

Transcriptomic Insights into Seed Germination Mechanisms of the Bamboo Qiongzhuea tumidinoda

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Material and Growth Conditions
2.2. RNA Sequencing and Transcriptomic Analysis
2.3. DEG Analysis
2.4. Weighted Gene Co-Expression Network Analysis (WGCNA)
2.5. The qPCR Analysis
3. Results
3.1. Transcriptome Assembly and Annotation
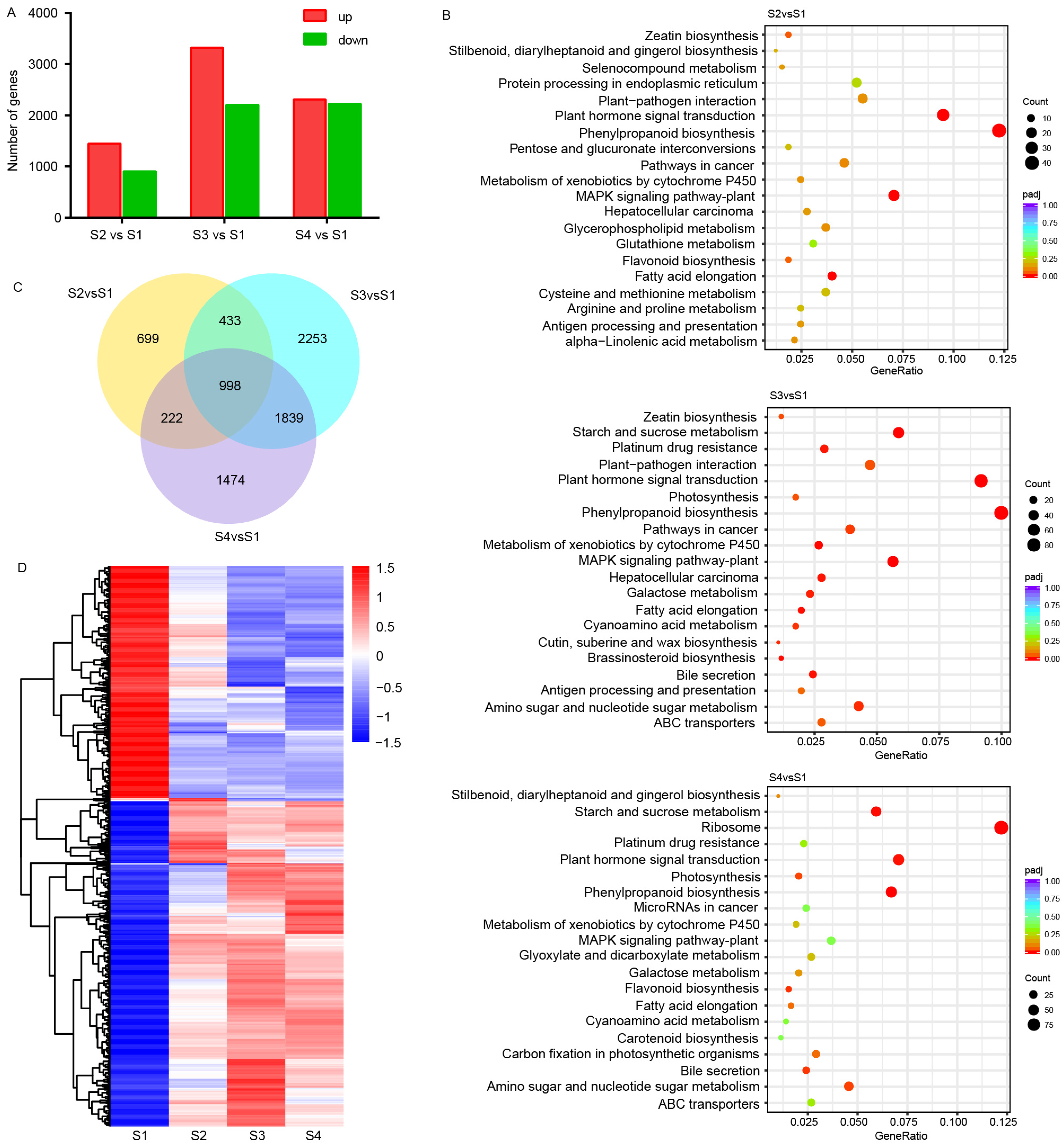
3.2. DEGs During Seed Germination
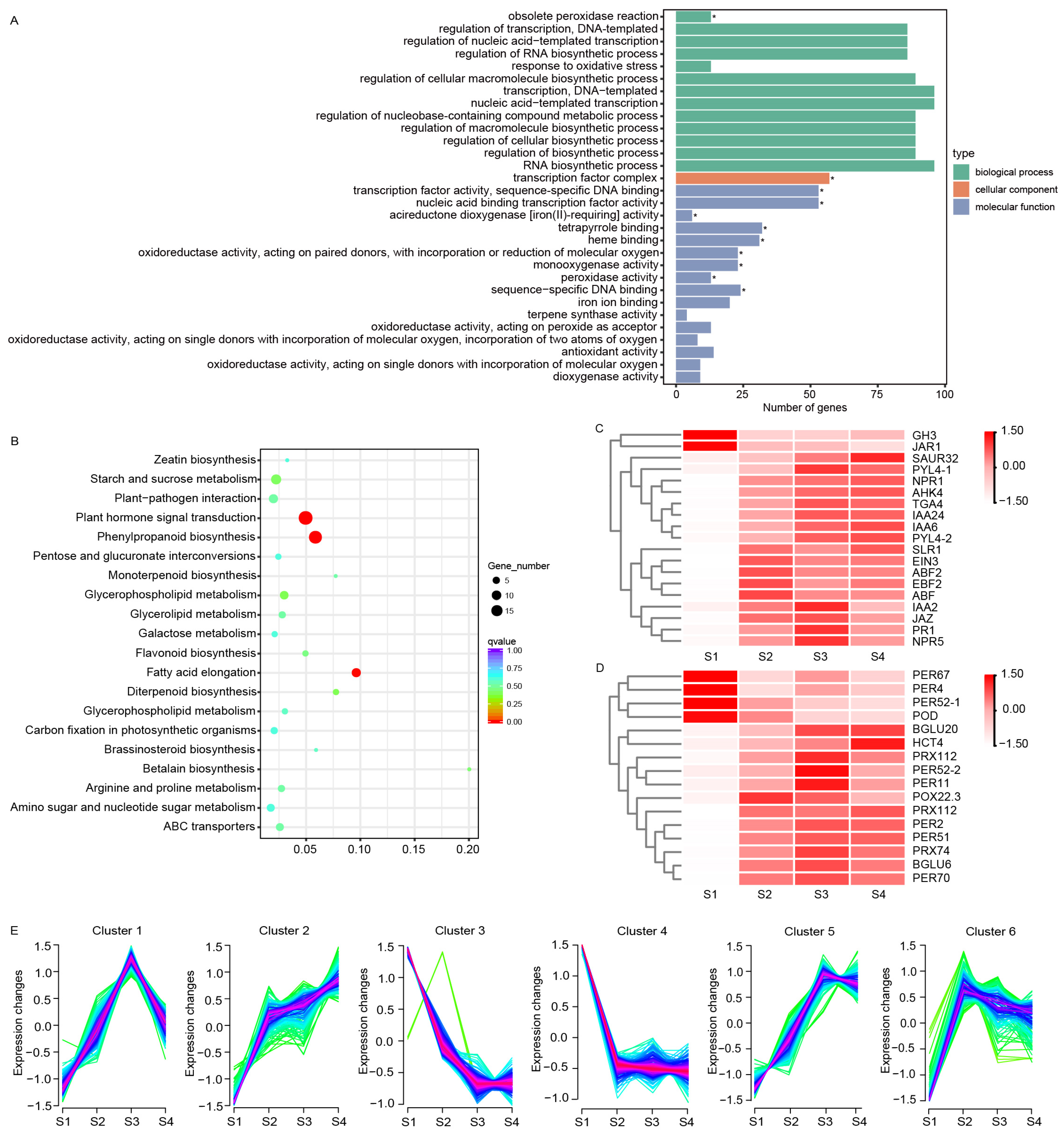
3.3. GO and KEGG Enrichment Analyses
3.4. Co-Expression Network Analysis with WGCNA
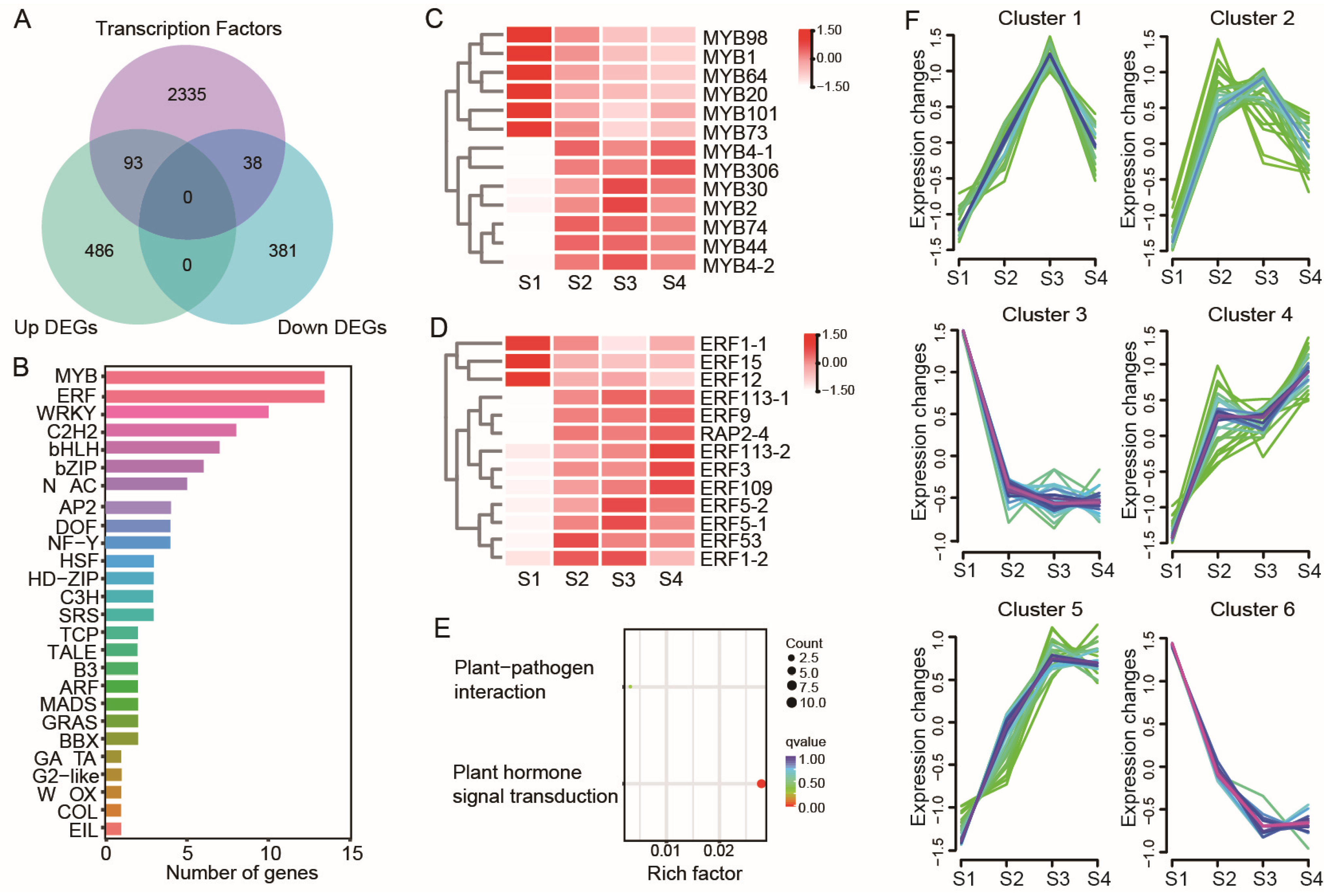
3.5. Analysis of Differentially Expressed Transcription Factors
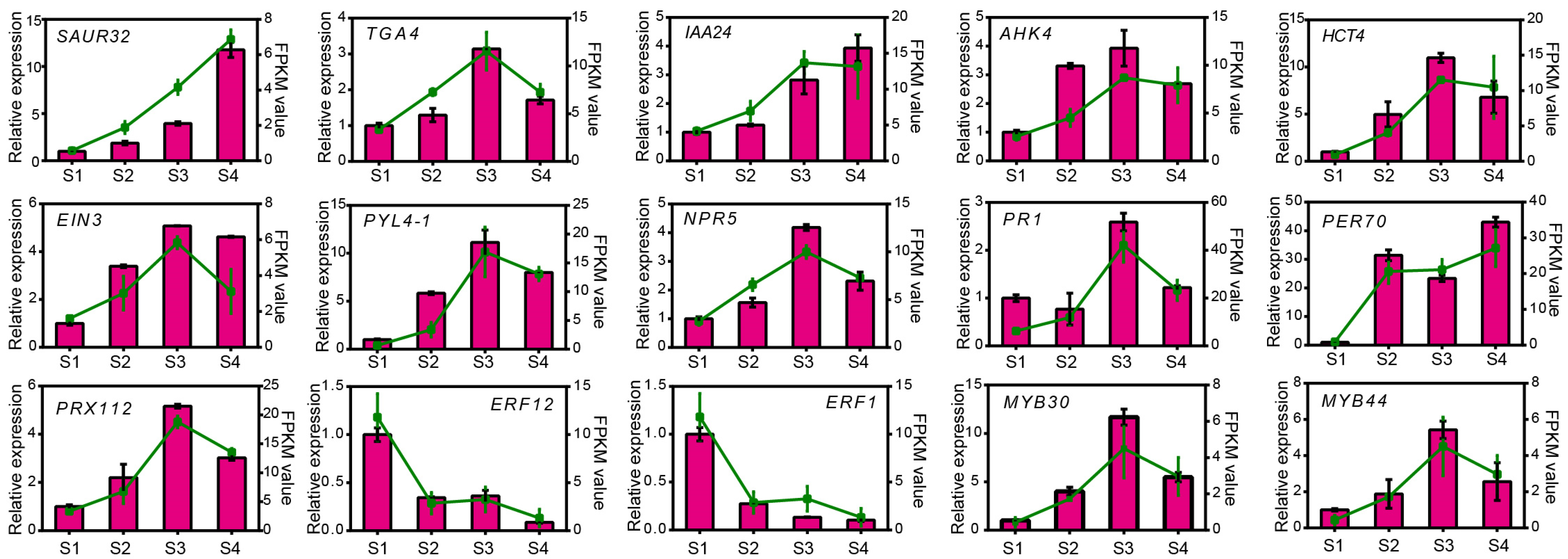
3.6. The qPCR Validation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Daszkowska-Golec, A. Arabidopsis seed germination under abiotic stress as a concert of action of phytohormones. Omics A J. Integr. Biol. 2011, 15, 763–774. [Google Scholar] [CrossRef] [PubMed]
- Penfield, S. Seed dormancy and germination. Curr. Biol. CB 2017, 27, R874–R878. [Google Scholar] [CrossRef] [PubMed]
- Finch-Savage, W.E.; Leubner-Metzger, G. Seed dormancy and the control of germination. New Phytol. 2006, 171, 501–523. [Google Scholar] [CrossRef]
- Zhao, M.; Zhang, H.; Yan, H.; Qiu, L.; Baskin, C.C. Mobilization and Role of Starch, Protein, and Fat Reserves during Seed Germination of Six Wild Grassland Species. Front. Plant Sci. 2018, 9, 234. [Google Scholar] [CrossRef]
- Zhang, K.; Wu, Z.; Tang, D.; Luo, K.; Lu, H.; Liu, Y.; Dong, J.; Wang, X.; Lv, C.; Wang, J. Comparative Transcriptome Analysis Reveals Critical Function of Sucrose Metabolism Related-Enzymes in Starch Accumulation in the Storage Root of Sweet Potato. Front. Plant Sci. 2017, 8, 914. [Google Scholar] [CrossRef]
