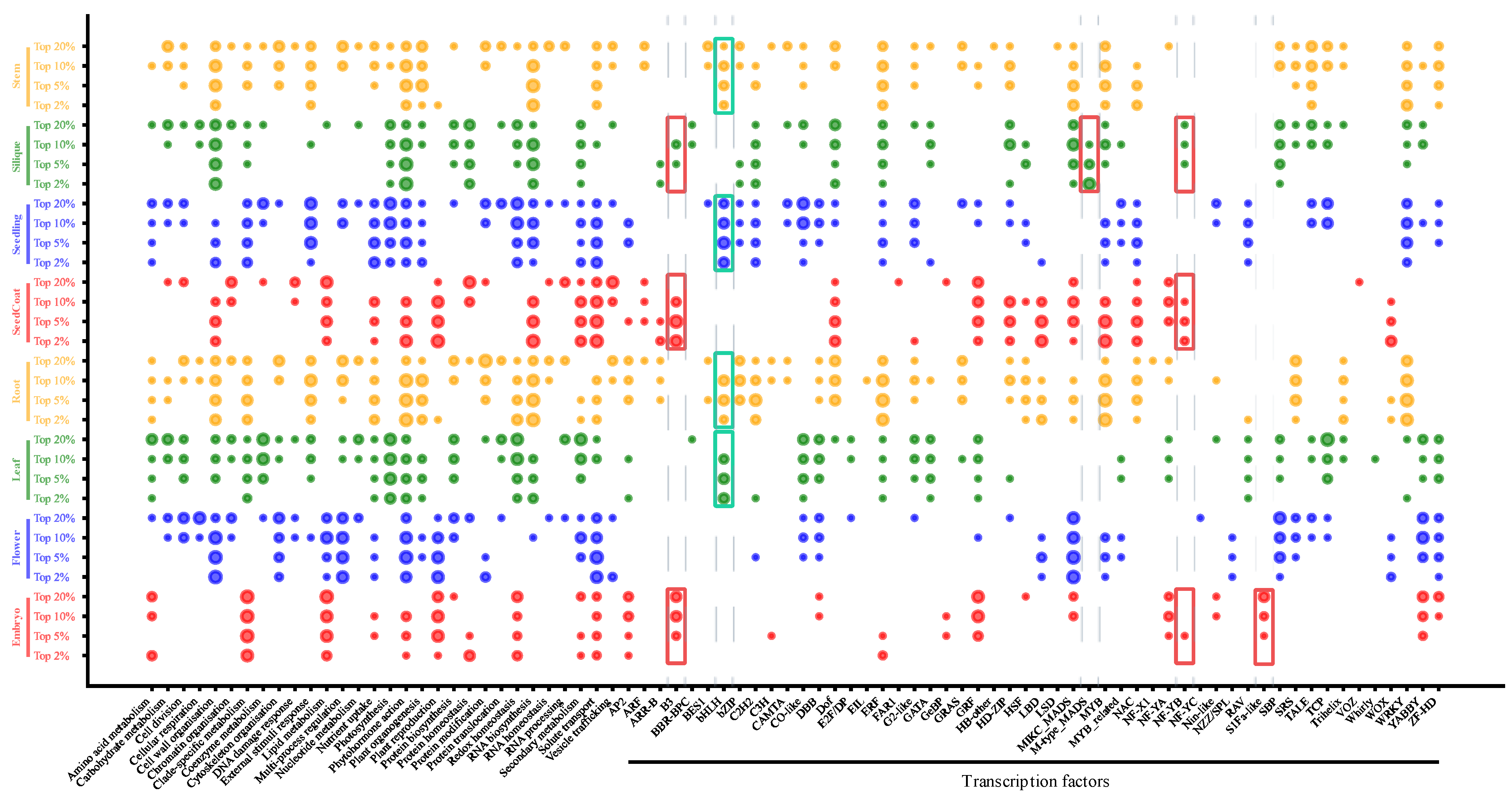
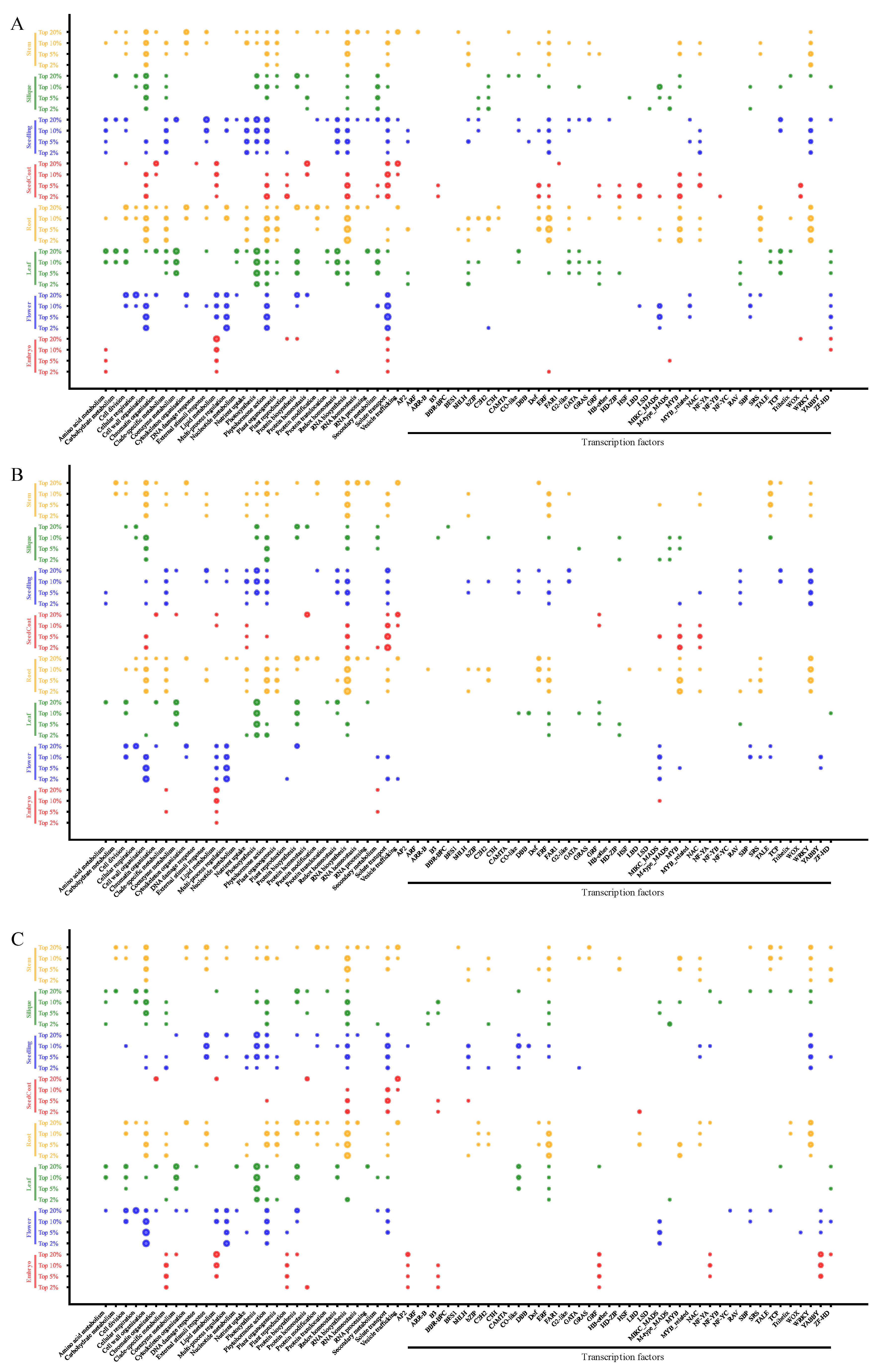
3.5. Enrichment Analysis of Gene Regulatory Pathways and TFs of SPM Genes
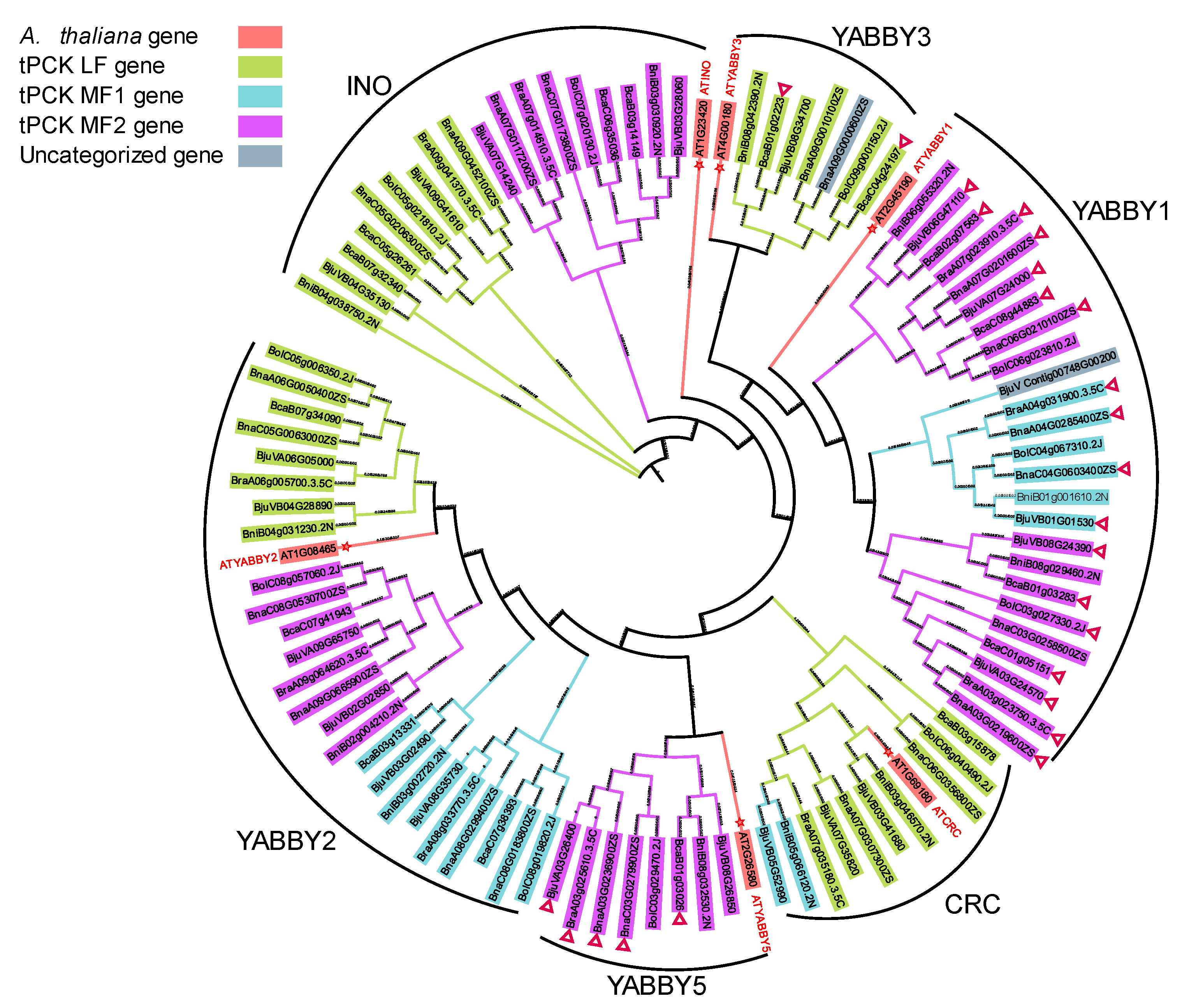
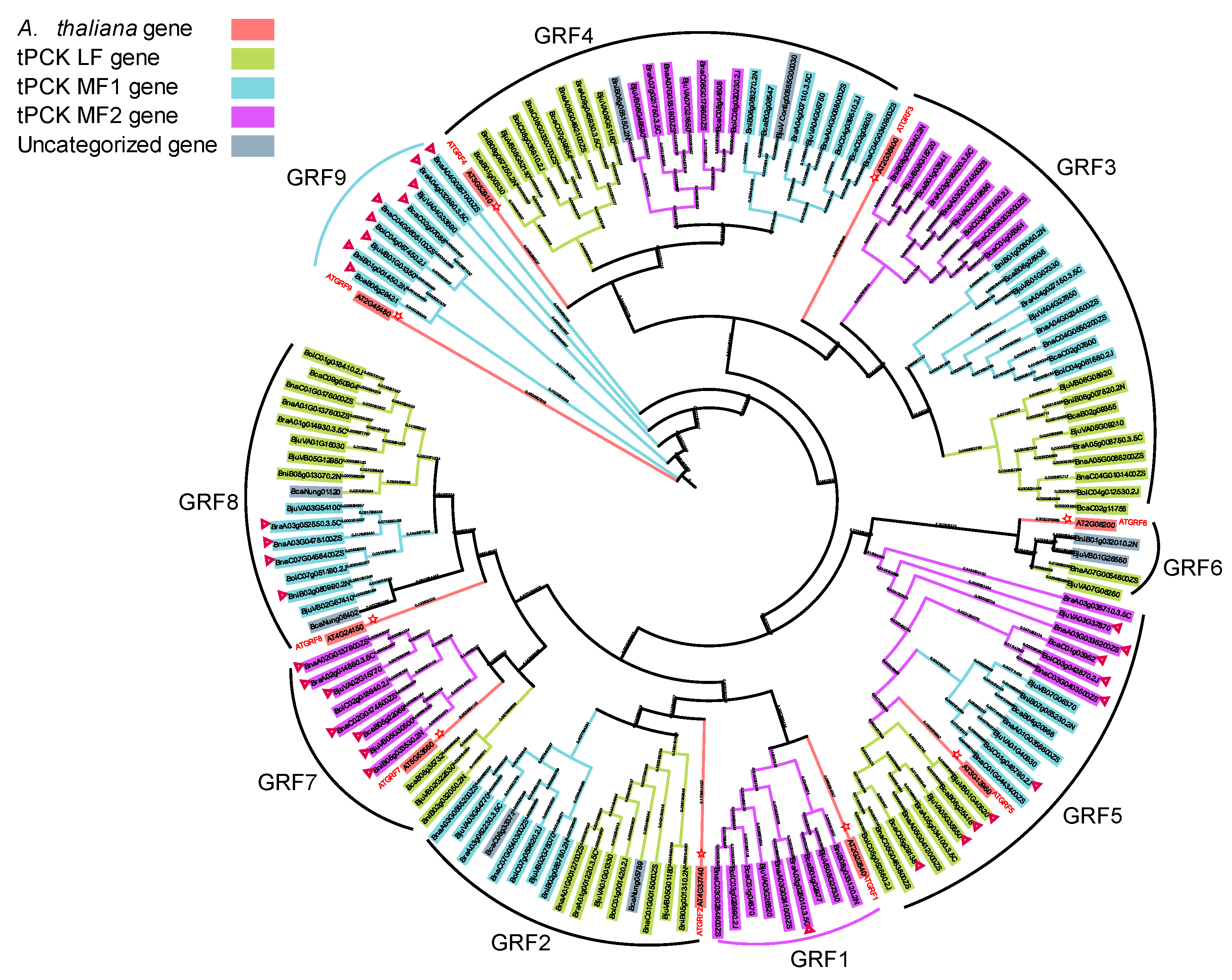
3.6. Phylogenetic Analysis of the YABBY and GRF TF Families in Brassica
3.6.1. YABBY1 and YABBY5 Dominantly Expressed in Embryo
3.6.2. GRF5, 7, 8 and 9 Dominantly Expressed in Embryo
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
| GEC | gene expression conservation |
| TPM | transcripts per million |
| SPM | specificity measure |
| JCC | Jaccard coefficient |
References
- Branca, F.; Cartea, E. Brassica. In Wild Crop Relatives: Genomic and Breeding Resources: Oilseeds; Kole, C., Ed.; Springer: Berlin/Heidelberg, Germany, 2011; pp. 17–36. [Google Scholar]
- Nagaharu, U. Genome analysis in Brassica with special reference to the experimental formation of B. napus and peculiar mode of fertilization. Jpn. J. Bot. 1935, 7, 389–452. [Google Scholar]
- Cai, X.; Chang, L.; Zhang, T.; Chen, H.; Zhang, L.; Lin, R.; Liang, J.; Wu, J.; Freeling, M.; Wang, X. Impacts of allopolyploidization and structural variation on intraspecific diversification in Brassica rapa. Genome Biol. 2021, 22, 166. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wang, Y.; Cai, C.; Ji, J.; Han, F.; Zhang, L.; Chen, S.; Zhang, L.; Yang, Y.; Tang, Q.; et al. Large-scale gene expression alterations introduced by structural variation drive morphotype diversification in Brassica oleracea. Nat. Genet. 2024, 56, 517–529. [Google Scholar] [CrossRef] [PubMed]
- Song, J.-M.; Guan, Z.; Hu, J.; Guo, C.; Yang, Z.; Wang, S.; Liu, D.; Wang, B.; Lu, S.; Zhou, R.; et al. Eight high-quality genomes reveal pan-genome architecture and ecotype differentiation of Brassica napus. Nat. Plants 2020, 6, 34–45. [Google Scholar] [CrossRef]
- Perumal, S.; Koh, C.S.; Jin, L.; Buchwaldt, M.; Higgins, E.E.; Zheng, C.; Sankoff, D.; Robinson, S.J.; Kagale, S.; Navabi, Z.-K.; et al. A high-contiguity Brassica nigra genome localizes active centromeres and defines the ancestral Brassica genome. Nat. Plants 2020, 6, 929–941. [Google Scholar] [CrossRef]
- Kang, L.; Qian, L.; Zheng, M.; Chen, L.; Chen, H.; Yang, L.; You, L.; Yang, B.; Yan, M.; Gu, Y.; et al. Genomic insights into the origin, domestication and diversification of Brassica juncea. Nat. Genet. 2021, 53, 1392–1402. [Google Scholar] [CrossRef]
