1. Introduction
The study of the dynamics of motion of molecular clusters is of great interest, since they can be used as structural elements of nanoelectromechanical devices [
1]. At the nanoscale, the most promising materials are carbon-based materials in the form of graphene, fullerene, nanotubes, etc. [
2,
3,
4]. Each allotropic form of carbon is interesting for its unique properties; for example, carbon nanotubes have ideal surface lattices, low density, and good electrical properties [
5,
6,
7]. Carbon nanotubes can be metallic or semiconducting [
8] and have high heat capacity, thermal conductivity, thermal stability and fire resistance [
9,
10,
11], and high tensile strength and Young’s modulus [
12,
13,
14]. Nanotubes find their application in electronics [
15], optics [
16], medicine [
17,
18,
19], and materials science [
20], allowing the production of nanodevices such as molecular probes, pipes, bearings, pumps, and so on.
Ferromagnetic nanoclusters are also of great scientific and technical importance. They can be used in different systems for magnetic cooling, mixing, identification, and acceleration of reactions of substances [
21,
22], as well as for biomedical purposes [
23,
24,
25], due to the high biocompatibility of the magnetic field and its ability to penetrate deep into living tissues without damaging them. However, their use is limited due to oxidation in air. At the same time, the use of ferromagnetic materials in combination with inert carbon materials allows one to overcome this limitation [
26]. In this regard, molecules with a rigid framework such as fullerenes and nanotori [
2,
3] are of particular interest.
A molecular torus is a nanotube with both ends connected together [
27]. The possibility of the existence of a whole family of nanotori with different numbers of carbon atoms from 80 and above has been theoretically predicted [
28]. Toroidal carbon nanotubes have been observed in various nanotube experiments [
3,
4,
6,
29]. However, nanotori have not been well studied experimentally due to a number of unsolved technological problems in their production in large quantities with given characteristics. Since nanotori are created from nanotubes, they share many of their properties, but at the same time, nanotori have their own unique properties due to their special geometric shape. The surface of carbon nanotori consists of carbon pentagons, hexagons, and heptagons [
4]. There are three approaches used to construct molecular tori [
30]. A generalized scheme for the generation of torodal molecules with different numbers of atoms is given in paper [
31]. An example of constructing a C
120 nanotorus using an initial structure of 12 carbon atoms is presented in paper [
32]. Nanotori have a geometric shape close to a disk shape, so they can form a columnar phase of liquid crystals [
33].
Carbon nanotori have good chemical and physical stability [
32,
34]. An analysis carried out using density functional theory computational methods showed that a carbon C
60 fullerene and C
120 nanotorus have the same chemical reactivity [
35]. A study of the fullerene of an endorally doped iron atom using theoretical density characteristics shows the stability of the C
59Fe, Fe@C
60, and Fe:C
60 fullerenes [
36,
37]. The best stability of endoral fullerene Fe@C
60 is observed when the iron atom is located under the center of the hexagonal ring [
36]. Despite the fact that hybridization occurs between the Fe atom and the C atoms of the framework, the magnetic moment of Fe@C
60 is close to the magnetic moment of an isolated Fe atom [
37]. Nanotori are also capable of holding atoms, molecules [
38,
39], and carbon [
40] and metal chains (Fe, Au, Cu) [
41] in the central and internal regions. Calculations of the electronic and magnetic properties of C
120 tori using density functional theory show that the composite structure containing C
120-Fe
10 iron has a magnetic moment the same as that of iron in the crystalline phase [
41]. In theoretical studies [
42,
43], C
120 +
nTi and Be:C
120 structures were investigated using density functional theory calculations as hydrogen-absorbing materials [
42,
43]. Calculations show that the simulated nanosystems have high structural and chemical stability.
The nature of the arrangement of carbon atoms in the nanotorus makes possible the existence of ring currents parallel to the torus plane [
44,
45,
46]. An unusually strong paramagnetic response has been theoretically predicted for some large carbon tori [
47,
48]. Due to the geometry of carbon nanthors and the features of their π-electron-dominated electronic structure, they appear to be ideal candidates for producing an enhanced magnetic response in the presence of an applied magnetic field. The influence of the magnetic field can be included in the π-orbital tight-binding Hamiltonian for carbon structures with metallic conductivity, as was done for nanotubes in paper [
49].