- Lai, Y.C.; Wang, S.Y.; Gao, H.Y.; Nguyen, K.M.; Nguyen, C.H.; Shih, M.C.; Lin, K.H. Physicochemical properties of starches and expression and activity of starch biosynthesis-related genes in sweet potatoes. Food Chem. 2016, 199, 556–564. [Google Scholar] [CrossRef]
- Lu, C.A.; Ho, T.H.; Ho, S.L.; Yu, S.M. Three novel MYB proteins with one DNA binding repeat mediate sugar and hormone regulation of alpha-amylase gene expression. Plant Cell 2002, 14, 1963–1980. [Google Scholar] [CrossRef]
- Gubler, F.; Kalla, R.; Roberts, J.K.; Jacobsen, J.V. Gibberellin-regulated expression of a myb gene in barley aleurone cells: Evidence for Myb transactivation of a high-pI alpha-amylase gene promoter. Plant Cell 1995, 7, 1879–1891. [Google Scholar] [CrossRef]
- Shu, K.; Liu, X.D.; Xie, Q.; He, Z.H. Two Faces of One Seed: Hormonal Regulation of Dormancy and Germination. Mol. Plant 2016, 9, 34–45. [Google Scholar] [CrossRef]
- Beaudoin, N.; Serizet, C.; Gosti, F.; Giraudat, J. Interactions between abscisic acid and ethylene signaling cascades. Plant Cell. 2000, 12, 1103–1115. [Google Scholar] [CrossRef]
- Sugliani, M.; Rajjou, L.; Clerkx, E.J.; Koornneef, M.; Soppe, W.J. Natural modifiers of seed longevity in the Arabidopsis mutants abscisic acid insensitive3-5 (abi3-5) and leafy cotyledon1-3 (lec1-3). New Phytol. 2009, 184, 898–908. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.; Dai, Y.; Zheng, C.; Yang, Y.; Chen, W.; Wang, Q.; Chandrasekaran, U.; Du, J.; Liu, W.; Shu, K. The ABI4-RbohD/VTC2 regulatory module promotes reactive oxygen species (ROS) accumulation to decrease seed germination under salinity stress. New Phytol. 2021, 229, 950–962. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Dou, L.; Gong, Z.; Wang, X.; Mao, T. BES1 hinders ABSCISIC ACID INSENSITIVE5 and promotes seed germination in Arabidopsis. New Phytol. 2019, 221, 908–918. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.G.; Lee, A.K.; Yoon, H.K.; Park, C.M. A membrane-bound NAC transcription factor NTL8 regulates gibberellic acid-mediated salt signaling in Arabidopsis seed germination. Plant J. Cell Mol. Biology 2008, 55, 77–88. [Google Scholar] [CrossRef]