- Yang, J.; Wang, J.; Li, Z.; Li, X.; He, Z.; Zhang, L.; Sha, T.; Lyu, X.; Chen, S.; Gu, Y.; et al. Genomic signatures of vegetable and oilseed allopolyploid Brassica juncea and genetic loci controlling the accumulation of glucosinolates. Plant Biotechnol. J. 2021, 19, 2619–2628. [Google Scholar] [CrossRef]
- Niu, Y.; Liu, Q.; He, Z.; Raman, R.; Wang, H.; Long, X.; Qin, H.; Raman, H.; Parkin, I.A.; Bancroft, I. A Brassica carinata pan-genome platform for Brassica crop improvement. Plant Commun. 2024, 5, 100725. [Google Scholar] [CrossRef]
- Song, X.; Wei, Y.; Xiao, D.; Gong, K.; Sun, P.; Ren, Y.; Yuan, J.; Wu, T.; Yang, Q.; Li, X.; et al. Brassica carinata genome characterization clarifies U’s triangle model of evolution and polyploidy in Brassica. Plant Physiol. 2021, 186, 388–406. [Google Scholar] [CrossRef]
- Yim, W.C.; Swain, M.L.; Ma, D.; An, H.; Bird, K.A.; Curdie, D.D.; Wang, S.; Ham, H.D.; Luzuriaga-Neira, A.; Kirkwood, J.S.; et al. The final piece of the Triangle of U: Evolution of the tetraploid Brassica carinata genome. Plant Cell 2022, 34, 4143–4172. [Google Scholar] [CrossRef]
- Town, C.D.; Cheung, F.; Maiti, R.; Crabtree, J.; Haas, B.J.; Wortman, J.R.; Hine, E.E.; Althoff, R.; Arbogast, T.S.; Tallon, L.J.; et al. Comparative Genomics of Brassica oleracea and Arabidopsis thaliana Reveal Gene Loss, Fragmentation, and Dispersal after Polyploidy. Plant Cell 2006, 18, 1348–1359. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.-W.; Lai, K.-N.; Tai, P.-Y.; Li, W.-H. Rates of Nucleotide Substitution in Angiosperm Mitochondrial DNA Sequences and Dates of Divergence Between Brassica and Other Angiosperm Lineages. J. Mol. Evol. 1999, 48, 597–604. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wang, H.; Wang, J.; Sun, R.; Wu, J.; Liu, S.; Bai, Y.; Mun, J.-H.; Bancroft, I.; Cheng, F.; et al. The genome of the mesopolyploid crop species Brassica rapa. Nat. Genet. 2011, 43, 1035–1039. [Google Scholar] [CrossRef]
- Cheng, F.; Wu, J.; Fang, L.; Sun, S.; Liu, B.; Lin, K.; Bonnema, G.; Wang, X. Biased Gene Fractionation and Dominant Gene Expression among the Subgenomes of Brassica rapa. PLoS ONE 2012, 7, e36442. [Google Scholar] [CrossRef]
