
Genome-Wide Identification of Selenium-Responsive MicroRNAs in Tea Plant (Camellia sinensis L. O. Kuntze)



Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Materials and Treatments
2.2. Total Se Content Testing and Data Analysis
2.3. Total RNA Isolation, Small RNA Library Construction and Sequencing
2.4. miRNA Analysis of Sequences
2.5. MiRNA Differential Expression Analysis, Target Genes Prediction and Functional Annotation
2.6. Quantitative Real Time PCR Analysis of miRNAs
3. Results
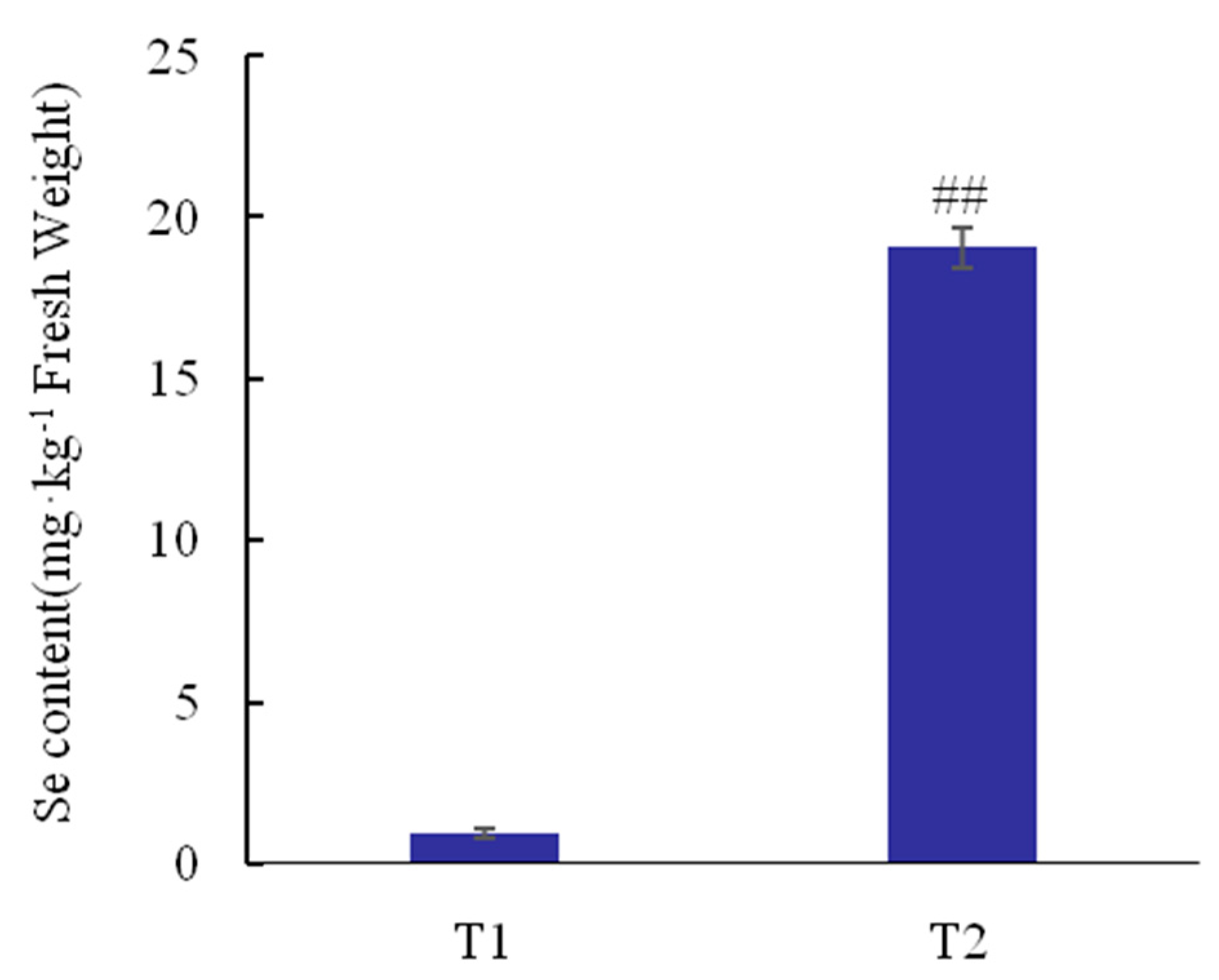
3.1. Total Se Content Analyses
3.2. Sequencing of miRNA in Tea Plant Roots
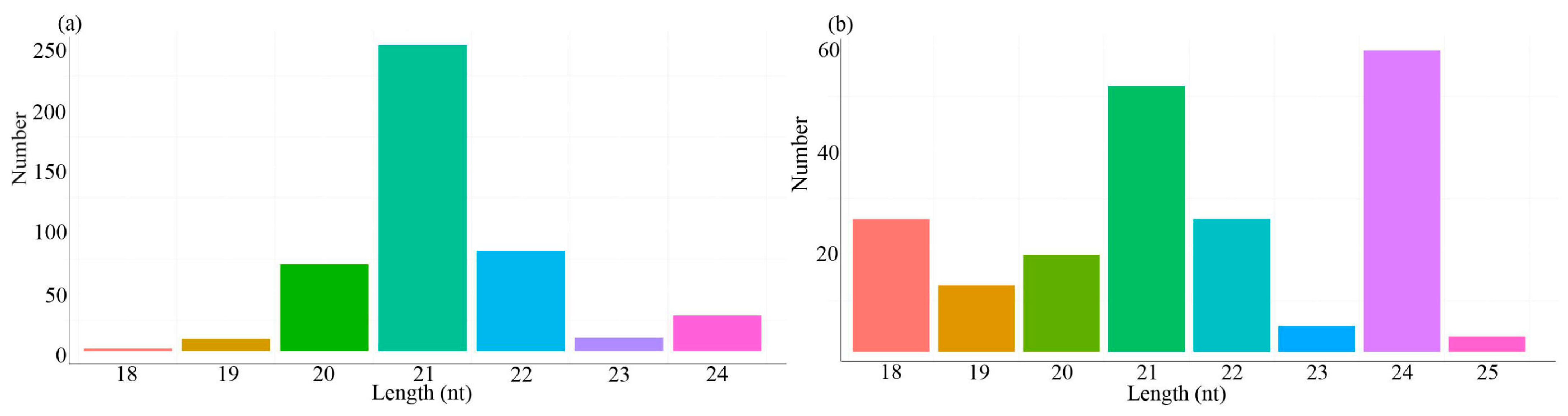
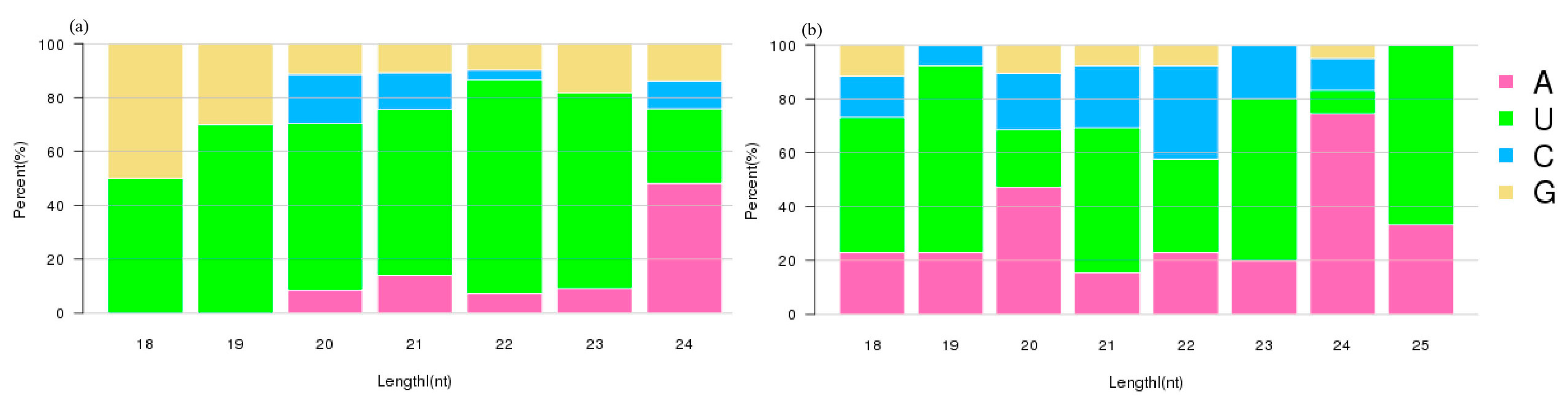
3.3. Identification of Tea Plant miRNAs
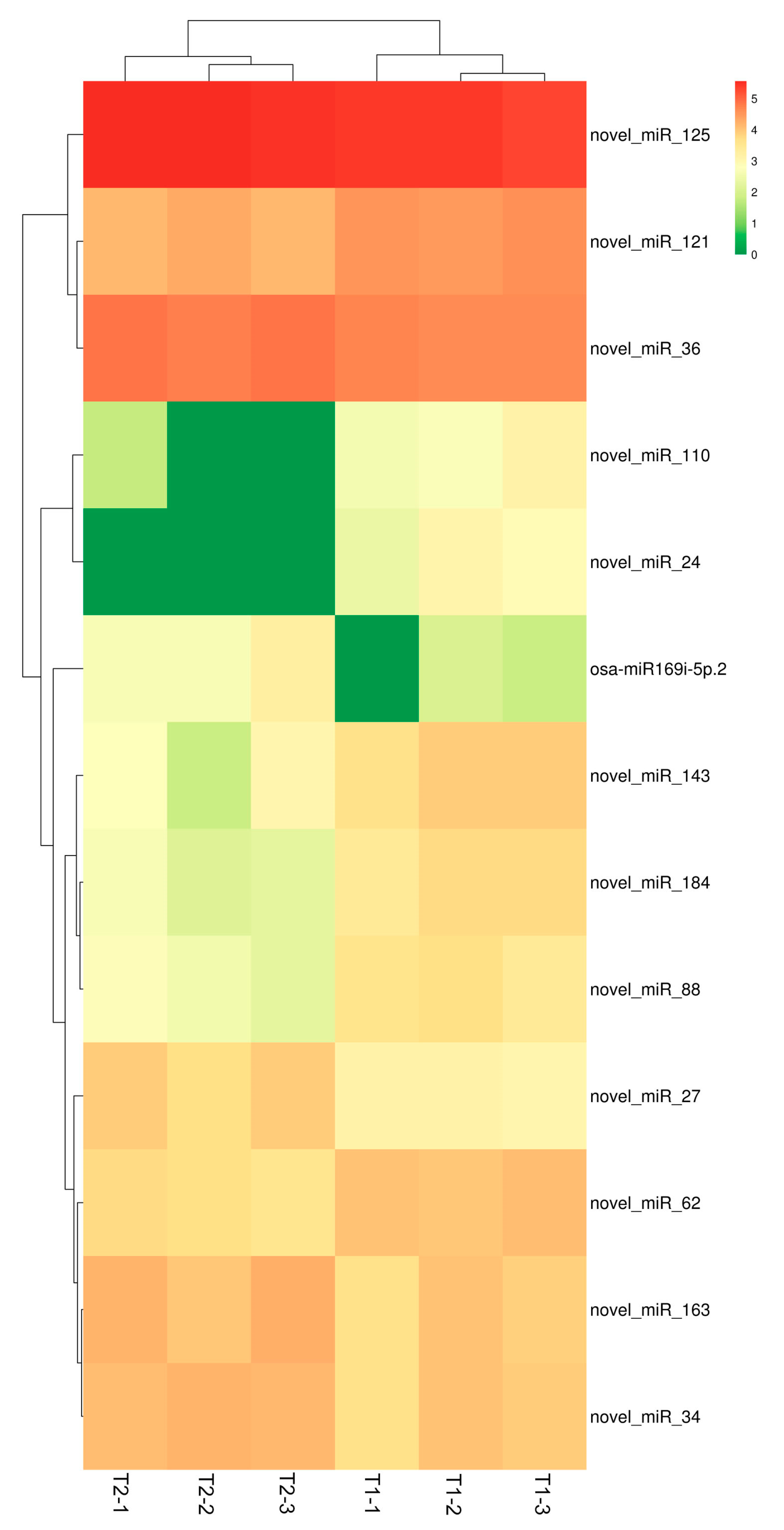
3.4. Analysis of Differentially Expressed miRNAs
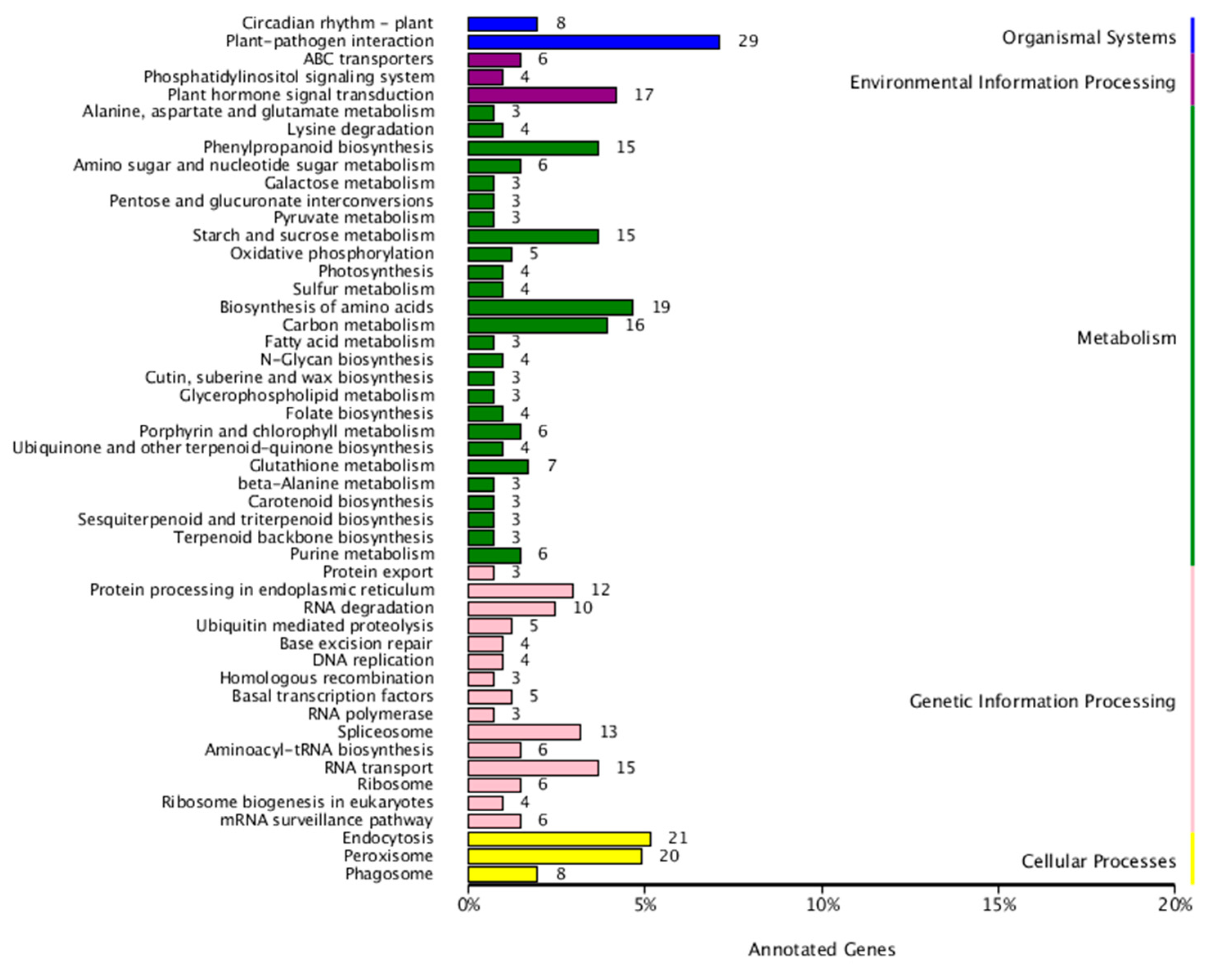
3.5. Identification of Target Genes of Differentially Expressed miRNAs
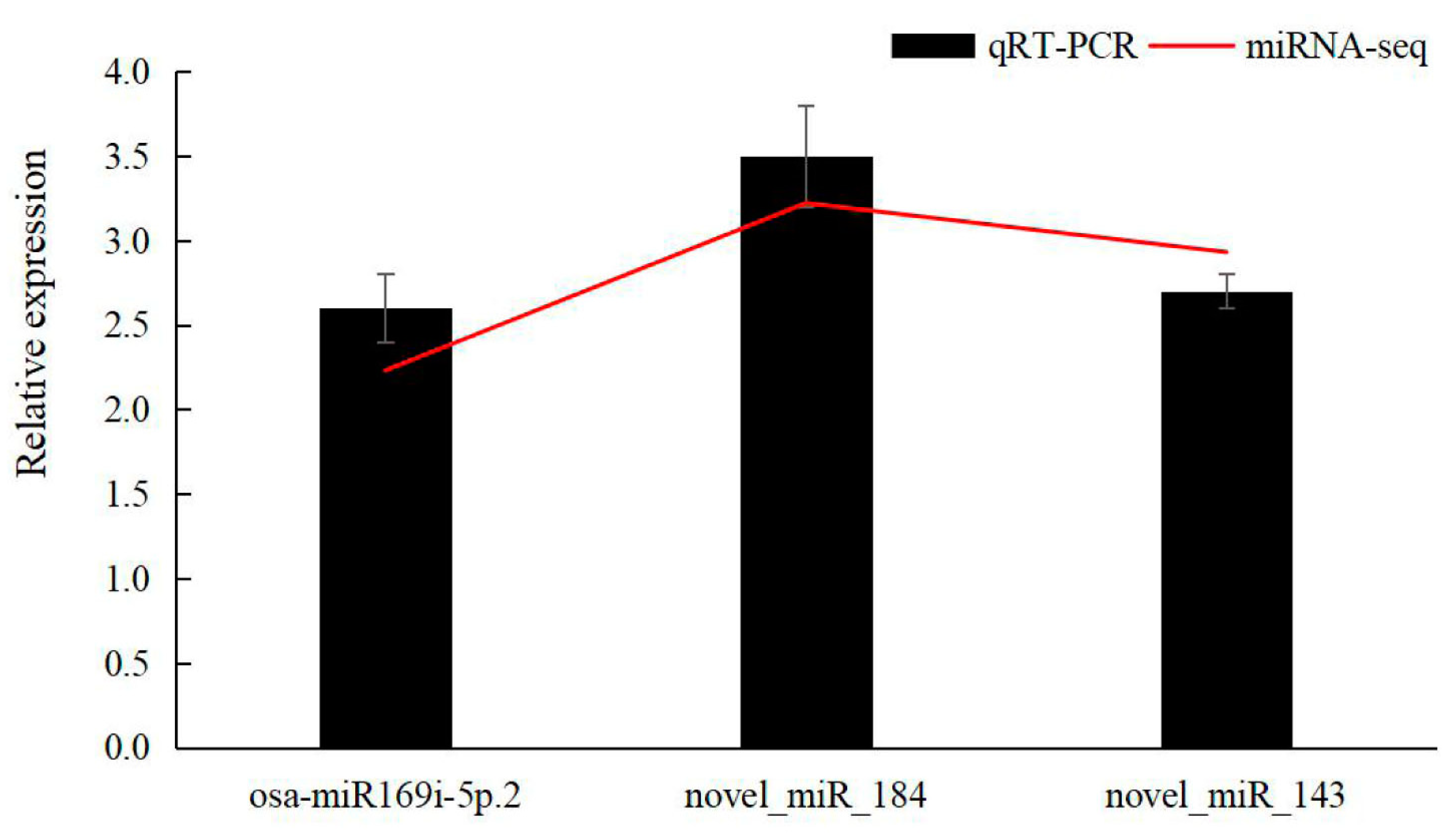
3.6. qRT-PCR Validation
4. Discussion
4.1. Se Content in Tea Plants
4.2. miRNAs Associated with Se Accumulation in Tea Plants
4.3. Pathways Associated with Se Uptake and Accumulation in Tea Plants
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Lee, R.C.; Feinbaum, R.L.; Ambros, V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell 1993, 75, 843. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.H.; Pan, X.P.; Cobb, G.P.; Anderson, T.A. Plant microRNA: A small regulatory molecule with big impact. Dev. Biol. 2006, 289, 3–16. [Google Scholar] [CrossRef] [PubMed]
- Mallory, A.C.; Vaucheret, H. Functions of microRNAs and related small RNAs in plants. Nat. Gene 2006, 38, S31–S36. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.Q.; Peng, J.; Qiu, C.X.; Yang, Z.M. Heavy metal-regulated new microRNAs from rice. J. Inorg. Biochem. 2009, 103, 282–287. [Google Scholar] [CrossRef] [PubMed]
- Shivaprasad, P.V.; Chen, H.M.; Patel, K.; Bond, D.M.; Santos, B.; Baulcombe, D.C. A microRNA superfamily regulates nucleotide binding site-leucine-rich repeats and other miRNAs. Plant Cell 2012, 24, 859–874. [Google Scholar] [CrossRef] [PubMed]