- Du, J.; Wang, S.; He, C.; Zhou, B.; Ruan, Y.L.; Shou, H. Identification of regulatory networks and hub genes controlling soybean seed set and size using RNA sequencing analysis. J. Exp. Bot. 2017, 68, 1955–1972. [Google Scholar] [CrossRef]
- Cadman, C.S.; Toorop, P.E.; Hilhorst, H.W.; Finch-Savage, W.E. Gene expression profiles of Arabidopsis Cvi seeds during dormancy cycling indicate a common underlying dormancy control mechanism. Plant J. Cell Mol. Biol. 2006, 46, 805–822. [Google Scholar] [CrossRef]
- Yu, Y.; Zhen, S.; Wang, S.; Wang, Y.; Cao, H.; Zhang, Y.; Li, J.; Yan, Y. Comparative transcriptome analysis of wheat embryo and endosperm responses to ABA and H2O2 stresses during seed germination. BMC Genom. 2016, 17, 97. [Google Scholar] [CrossRef]
- Guo, B.; Chen, Y.; Zhang, G.; Xing, J.; Hu, Z.; Feng, W.; Yao, Y.; Peng, H.; Du, J.; Zhang, Y.; et al. Comparative proteomic analysis of embryos between a maize hybrid and its parental lines during early stages of seed germination. PLoS ONE. 2013, 8, e65867. [Google Scholar] [CrossRef]
- Narsai, R.; Secco, D.; Schultz, M.D.; Ecker, J.R.; Lister, R.; Whelan, J. Dynamic and rapid changes in the transcriptome and epigenome during germination and in developing rice (Oryza sativa) coleoptiles under anoxia and re-oxygenation. Plant J. Cell Mol. Biol. 2017, 89, 805–824. [Google Scholar] [CrossRef]
- Yi, Y.; Wenyuan, D.; Yuequn, Q.; Wen, L.; Jiajun, Y.; Yan, H. Transformation of Nutritional Compositions in Chimonobambusa tumidissinoda Shoots During Growth Process. J. Northeast. For. Univ. 2015, 43, 80–87. [Google Scholar]
- Singh, S.R.; Singh, R.; Kalia, S.; Dalal, S.; Dhawan, A.K.; Kalia, R.K. Limitations, progress and prospects of application of biotechnological tools in improvement of bamboo-a plant with extraordinary qualities. Physiol. Mol. Biol. Plants 2013, 19, 21–41. [Google Scholar] [CrossRef] [PubMed]