- Wang, Z.; Yang, J.; Cheng, F.; Li, P.; Xin, X.; Wang, W.; Yu, Y.; Zhang, D.; Zhao, X.; Yu, S.; et al. Subgenome dominance and its evolutionary implications in crop domestication and breeding. Hortic. Res. 2022, 9, uhac090. [Google Scholar] [CrossRef]
- Yang, H.; Liu, J.; Huang, S.; Guo, T.; Deng, L.; Hua, W. Selection and evaluation of novel reference genes for quantitative reverse transcription PCR (qRT-PCR) based on genome and transcriptome data in Brassica napus L. Gene 2014, 538, 113–122. [Google Scholar] [CrossRef]
- Privitera, G.F.; Treccarichi, S.; Nicotra, R.; Branca, F.; Pulvirenti, A.; Lo Piero, A.R.; Sicilia, A. Comparative transcriptome analysis of B. oleracea L. var. italica and B. macrocarpa Guss. genotypes under drought stress: De novo vs reference genome assembly. Plant Stress 2024, 14, 100657. [Google Scholar] [CrossRef]
- Singh, K.P.; Kumari, P.; Yadava, D.K. Development of de-novo transcriptome assembly and SSRs in allohexaploid Brassica with functional annotations and identification of heat-shock proteins for thermotolerance. Front. Genet. 2022, 13, 958217. [Google Scholar] [CrossRef]
- An, H.; Qi, X.; Gaynor, M.L.; Hao, Y.; Gebken, S.C.; Mabry, M.E.; McAlvay, A.C.; Teakle, G.R.; Conant, G.C.; Barker, M.S.; et al. Transcriptome and organellar sequencing highlights the complex origin and diversification of allotetraploid Brassica napus. Nat. Commun. 2019, 10, 2878. [Google Scholar] [CrossRef]
- Parkin, I.A.P.; Koh, C.; Tang, H.; Robinson, S.J.; Kagale, S.; Clarke, W.E.; Town, C.D.; Nixon, J.; Krishnakumar, V.; Bidwell, S.L.; et al. Transcriptome and methylome profiling reveals relics of genome dominance in the mesopolyploid Brassica oleracea. Genome Biol. 2014, 15, R77. [Google Scholar] [CrossRef]
- Tong, C.; Wang, X.; Yu, J.; Wu, J.; Li, W.; Huang, J.; Dong, C.; Hua, W.; Liu, S. Comprehensive analysis of RNA-seq data reveals the complexity of the transcriptome in Brassica rapa. BMC Genom. 2013, 14, 689. [Google Scholar] [CrossRef] [PubMed]
- Ziegler, D.J.; Khan, D.; Kalichuk, J.L.; Becker, M.G.; Belmonte, M.F. Transcriptome landscape of the early Brassica napus seed. J. Integr. Plant Biol. 2019, 61, 639–650. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Liang, J.; Lin, R.; Zhang, L.; Zhang, Z.; Wu, J.; Wang, X. Single-cell transcriptome reveals differentiation between adaxial and abaxial mesophyll cells in Brassica rapa. Plant Biotechnol. J. 2022, 20, 2233–2235. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Feng, D.; Liu, M.; Qin, R.; Li, Y.; Lu, Y.; Zhang, X.; Wang, Y.; Shen, S.; Ma, W.; et al. Single-cell transcriptome reveals dominant subgenome expression and transcriptional response to heat stress in Chinese cabbage. Genome Biol. 2022, 23, 262. [Google Scholar] [CrossRef]
- Gao, P.; Quilichini, T.D.; Yang, H.; Li, Q.; Nilsen, K.T.; Qin, L.; Babic, V.; Liu, L.; Cram, D.; Pasha, A.; et al. Evolutionary divergence in embryo and seed coat development of U’s Triangle Brassica species illustrated by a spatiotemporal transcriptome atlas. New Phytol. 2022, 233, 30–51. [Google Scholar] [CrossRef]
- Wang, K.; Wang, D.; Zheng, X.; Qin, A.; Zhou, J.; Guo, B.; Chen, Y.; Wen, X.; Ye, W.; Zhou, Y.; et al. Multi-strategic RNA-seq analysis reveals a high-resolution transcriptional landscape in cotton. Nat. Commun. 2019, 10, 4714. [Google Scholar] [CrossRef]
- Wu, Q.; Tong, W.; Zhao, H.; Ge, R.; Li, R.; Huang, J.; Li, F.; Wang, Y.; Mallano, A.I.; Deng, W.; et al. Comparative transcriptomic analysis unveils the deep phylogeny and secondary metabolite evolution of 116 Camellia plants. Plant J. 2022, 111, 406–421. [Google Scholar] [CrossRef]
- Chen, J.; Hu, Y.; Zhao, T.; Huang, C.; Chen, J.; He, L.; Dai, F.; Chen, S.; Wang, L.; Jin, S.; et al. Comparative transcriptomic analysis provides insights into the genetic networks regulating oil differential production in oil crops. BMC Biol. 2024, 22, 110. [Google Scholar] [CrossRef]
- Libourel, C.; Keller, J.; Brichet, L.; Cazalé, A.-C.; Carrère, S.; Vernié, T.; Couzigou, J.-M.; Callot, C.; Dufau, I.; Cauet, S.; et al. Comparative phylotranscriptomics reveals ancestral and derived root nodule symbiosis programmes. Nat. Plants 2023, 9, 1067–1080. [Google Scholar] [CrossRef]
- Cheng, F.; Mandáková, T.; Wu, J.; Xie, Q.; Lysak, M.A.; Wang, X. Deciphering the Diploid Ancestral Genome of the Mesohexaploid Brassica rapa. Plant Cell 2013, 25, 1541–1554. [Google Scholar] [CrossRef]
- Chen, H.; Wang, T.; He, X.; Cai, X.; Lin, R.; Liang, J.; Wu, J.; King, G.; Wang, X. BRAD V3.0: An upgraded Brassicaceae database. Nucleic Acids Res. 2021, 50, D1432–D1441. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Paggi, J.M.; Park, C.; Bennett, C.; Salzberg, S.L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 2019, 37, 907–915. [Google Scholar] [CrossRef] [PubMed]