- Phillips, J.R.; Dalmay, T.; Bartels, D. The role of small RNAs in abiotic stress. Febs Lett. 2007, 581, 3592–3597. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Zeng, J.H.Q.; Song, J.; Feng, S.J.; Yang, Z.M. miRNA778 and SUVH6 are involved in phosphate homeostasis in Arabidopsis. Plant Sci. 2015, 238, 273–285. [Google Scholar] [CrossRef]
- Zeng, H.Q.; Zhu, Y.Y.; Huang, S.Q.; Yang, Z.M. Analysis of phosphorus-deficient responsive miRNAs and cis-elements from soybean (Glycine max L.). J. Plant Physiol. 2010, 167, 1289–1297. [Google Scholar] [CrossRef]
- Lyu, C.Y.; Sha, A.H. Response to phosphorus deficiency regulated by microRNA168 in soybean plant. Chin. J. Oil Crop Sci. 2017, 39, 321–325. [Google Scholar] [CrossRef]
- He, H.; Liang, G.; Li, Y.; Wang, F.; Yu, D. Two young microRNAs originating from target duplication mediate nitrogen starvation adaptation via regulation of glucosinolate synthesis in Arabidopsis thaliana. Plant Physiol. 2014, 164, 853–865. [Google Scholar] [CrossRef]
- Liu, Z.W.; Li, H.; Liu, J.X.; Wang, Y.; Zhuang, J. Integrative transcriptome, proteome, and microRNA analysis reveals the effects of nitrogen sufficiency and deficiency conditions on theanine metabolism in the tea plant (Camellia sinensis). Hortic. Res. 2020, 7, 1865–1877. [Google Scholar] [CrossRef] [PubMed]
- Matthewman, C.A.; Kawashima, C.G.; Húska, D.; Csorba, T.; Dalmay, T.; Kopriva, S. miR395 is a general component of thesulfate assimilation regulatory network in Arabidopsis. Febs Lett. 2012, 586, 3242–3324. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.X.; Wang, H.M.; Liu, Q.L.; Meng, C.X. Summary of selenium in soil and plant. J. Hebei Agric. Sci. 2008, 12, 43–45. [Google Scholar] [CrossRef]
- Wang, Y.D.; Wang, X.; Ngai, S.M.; Wong, Y.S. Comparative proteomics analysis of selenium responses in selenium-enrichedricegrains. J. Proteome Res. 2013, 12, 808–820. [Google Scholar] [CrossRef] [PubMed]
- Fordyce, F. Selenium geochemistry and health. AMBIO A J. Hum. Environ. 2007, 36, 94–97. [Google Scholar] [CrossRef]
- Paul, N.; Williams, E.L.; Sun, G.X.; Scheckel, K.; Zhu, Y.G.; Feng, X.B.; Zhu, J.M.; Carey, A.M.; Adomako, E.; Lawgali, Y.; et al. Selenium Characterisation in the Global Rice Supply Chain. Environ. Sci. Technol. 2009, 43, 6024–6030. [Google Scholar] [CrossRef]
- Rayman, M.P. Selenium and human health. Lancet 2012, 379, 1256–1268. [Google Scholar] [CrossRef]