- Haas, B.J.; Papanicolaou, A.; Yassour, M.; Grabherr, M.; Blood, P.D.; Bowden, J.; Couger, M.B.; Eccles, D.; Li, B.; Lieber, M.; et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 2013, 8, 1494–1512. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Tarazona, S.; García-Alcalde, F.; Dopazo, J.; Ferrer, A.; Conesa, A. Differential expression in RNA-seq: A matter of depth. Genome Res. 2011, 21, 2213–2223. [Google Scholar] [CrossRef]
- Chen, C.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.; Xia, R. TBtools: An Integrative Toolkit Developed for Interactive Analyses of Big Biological Data. Mol. Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef]
- Zhang, H.; Jin, J.; Tang, L.; Zhao, Y.; Gu, X.; Gao, G.; Luo, J. PlantTFDB 2.0: Update and improvement of the comprehensive plant transcription factor database. Nucleic Acids Res. 2011, 39, D1114–D1117. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef]
- Huang, F.; Liu, T.; Hou, X. Isolation and Functional Characterization of a Floral Repressor, BcMAF1, From Pak-choi (Brassica rapa ssp. Chinensis). Front. Plant Sci. 2018, 9, 290. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Han, C.; Yang, P. Studies on the molecular mechanisms of seed germination. Proteomics 2015, 15, 1671–1679. [Google Scholar] [CrossRef] [PubMed]
- Duan, X.; Jiang, W.; Wu, K.; Chen, J.; Li, Y.; Tao, Z. Integrating Transcriptomics and Hormones Dynamics Reveal Seed Germination and Emergence Process in Polygonatum cyrtonema Hua. Int. J. Mol. Sci. 2023, 24, 3792. [Google Scholar] [CrossRef] [PubMed]
- Park, S.Y.; Fung, P.; Nishimura, N.; Jensen, D.R.; Fujii, H.; Zhao, Y.; Lumba, S.; Santiago, J.; Rodrigues, A.; Chow, T.F.; et al. Abscisic acid inhibits type 2C protein phosphatases via the PYR/PYL family of START proteins. Science 2009, 324, 1068–1071. [Google Scholar] [CrossRef]