- Bray, N.L.; Pimentel, H.; Melsted, P.; Pachter, L. Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 2016, 34, 525–527. [Google Scholar] [CrossRef]
- Julca, I.; Ferrari, C.; Flores-Tornero, M.; Proost, S.; Lindner, A.-C.; Hackenberg, D.; Steinbachová, L.; Michaelidis, C.; Gomes Pereira, S.; Misra, C.S.; et al. Comparative transcriptomic analysis reveals conserved programmes underpinning organogenesis and reproduction in land plants. Nat. Plants 2021, 7, 1143–1159. [Google Scholar] [CrossRef]
- Lohse, M.; Nagel, A.; Herter, T.; May, P.; Schroda, M.; Zrenner, R.; Tohge, T.; Fernie, A.R.; Stitt, M.; Usadel, B. Mercator: A fast and simple web server for genome scale functional annotation of plant sequence data. Plant Cell Environ. 2014, 37, 1250–1258. [Google Scholar] [CrossRef]
- Tian, F.; Yang, D.-C.; Meng, Y.-Q.; Jin, J.; Gao, G. PlantRegMap: Charting functional regulatory maps in plants. Nucleic Acids Res. 2019, 48, D1104–D1113. [Google Scholar] [CrossRef]
- Quinn, G.P.; Keough, M.J. Experimental Design and Data Analysis for Biologists; Cambridge University Press: Cambridge, UK, 2002. [Google Scholar]
- Edgar, R.C. MUSCLE: A multiple sequence alignment method with reduced time and space complexity. BMC Bioinform. 2004, 5, 113. [Google Scholar] [CrossRef]
- Nguyen, L.-T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Mol. Biol. Evol. 2014, 32, 268–274. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive Tree of Life (iTOL) v6: Recent updates to the phylogenetic tree display and annotation tool. Nucleic Acids Res. 2024, 52, W78–W82. [Google Scholar] [CrossRef]
- Bird, K.A.; Niederhuth, C.E.; Ou, S.; Gehan, M.; Pires, J.C.; Xiong, Z.; VanBuren, R.; Edger, P.P. Replaying the evolutionary tape to investigate subgenome dominance in allopolyploid Brassica napus. New Phytol. 2021, 230, 354–371. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Liu, D.; Wang, X.; Ji, C.; Cheng, F.; Liu, B.; Hu, Z.; Chen, S.; Pental, D.; Ju, Y.; et al. The genome sequence of allopolyploid Brassica juncea and analysis of differential homoeolog gene expression influencing selection. Nat. Genet. 2016, 48, 1225–1232. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, A.; Kagaya, Y.; Toyoshima, R.; Kagaya, M.; Takeda, S.; Hattori, T. Arabidopsis NF-YB subunits LEC1 and LEC1-LIKE activate transcription by interacting with seed-specific ABRE-binding factors. Plant J. 2009, 58, 843–856. [Google Scholar] [CrossRef]