- Du, Q.Z.; Shen, X.R.; Fang, X.H. Analysis on selenium compositions in tea leaves. J. Tea Sci. 1991, 11, 133–137. [Google Scholar]
- Zhao, H.; Huang, J.; Li, Y.; Song, X.W.; Luo, J.L.; Yu, Z.; Ni, D.J. Natural variation of selenium concentration in diverse tea plant (Camellia sinensis) accessions at seedling stage. Sci. Hortic. 2016, 198, 163–169. [Google Scholar] [CrossRef]
- Cao, D.; Liu, Y.L.; Ma, L.L.; Jin, X.F.; Guo, G.Y.; Tan, R.R.; Liu, Z.; Ye, F.; Liu, W. Transcriptome of differentially expressed genes involved in selenium accumulation in tea plant (Camellia sinensis). PLoS ONE 2018, 13, E0197506. [Google Scholar] [CrossRef]
- Cao, D.; Liu, Y.L.; Ma, L.L.; Liu, Z.H.; Li, J.; Wen, B.B.; Zhang, X.N.; Yin, P.; Jin, X.F.; Huang, J.A. Genome-wide identificationand characterization of phosphate transporter gene family members in tea plants (Camellia sinensis L.O. Kuntze) underdifferent selenite levels. Plant Physiol. Biochem. 2021, 166, 668–676. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhu, X.J.; Chen, X.; Song, C.N.; Zou, Z.W.; Wang, Y.H.; Wang, M.L.; Fang, W.P.; Li, X.H. Identification and characterization of cold-responsive microRNAs in tea plant (Camellia sinensis) and their targets using high-throughput sequencing and degradome analysis. BMC Plant Biol. 2014, 14, 271. [Google Scholar] [CrossRef] [PubMed]
- Jeyaraj, A.; Elango, T.; Li, X.H.; Guo, G.Y. Utilization of microRNAs and their regulatory functions for improving biotic stress tolerance in tea plant [Camellia sinensis (L.)O. Kuntze]. RNA Biol. 2020, 17, 1365–1382. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Lin, Q.Q.; Yan, M.L.; Wang, M.L.; Wang, P.; Zhao, H.; Wang, Y.; Ni, D.J.; Guo, F. Relationship between secondary metabolism and mirna for important flavor compounds in different tissues of tea plant (Camellia sinensis) as revealed by genome-wide mirna analysis. J. Agric. Food Chem. 2021, 69, 2001–2012. [Google Scholar] [CrossRef] [PubMed]
- Çakır, Ö.; Turgutkara, N.; Arı, S.; Zhang, B.H. De novo transcriptome assembly and comparative analysis elucidate complicated mechanism regulating Astragalus chrysochlorus response to selenium stimuli. PLoS ONE 2015, 10, E0135677. [Google Scholar] [CrossRef]