Typically, the nanotorus is studied as a single particle or in interaction with atoms or other large molecules. However, larger associations of nanotori have not been well studied. Previously, the authors, using the molecular dynamics method, showed that complex rotational–translational movements of some nanotori can lead to a loss of stability of the column, as a result of which it is divided into individual nanotori or fragments of smaller sizes [
50,
51]. At the same time, even for stable clusters, a change in molecular geometry (doping with iron atoms) and an external force (such as a magnetic field) can unpredictably change the processes of interaction of nanotori in the columnar phase. Stable structures of this kind can exhibit collective gyroscopic effects, which can significantly enhance resistance to external influences. In this regard, the stability of such structures in the presence and absence of a magnetic field is of interest. The most convenient approach for studying large associations of molecules is using the methods of classical molecular dynamics. The studies using the molecular dynamics method and continuum approximation of the oscillatory characteristics of a toroidal C
120 molecule located inside a nanotube, as well as a nanotube passing through the inner region of a nanotorus, have shown that the oscillation frequencies are in the megahertz range [
52,
53]. The interaction between two nanotori of different diameters was studied semi-analytically using the continuum approximation, which showed the presence of oscillatory motion [
54].The study of nanotori using classical molecular dynamics methods showed their high resistance to mechanical stress and deformation, and also revealed the flow of molecular current in the most deformed areas of the atomic framework [
55]. Therefore, carbon nanotori can also be used to improve the tribological properties of lubricants [
56]. Thus, carbon nanotori are nanodevices with many outstanding properties that allow them to be used as gigahertz or megahertz oscillators, traps for atoms and molecules, and so on. The functionality of these nanoscale devices is determined by their molecular structures, their sizes, and what atoms or molecules are included in their structures.
In this paper, we numerically studied the dynamic state of a cluster located in the columnar phase and consisting of several toroidal molecules. The interaction between molecules under the influence of a magnetic field is modeled using classical molecular dynamics methods [
57,
58,
59]. The influence of temperature and the magnitude and direction of a magnetic field on the collective dynamics of a molecular cluster that has a different number of nanotori and consists of molecules with a different number of atoms is parametrically analyzed.
2. Research Methodology and Mathematical Formulation of the Problem
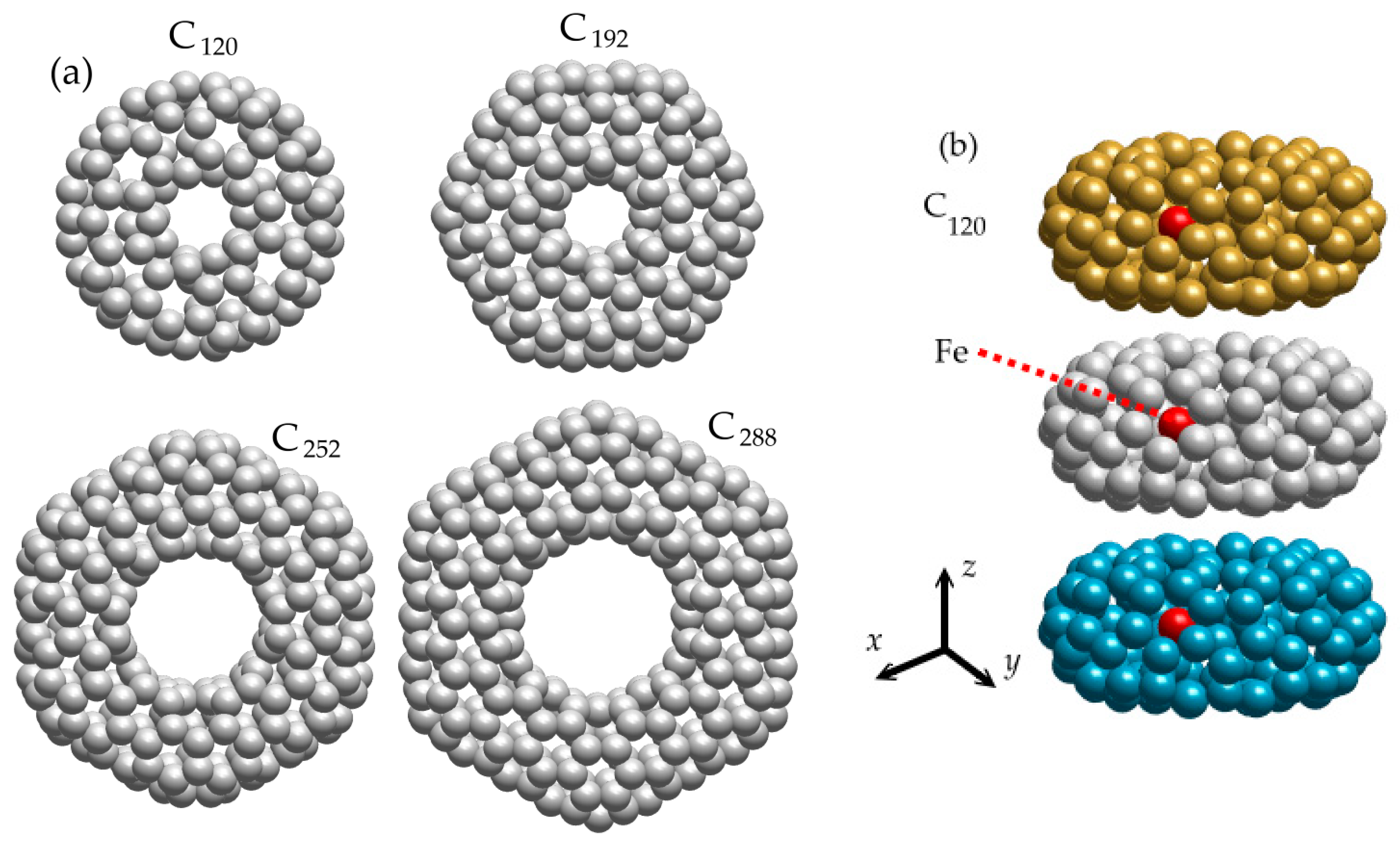
We have considered the group interaction of identical carbon nanotori, which can have different numbers of carbon atoms
n1 = (
n − 1) = 120, 192, 252, 288 (
Figure 1a). All molecules except C
120 (
D5d) belong to point group symmetries
D6h [
31,
32,
60]. The C
192 and C
252 nanotori have an armchair structure, and C
288 has a zigzag structure. In our study, each nanotorus has an additional iron atom (Fe, 56 amu) located within the interior of the nanotorus itself (
Figure 1b). Nanotori are considered as non-deformable objects that interact with each other through van der Waals forces. We chose nanotori in which an external magnetic field induces weak paratropic ring currents [
47], so we have neglected this in our mathematical model. Note that the collective behavior of toroidal molecules possessing high paramagnetic or diamagnetic moments may differ from the behavior of nanotori described in this article and requires additional study.