- Casanova-Sáez, R.; Voß, U. Auxin Metabolism Controls Developmental Decisions in Land Plants. Trends Plant Sci. 2019, 24, 741–754. [Google Scholar] [CrossRef]
- Yang, Q.X.; Chen, D.; Zhao, Y.; Zhang, X.Y.; Zhao, M.; Peng, R.; Sun, N.X.; Baldwin, T.C.; Yang, S.C.; Liang, Y.L. RNA-seq analysis reveals key genes associated with seed germination of Fritillaria taipaiensis P.Y.Li by cold stratification. Front. Plant Sci. 2022, 13, 1021572. [Google Scholar] [CrossRef]
- Zhang, N.; Huang, X.; Bao, Y.; Wang, B.; Zeng, H.; Cheng, W.; Tang, M.; Li, Y.; Ren, J.; Sun, Y. Genome-wide identification of SAUR genes in watermelon (Citrullus lanatus). Physiol. Mol. Biol. Plants Int. J. Funct. Plant Biol. 2017, 23, 619–628. [Google Scholar] [CrossRef]
- He, Y.; Liu, Y.; Li, M.; Lamin-Samu, A.T.; Yang, D.; Yu, X.; Izhar, M.; Jan, I.; Ali, M.; Lu, G. The Arabidopsis SMALL AUXIN UP RNA32 Protein Regulates ABA-Mediated Responses to Drought Stress. Front. Plant Sci. 2021, 12, 625493. [Google Scholar] [CrossRef]
- Markakis, M.N.; Boron, A.K.; Van Loock, B.; Saini, K.; Cirera, S.; Verbelen, J.P.; Vissenberg, K. Characterization of a small auxin-up RNA (SAUR)-like gene involved in Arabidopsis thaliana development. PLoS ONE 2013, 8, e82596. [Google Scholar] [CrossRef]
- Sun, M.; Tuan, P.A.; Izydorczyk, M.S.; Ayele, B.T. Ethylene regulates post-germination seedling growth in wheat through spatial and temporal modulation of ABA/GA balance. J. Exp. Bot. 2020, 71, 1985–2004. [Google Scholar] [CrossRef]
- Zhang, L.; Li, Z.; Quan, R.; Li, G.; Wang, R.; Huang, R. An AP2 domain-containing gene, ESE1, targeted by the ethylene signaling component EIN3 is important for the salt response in Arabidopsis. Plant Physiol. 2011, 157, 854–865. [Google Scholar] [CrossRef]