- Elberse, I.A.M.; VanDamme, J.M.M.; VanTienderen, P.H. Plasticity of growth characteristics in wild barley (Hordeum spontaneum) in response to nutrient limitation. J. Ecol. 2003, 91, 371–382. [Google Scholar] [CrossRef]
- Cao, D.; Ma, L.L.; Liu, Y.L.; Gong, Z.M.; Jin, X.F. Absorption and accumulation characteristics of selenium in tea plant (Camellia sinensis) and expression analysis of genes related to selenium regulation. J. Tea Sci. 2020, 40, 77–84. [Google Scholar] [CrossRef]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10, R25. [Google Scholar] [CrossRef]
- Li, B.; Ruotti, V.; Stewart, R.M.; Thomson, J.A.; Dewey, C.N. RNA-Seq gene expression estimation with read mapping uncertainty. Bioinformatics 2009, 26, 493–500. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. ModeratedestimationoffoldchangeanddispersionforRNA-seqdatawithDESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Allen, E.; Xie, Z.; Gustafson, A.M.; Carrington, J.C. microRNA-directed phasing during trans-acting siRNA biogenesis in plants. Cell 2005, 121, 207–221. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.Y.; Li, J.Q.; Wu, S.F.; Zhu, Y.P.; Chen, Y.W.; He, F.C. Integrated nr database in protein annotation system and its Localization. Comput. Eng. 2006, 32, 71–74. [Google Scholar] [CrossRef]
- Koonin, E.V.; Fedorova, N.D.; Jackson, J.D.; Jacobs, A.R.; Krylov, D.M.; Makarova, K.S.; Mazumder, R.; Mekhedov, S.; Nikolskaya, A.N.; Rao, B.S.; et al. A comprehensive evolutionary classification of proteins encoded in complete eukaryotic genomes. Genome Biol. 2004, 5, R7. [Google Scholar] [CrossRef] [PubMed]
- Tatusov, R.L.; Galperin, M.Y.; Natale, D.A.; Koonin, E.V. The COG database: A tool for genome scale analysis of protein functions and evolution. Nucleic Acids Res. 2000, 28, 33–36. [Google Scholar] [CrossRef]
- Eddy, S.R. Profile hidden Markov models. Bioinformatics 1998, 14, 755–763. [Google Scholar] [CrossRef] [PubMed]
- Apweiler, R.; Bairoch, A.; Wu, C.H.; Yeh, L.S.L. UniProt: The Universal Protein Knowledgebase. Nucleic Acids Res. 2017, 45, D158–D169. [Google Scholar] [CrossRef]
- Minoru, K.; Susumu, G.; Shuichi, K.; Yasushi, O.; Masahiro, H. The KEGG resource for deciphering the genome. Nucleic Acids Res. 2004, 32, D277. [Google Scholar] [CrossRef]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Cherry, J.M. Gene ontology: Tool for the unification of biology. The gene ontology consortium. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef]