- Romanel, E.A.C.; Schrago, C.G.; Couñago, R.M.; Russo, C.A.M.; Alves-Ferreira, M. Evolution of the B3 DNA Binding Superfamily: New Insights into REM Family Gene Diversification. PLoS ONE 2009, 4, e5791. [Google Scholar] [CrossRef]
- Becker, A.; Theißen, G. The major clades of MADS-box genes and their role in the development and evolution of flowering plants. Mol. Phylogenet. Evol. 2003, 29, 464–489. [Google Scholar] [CrossRef]
- Sheng, X.-G.; Zhao, Z.-Q.; Wang, J.-S.; Yu, H.-F.; Shen, Y.-S.; Zeng, X.-Y.; Gu, H.-H. Genome wide analysis of MADS-box gene family in Brassica oleracea reveals conservation and variation in flower development. BMC Plant Biol. 2019, 19, 106. [Google Scholar] [CrossRef]
- Masiero, S.; Colombo, L.; Grini, P.E.; Schnittger, A.; Kater, M.M. The Emerging Importance of Type I MADS Box Transcription Factors for Plant Reproduction. Plant Cell 2011, 23, 865–872. [Google Scholar] [CrossRef]
- Boulard, C.; Fatihi, A.; Lepiniec, L.; Dubreucq, B. Regulation and evolution of the interaction of the seed B3 transcription factors with NF-Y subunits. Biochim. Biophys. Acta (BBA)-Gene Regul. Mech. 2017, 1860, 1069–1078. [Google Scholar] [CrossRef]
- Siefers, N.; Dang, K.K.; Kumimoto, R.W.; Bynum, W.E.T.; Tayrose, G.; Holt, B.F., 3rd. Tissue-specific expression patterns of Arabidopsis NF-Y transcription factors suggest potential for extensive combinatorial complexity. Plant Physiol. 2009, 149, 625–641. [Google Scholar] [CrossRef]
- Yong, H.-Y.; Zou, Z.; Kok, E.-P.; Kwan, B.-H.; Chow, K.; Nasu, S.; Nanzyo, M.; Kitashiba, H.; Nishio, T. Comparative Transcriptome Analysis of Leaves and Roots in Response to Sudden Increase in Salinity in Brassica napus by RNA-seq. BioMed Res. Int. 2014, 2014, 467395. [Google Scholar] [CrossRef]
- Tahmasebi, A.; Ashrafi-Dehkordi, E.; Shahriari, A.G.; Mazloomi, S.M.; Ebrahimie, E. Integrative meta-analysis of transcriptomic responses to abiotic stress in cotton. Prog. Biophys. Mol. Biol. 2019, 146, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Niu, Y.; Dong, H.; Jia, Y.; Wang, Y. Characterization of the Function of Two S1Fa-Like Family Genes from Populus trichocarpa. Front. Plant Sci. 2021, 12, 753099. [Google Scholar] [CrossRef] [PubMed]