- Yamada, H.; Suzuki, T.; Terada, K.; Takei, K.; Ishikawa, K.; Miwa, K.; Yamashino, T.; Mizuno, T. The Arabidopsis AHK4 histidine kinase is a cytokinin-binding receptor that transduces cytokinin signals across the membrane. Plant Cell Physiol. 2001, 42, 1017–1023. [Google Scholar] [CrossRef] [PubMed]
- Budimir, J.; Treffon, K.; Nair, A.; Thurow, C.; Gatz, C. Redox-active cysteines in TGACG-BINDING FACTOR 1 (TGA1) do not play a role in salicylic acid or pathogen-induced expression of TGA1-regulated target genes in Arabidopsis thaliana. New Phytol. 2021, 230, 2420–2432. [Google Scholar] [CrossRef] [PubMed]
- Canales, J.; Contreras-López, O.; Álvarez, J.M.; Gutiérrez, R.A. Nitrate induction of root hair density is mediated by TGA1/TGA4 and CPC transcription factors in Arabidopsis thaliana. Plant J. Cell Mol. Biol. 2017, 92, 305–316. [Google Scholar] [CrossRef] [PubMed]
- Besseau, S.; Hoffmann, L.; Geoffroy, P.; Lapierre, C.; Pollet, B.; Legrand, M. Flavonoid accumulation in Arabidopsis repressed in lignin synthesis affects auxin transport and plant growth. Plant Cell 2007, 19, 148–162. [Google Scholar] [CrossRef]
- Yu, A.; Zhao, J.; Wang, Z.; Cheng, K.; Zhang, P.; Tian, G.; Liu, X.; Guo, E.; Du, Y.; Wang, Y. Transcriptome and metabolite analysis reveal the drought tolerance of foxtail millet significantly correlated with phenylpropanoids-related pathways during germination process under PEG stress. BMC Plant Biol. 2020, 20, 274. [Google Scholar] [CrossRef]
- Appelhagen, I.; Thiedig, K.; Nordholt, N.; Schmidt, N.; Huep, G.; Sagasser, M.; Weisshaar, B. Update on transparent testa mutants from Arabidopsis thaliana: Characterisation of new alleles from an isogenic collection. Planta 2014, 240, 955–970. [Google Scholar] [CrossRef]
- Jiang, J.; Zhu, S.; Yuan, Y.; Wang, Y.; Zeng, L.; Batley, J.; Wang, Y.P. Transcriptomic comparison between developing seeds of yellow- and black-seeded Brassica napus reveals that genes influence seed quality. BMC Plant Biol. 2019, 19, 203. [Google Scholar] [CrossRef]