- Chen, X.; Mao, X.; Huang, D.; Wu, J.; Dong, S.; Lei, K.; Ge, G.; Li, C.Y.; Wei, L. KOBAS2.0: A web server for annotation andidentification of enriched pathways and diseases. Nucleic Acids Res. 2011, 39, 316–322. [Google Scholar] [CrossRef]
- Zhang, L.B.; Zou, J.; Li, S.S.; Wang, B.S.; Raboanatahiry, N.; Li, M.T. Characterization and expression profiles of miRNAs in the triploid hybrids of Brassica napus and Brassica rapa. BMC Genom. 2019, 20, 649. [Google Scholar] [CrossRef]
- Guo, N.; Zhang, Y.J.; Sun, X.; Fan, H.H.; Gao, J.S.; Chao, Y.P.; Liu, A.F.; Yu, X.T.; Cai, Y.P.; Un, Y. Genome-wide identificationof differentially expressed miRNAs induced by ethephon treatment in abscission layer cells of cotton (Gossypium hirsutum). Gene 2018, 676, 263–268. [Google Scholar] [CrossRef] [PubMed]
- Li, P.R.; Su, T.B.; Zhang, D.S.; Wang, W.H.; Zhang, F.L. Genome-wide analysis of changes in miRNA and target gene expression reveals key roles in heterosis for Chinese cabbage biomass. Hortic. Res. 2021, 8, 39. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.; Sheng, J.C.; Xu, J.; An, X.X.; Hu, Q.H. Antioxidant activities of crude tea polyphenols, polysaccharides and proteins ofselenium-enriched tea and regular green tea. Eur. Food Res. Technol. 2007, 225, 843–848. [Google Scholar] [CrossRef]
- Hu, Q.; Pan, G.; Zhu, J. Effect of fertilization on selenium content of tea and the nutritional function of Se-enriched tea in rats. Plant Soil 2002, 238, 91–95. [Google Scholar] [CrossRef]
- Ye, Y.Y.; Yan, W.; Peng, L.J.; Zhou, J.J.; He, J.L.; Zhang, N.; Cheng, S.Y.; Cai, J. Insights into the key quality components in Se-Enriched green tea and their relationship with Selenium. Food Res. Int. 2023, 165, 112460. [Google Scholar] [CrossRef]
- Zhao, S.Q.; Wang, X.W.; Yan, X.M.; Guo, L.X.; Mi, X.Z.; Xu, Q.S.; Zhu, J.Y.; Wu, A.L.; Liu, L.L.; Wei, C.L. Revealing of the microRNA involved regulatory gene networks on terpenoid biosynthesis in Camellia sinensis in different growing time points. J. Agr. Food Chem. 2018, 66, 12604–12616. [Google Scholar] [CrossRef]