- Anwar, A.; Zhang, S.; Wang, L.-X.; Wang, F.; He, L.; Gao, J. Genome-Wide Identification and Characterization of Chinese Cabbage S1fa Transcription Factors and Their Roles in Response to Salt Stress. Antioxidants 2022, 11, 1782. [Google Scholar] [CrossRef] [PubMed]
- Finet, C.; Floyd, S.K.; Conway, S.J.; Zhong, B.; Scutt, C.P.; Bowman, J.L. Evolution of the YABBY gene family in seed plants. Evol. Dev. 2016, 18, 116–126. [Google Scholar] [CrossRef]
- Eckardt, N.A. YABBY Genes and the Development and Origin of Seed Plant Leaves. Plant Cell 2010, 22, 2103. [Google Scholar] [CrossRef]
- Izhaki, A.; Bowman, J.L. KANADI and Class III HD-Zip Gene Families Regulate Embryo Patterning and Modulate Auxin Flow during Embryogenesis in Arabidopsis. Plant Cell 2007, 19, 495–508. [Google Scholar] [CrossRef]
- Montgomery, T.A.; Yoo, S.J.; Fahlgren, N.; Gilbert, S.D.; Howell, M.D.; Sullivan, C.M.; Alexander, A.; Nguyen, G.; Allen, E.; Ahn, J.H.; et al. AGO1-miR173 complex initiates phased siRNA formation in plants. Proc. Natl. Acad. Sci. USA 2008, 105, 20055–20062. [Google Scholar] [CrossRef]
- Zhang, D.; Sun, W.; Singh, R.; Zheng, Y.; Cao, Z.; Li, M.; Lunde, C.; Hake, S.; Zhang, Z. GRF-interacting factor1 Regulates Shoot Architecture and Meristem Determinacy in Maize. Plant Cell 2018, 30, 360–374. [Google Scholar] [CrossRef]
- Omidbakhshfard, M.A.; Proost, S.; Fujikura, U.; Mueller-Roeber, B. Growth-Regulating Factors (GRFs): A Small Transcription Factor Family with Important Functions in Plant Biology. Mol. Plant 2015, 8, 998–1010. [Google Scholar] [CrossRef]
- Wang, L.; Gu, X.; Xu, D.; Wang, W.; Wang, H.; Zeng, M.; Chang, Z.; Huang, H.; Cui, X. miR396-targeted AtGRF transcription factors are required for coordination of cell division and differentiation during leaf development in Arabidopsis. J. Exp. Bot. 2010, 62, 761–773. [Google Scholar] [CrossRef]
- Lamesch, P.; Berardini, T.Z.; Li, D.; Swarbreck, D.; Wilks, C.; Sasidharan, R.; Muller, R.; Dreher, K.; Alexander, D.L.; Garcia-Hernandez, M.; et al. The Arabidopsis Information Resource (TAIR): Improved gene annotation and new tools. Nucleic Acids Res. 2011, 40, D1202–D1210. [Google Scholar] [CrossRef] [PubMed]