- Taylor, L.P.; Grotewold, E. Flavonoids as developmental regulators. Curr. Opin. Plant Biology. 2005, 8, 317–323. [Google Scholar] [CrossRef]
- Guo, H.; Lyv, Y.; Zheng, W.; Yang, C.; Li, Y.; Wang, X.; Chen, R.; Wang, C.; Luo, J.; Qu, L. Comparative Metabolomics Reveals Two Metabolic Modules Affecting Seed Germination in Rice (Oryza sativa). Metabolites. 2021, 11, 880. [Google Scholar] [CrossRef]
- Hoffmann, L.; Besseau, S.; Geoffroy, P.; Ritzenthaler, C.; Meyer, D.; Lapierre, C.; Pollet, B.; Legrand, M. Silencing of hydroxycinnamoyl-coenzyme A shikimate/quinate hydroxycinnamoyltransferase affects phenylpropanoid biosynthesis. Plant Cell. 2004, 16, 1446–1465. [Google Scholar] [CrossRef]
- Hoffmann, L.; Maury, S.; Martz, F.; Geoffroy, P.; Legrand, M. Purification, cloning, and properties of an acyltransferase controlling shikimate and quinate ester intermediates in phenylpropanoid metabolism. J. Biol. Chem. 2003, 278, 95–103. [Google Scholar] [CrossRef] [PubMed]
- Fang, L.; Xu, X.; Li, J.; Zheng, F.; Li, M.; Yan, J.; Li, Y.; Zhang, X.; Li, L.; Ma, G.; et al. Transcriptome analysis provides insights into the non-methylated lignin synthesis in Paphiopedilum armeniacum seed. BMC Genom. 2020, 21, 524. [Google Scholar] [CrossRef] [PubMed]
- Meng, A.; Wen, D.; Zhang, C. Dynamic Changes in Seed Germination under Low-Temperature Stress in Maize. Int. J. Mol. Sci. 2022, 23, 5495. [Google Scholar] [CrossRef]
- Deng, F.; He, Q.; Zhao, Z. Suppressing a Peroxidase Gene Reduces Survival in the Wheat Aphid Sitobion avenae. Arch. Insect Biochem. Physiol. 2016, 93, 86–95. [Google Scholar] [CrossRef]
- Howell, K.A.; Narsai, R.; Carroll, A.; Ivanova, A.; Lohse, M.; Usadel, B.; Millar, A.H.; Whelan, J. Mapping metabolic and transcript temporal switches during germination in rice highlights specific transcription factors and the role of RNA instability in the germination process. Plant Physiol. 2009, 149, 961–980. [Google Scholar] [CrossRef]