- Jeyaraj, A.; Wang, X.W.; Wang, S.S.; Liu, S.R.; Wei, C.L. Identification of regulatory networks of microRNAs and their targets in response to colletotrichum gloeosporioides in tea plant (Camellia sinensis L.). Front. Plant Sci. 2019, 10, 1096. [Google Scholar] [CrossRef]
- Chen, L.B.; Xia, L.F.; Liu, Y.; Sun, Y.N.; Jiang, H.B.; Tian, Y.L.; Chen, L. Screening of miRNA related to anthocyanin synthesis in tea cultivar ‘Zijuan’ based on high throughput sequencing. J. Tea Sci. 2019, 39, 681–691. [Google Scholar]
- Mi, S.; Cai, T.; Hu, Y.; Chen, Y.; Hodges, E.; Ni, F.; Wu, L.; Shan, L.; Zhou, H.; Ling, C. Sorting of small RNAs into Arabidopsis argonaute complexes is directed by the 5′terminal nucleotide. Cell 2008, 133, 116–127. [Google Scholar] [CrossRef]
- Mroczek-Zdyrska, M.; Magorzata, W. The Influence of selenium on root growth and oxidative stress induced by lead in viciafabal. minor plants. Biol. Trace Elem. Res. 2012, 147, 320–328. [Google Scholar] [CrossRef]
- Xu, M.Y.; Zhu, J.X.; Zhang, M.; Wang, L. Advances on plant miR169/NF-YA regulation modules. Hereditas 2016, 38, 700–706. [Google Scholar] [CrossRef] [PubMed]
- Rao, S.; Balyan, S.; Jha, S.; Mathur, S. Novel insights into expansion and functional diversification of MIR169 family in tomato. Planta 2020, 251, 55. [Google Scholar] [CrossRef] [PubMed]
- Jiao, Z.Y.; Lian, C.L.; Han, S.; Huang, M.B.; Shen, C.; Li, Q.; Niu, M.X.; Yu, X.; Yin, W.L.; Xia, X.L. PtmiR169o plays a positive role in regulating drought tolerance and growth by targeting the PtNF-YA6 gene in poplar. Environ. Exp. Bot. 2021, 189, 104549. [Google Scholar] [CrossRef]
- Zhao, M.; Ding, H.; Zhu, J.K.; Zhang, F.S.; Li, W.X. Involvement of miR169 in the nitrogen-starvation responses in Arabidopsis. New Phytol. 2011, 190, 906–915. [Google Scholar] [CrossRef] [PubMed]
- Luan, M.D.; Xu, M.Y.; Lu, Y.M.; Zhang, Q.X.; Zhang, L.; Zhang, C.Y.; Fan, Y.L.; Lang, Z.H.; Wang, L. Family-wide survey of miR169s and NF-YAs and their expression profiles response to abiotic stress in maize roots. PLoS ONE 2014, 9, E91369. [Google Scholar] [CrossRef] [PubMed]
- Buchner, P.; Parmar, S.; Kriegel, A. The sulfate transporter family in wheat: Tissue-specific gene expression inrelation tonutrition. Mol. Plant 2010, 3, 374–389. [Google Scholar] [CrossRef]