- Wendel, J.F. The wondrous cycles of polyploidy in plants. Am. J. Bot. 2015, 102, 1753–1756. [Google Scholar] [CrossRef] [PubMed]
- Rapp, R.A.; Udall, J.A.; Wendel, J.F. Genomic expression dominance in allopolyploids. BMC Biol. 2009, 7, 18. [Google Scholar] [CrossRef] [PubMed]
- Bottani, S.; Zabet, N.R.; Wendel, J.F.; Veitia, R.A. Gene Expression Dominance in Allopolyploids: Hypotheses and Models. Trends Plant Sci. 2018, 23, 393–402. [Google Scholar] [CrossRef]
- Edger, P.P.; Smith, R.; McKain, M.R.; Cooley, A.M.; Vallejo-Marin, M.; Yuan, Y.; Bewick, A.J.; Ji, L.; Platts, A.E.; Bowman, M.J.; et al. Subgenome Dominance in an Interspecific Hybrid, Synthetic Allopolyploid, and a 140-Year-Old Naturally Established Neo-Allopolyploid Monkeyflower. Plant Cell 2017, 29, 2150–2167. [Google Scholar] [CrossRef]
- Freeling, M. Bias in Plant Gene Content Following Different Sorts of Duplication: Tandem, Whole-Genome, Segmental, or by Transposition. Annu. Rev. Plant Biol. 2009, 60, 433–453. [Google Scholar] [CrossRef]
- Osborn, T.C.; Chris Pires, J.; Birchler, J.A.; Auger, D.L.; Jeffery Chen, Z.; Lee, H.-S.; Comai, L.; Madlung, A.; Doerge, R.W.; Colot, V.; et al. Understanding mechanisms of novel gene expression in polyploids. Trends Genet. 2003, 19, 141–147. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, H.; Cai, X.; Wu, J.; Hou, X.; Wang, X. Interspecies Conservation of Gene Expression Patterns in Brassica Reproductive Organs Unveiled by Comparative Transcriptomics. Horticulturae 2025, 11, 427. https://doi.org/10.3390/horticulturae11040427
Chen H, Cai X, Wu J, Hou X, Wang X. Interspecies Conservation of Gene Expression Patterns in Brassica Reproductive Organs Unveiled by Comparative Transcriptomics. Horticulturae. 2025; 11(4):427. https://doi.org/10.3390/horticulturae11040427
Chicago/Turabian StyleChen, Haixu, Xu Cai, Jian Wu, Xilin Hou, and Xiaowu Wang. 2025. "Interspecies Conservation of Gene Expression Patterns in Brassica Reproductive Organs Unveiled by Comparative Transcriptomics" Horticulturae 11, no. 4: 427. https://doi.org/10.3390/horticulturae11040427
APA StyleChen, H., Cai, X., Wu, J., Hou, X., & Wang, X. (2025). Interspecies Conservation of Gene Expression Patterns in Brassica Reproductive Organs Unveiled by Comparative Transcriptomics. Horticulturae, 11(4), 427. https://doi.org/10.3390/horticulturae11040427