- Washio, K. Functional dissections between GAMYB and Dof transcription factors suggest a role for protein-protein associations in the gibberellin-mediated expression of the RAmy1A gene in the rice aleurone. Plant Physiol. 2003, 133, 850–863. [Google Scholar] [CrossRef]
- Zhao, J.; He, Y.; Zhang, H.; Wang, Z. Advances in the molecular regulation of seed germination in plants. Seed Biol. 2024, 3, e006. [Google Scholar] [CrossRef]
- Yu, H.; Teng, Z.; Liu, B.; Lv, J.; Chen, Y.; Qin, Z.; Peng, Y.; Meng, S.; He, Y.; Duan, M.; et al. Transcription factor OsMYB30 increases trehalose content to inhibit α-amylase and seed germination at low temperature. Plant Physiol. 2024, 194, 1815–1833. [Google Scholar] [CrossRef]
- Li, J.; Bai, Y.; Xie, Y.; Gao, J. Ultrastructure change and transcriptome analysis of GA3 treatment on seed germination of moso bamboo (Phyllostachys edulis). Plant Signal. Behav. 2022, 17, 2091305. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, F.; Wang, J.; Zhang, X.; Lin, S. Transcriptomic Insights into Seed Germination Mechanisms of the Bamboo Qiongzhuea tumidinoda. Horticulturae 2025, 11, 430. https://doi.org/10.3390/horticulturae11040430
Huang F, Wang J, Zhang X, Lin S. Transcriptomic Insights into Seed Germination Mechanisms of the Bamboo Qiongzhuea tumidinoda. Horticulturae. 2025; 11(4):430. https://doi.org/10.3390/horticulturae11040430
Chicago/Turabian StyleHuang, Feiyi, Jiaxin Wang, Xu Zhang, and Shuyan Lin. 2025. "Transcriptomic Insights into Seed Germination Mechanisms of the Bamboo Qiongzhuea tumidinoda" Horticulturae 11, no. 4: 430. https://doi.org/10.3390/horticulturae11040430
APA StyleHuang, F., Wang, J., Zhang, X., & Lin, S. (2025). Transcriptomic Insights into Seed Germination Mechanisms of the Bamboo Qiongzhuea tumidinoda. Horticulturae, 11(4), 430. https://doi.org/10.3390/horticulturae11040430