- Freeman, J.L.; Tamaoki, M.; Stushnoff, C.; Yuinn, C.F.; Cappa, J.J.; Devonshire, J.; Fakra, S.C.; Marcus, M.A.; McGrath, S.P.; Hoewyk, D.V.; et al. Molecular mechanisms of selenium tolerance and hyperaccumulation in Stanleya pinnata. Plant Physiol. 2010, 153, 1630–1652. [Google Scholar] [CrossRef]
- Hsieh, L.C.; Lin, S.I.; Kuo, H.F.; Chiou, T.J. Uncovering small RNA-mediated responses to phosphate deficiency in Arabidopsis by deep sequencing. Plant Physiol. 2009, 151, 2120. [Google Scholar] [CrossRef]
- Han, L.H.; Liu, C.; Zhao, M.Y.; Hu, L.J.; Hu, Y.S. Genomic identification and characterization analysis of the phosphate transporter protein 1 family gene in pineapple. Guihaia 2021, 41, 1955–1963. [Google Scholar] [CrossRef]
- Byrne, S.L.; Durandeau, K.; Nagy, I.; Barth, S. Identification of ABC transporters from Lolium perenne L. that are regulated by toxic levels of selenium. Planta 2010, 231, 901–911. [Google Scholar] [CrossRef]
- Ren, H.; Li, X.M.; Guo, L.; Wang, L.; Hao, X.Y.; Zeng, J.M. Integrative transcriptome and proteome analysis reveals the absorption and metabolism of selenium in tea plants [Camellia sinensis (L.) O. Kuntze]. Front. Plant Sci. 2022, 13, 848349. [Google Scholar] [CrossRef]
- Zhang, L.H.; Hu, B.; Deng, K.; Gao, X.K.; Sun, G.X.; Zhang, Z.L.; Li, P.; Wang, W.; Li, H.; Zhang, Z.H.; et al. NRT1.1B improves selenium concentrations in rice grains by facilitating selenomethinone translocation. Plant Biotechnol. J. 2019, 17, 1058–1068. [Google Scholar] [CrossRef]
- Li, Q.B.; Li, L.; Hu, C.X.; Tan, Q.L.; Sun, X.C. Metabolism response and molybdenum tolerance mechanism of winter wheat to excess molybdenum stress. J. Huazhong Agric. Univ. 2021, 40, 54–61. [Google Scholar] [CrossRef]
- Tian, M.; Xu, X.Y.; Liu, F.X.; Fan, X.; Pan, S.Y. Untargeted metabolomics reveals predominant alterations in primary metabolites of broccoli sprouts in response to pre-harvest selenium treatment. Food Res. Int. 2018, 111, 205–211. [Google Scholar] [CrossRef]
- Duan, N. Growth and Physiological and Transcriptomic Studies in Response to Nitrogen Addition and Drought Stressin Nitraria tangutorum. 2019. Available online: https://kns.cnki.net/KCMS/detail/detail.aspx?dbname=CDFDLAST2020&filename=1019211118.nh (accessed on 6 June 2019).
- Li, H.F.; Zhao, F.J. Selenium uptake, translocation and speciation in wheat supplied with selenate or selenite. New Phytol. 2008, 178, 92–102. [Google Scholar] [CrossRef]
- Chao, D.Y.; Baraniecka, P.; Danku, J.; Koprivova, A.; Lahner, B.; Luo, H.B.; Yakubova, E.; Dilkes, B.; Kopriva, S.; Salt, D. Variation in sulfur and selenium accumulation is controlled by naturally occurring isoforms of the key sulfur assimilation enzyme adenosine 5′-phosphosulfate reductase 2 across the Arabidopsis species range. Plant Physiol. 2014, 163, 1593–1608. [Google Scholar] [CrossRef]
- Kopriva, J.K.; Malagoli, M.; Takahashi, H. Sulfur nutrition: Impacts on plant development, metabolism, and stress responses. J. Exp. Bot. 2019, 70, 4069–4073. [Google Scholar] [CrossRef]
- Chan, K.X.; Phua, S.Y.; Breusegem, F.V. Secondary sulfur metabolism in cellular signalling and oxidative stress responses. J. Exp. Bot. 2019, 70, 4237–4250. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Samples | Raw Data | ‘N’-Containing Reads | Length < 18 | Length > 30 | Clean-Reads | Q30 (%) |
|---|---|---|---|---|---|---|
| T1-1 | 18,596,150 | 55 | 4,273,479 | 629,887 | 13,692,729 | 98.86 |
| T1-2 | 17,112,260 | 40 | 5,855,775 | 426,823 | 10,829,622 | 98.77 |
| T1-3 | 20,020,521 | 50 | 3,045,724 | 1,440,684 | 15,534,063 | 98.73 |
| T2-1 | 17,986,979 | 44 | 2,127,462 | 1,666,703 | 14,192,770 | 98.79 |
| T2-2 | 18,063,864 | 56 | 5,012,324 | 837,104 | 12,214,380 | 98.86 |
| T2-3 | 20,471,991 | 42 | 4,127,683 | 899,397 | 15,444,869 | 98.86 |
| Samples | Known miRNAs | Novel miRNAs | Total |
|---|---|---|---|
| T1-1 | 260 | 164 | 424 |
| T1-2 | 267 | 187 | 450 |
| T1-3 | 261 | 189 | 450 |
| T2-1 | 230 | 153 | 383 |
| T2-2 | 253 | 163 | 416 |
| T2-3 | 216 | 158 | 374 |
| Total | 455 | 203 | 658 |
| Type | COG | GO | KEGG | KOG | Pfam | Swissprot | EggNOG | Nr |
|---|---|---|---|---|---|---|---|---|
| T1 vs. T2 | 914 | 1003 | 866 | 1107 | 1836 | 1788 | 2094 | 2162 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cao, D.; Li, J.; Ma, L.; Liu, Y.; Huang, J.; Jin, X. Genome-Wide Identification of Selenium-Responsive MicroRNAs in Tea Plant (Camellia sinensis L. O. Kuntze). Horticulturae 2023, 9, 1278. https://doi.org/10.3390/horticulturae9121278
Cao D, Li J, Ma L, Liu Y, Huang J, Jin X. Genome-Wide Identification of Selenium-Responsive MicroRNAs in Tea Plant (Camellia sinensis L. O. Kuntze). Horticulturae. 2023; 9(12):1278. https://doi.org/10.3390/horticulturae9121278
Chicago/Turabian StyleCao, Dan, Juan Li, Linlong Ma, Yanli Liu, Jianan Huang, and Xiaofang Jin. 2023. "Genome-Wide Identification of Selenium-Responsive MicroRNAs in Tea Plant (Camellia sinensis L. O. Kuntze)" Horticulturae 9, no. 12: 1278. https://doi.org/10.3390/horticulturae9121278
APA StyleCao, D., Li, J., Ma, L., Liu, Y., Huang, J., & Jin, X. (2023). Genome-Wide Identification of Selenium-Responsive MicroRNAs in Tea Plant (Camellia sinensis L. O. Kuntze). Horticulturae, 9(12), 1278. https://doi.org/10.3390/horticulturae9121278