2.1. Mathematical Formulation of the Problem
Since tori are frame molecular structures, their movement and interaction with each other can be described as the movement of non-deformable molecular bodies performing translational movements and simultaneously rotating around their own centers of mass. In this case, the impact of one object on another object is determined by the sums of all possible pair interactions of individual atoms that make up the molecular objects under consideration. This is the so-called atom–atom interaction approach, which has its advantages. Firstly, it is an opportunity to write down formally strict equations of motion of objects. Secondly, most of the resulting equations are resolved with respect to the sought functions. This makes it possible to apply the entire set of previously developed schemes for the numerical integration of equations that determine the mechanical motion of the objects under consideration. Nevertheless, this approach has disadvantages. The atoms that make up the framework structures under consideration are in a bound state. Therefore, pairwise interactions of bonded atoms belonging to different nanotori can be determined only through the effective parameters of the spherical potential used. These parameters, as a rule, differ significantly from the corresponding parameters of free atoms. However, it is possible to determine the values of these parameters from some integral characteristics of the interaction of carbon objects that have a completely identical or even similar crystal structure. In this way, the integral molecular interaction can be approximated to the true interaction, despite the simple form of the potential used.
The translational motions of nanotori are determined by the equations
Here,
M is the mass of the torus;
is the velocity vector of the center of mass of the
ith torus;
U is the Lennard-Jones interatomic pair potential [
61];
is the distance between the
kth and
mth atoms in the
ith and
jth tori;
is the radius vector of the position of the center of mass of the
kth torus;
n is the total number of carbon and iron atoms in the molecular torus Fe@C
n–1;
N is the number of nanotori. The pair interaction potential
U is calculated according to the Lorentz–Berthelot mixing rule [
61], i.e., its value depends on which pair of atoms it is determined for. These could be two carbon atoms, a carbon atom and an iron atom, or two iron atoms.
Vector Equation (1) is integrated under the following initial conditions:
where
is the initial thermal velocity of an individual molecule, and
is its initial position.
The rotation of the
ith molecule around its center of mass is determined from the equation for angular momentum in the following form:
Here, Ki = Ji ωi is the angular moment of the ith torus; ωi = (ωix, ωiy, ωiz) is the angular velocity of the molecule; Mi is the moment of the external forces of the ith molecule; is the torque caused by the magnetic field; Ji is the inertia tensor determined by the nature of the mass distribution in space, i.e., coordinates of the centers of the atoms that make up the body. If we encounter an iron atom, then mk = m(Fe). In all other cases, mk = m(C). If these coordinates are measured from the center of mass of the molecular system, the components of the symmetric inertia tensor are determined by Equation (7).
Vectors of moments of forces acting on the
ith torus are determined by the following equations:
Here, rik = (xik, yik, zik) is the radius vector that determines the position of the kth atom in the ith molecular torus.
To close the model of this level, it is necessary to have data on the magnetic moments of iron atoms contained inside nanotori. As a first approximation, we assume that an iron atom embedded in a torus molecule has the same magnetic moment as a free iron atom. Then, each nanotorus will have its own magnetic moment, coinciding in magnitude with the corresponding moment of the iron atom. It can be directed arbitrarily relative to the main axes of the nanotorus.
We suppose that the C
120 nanocage (like the other high-molecular-weight carbon systems considered here) is endorally doped with one iron atom. Calculations carried out using density functional theory [
36,
37] show that the endoral position of the iron atom, which corresponds to the best stability of the Fe@C
60 system, is located under the center of the hexagonal ring. There is also the distance at which the Fe atom is located from the fullerene surface. In addition, in the same work, the total magnetic moment of the Fe@C
60 system was determined. It turned out to be very close to the magnetic moment of an isolated dopant atom. The nanotori under consideration have a similar carbon-based molecular framework to C
60. Therefore, it would be appropriate to assume that the iron atom is endorally attached to this base in a similar way and the nature of the hybridization of the orbitals remains similar. Thus, the data on the total magnetic moment for the Fe@C
60 and Fe@C
120 complexes are close, as are the data on the distance at which the iron atom is separated from the carbon surface. With such a preliminary analysis, it is already possible to perform a calculation using the model of atom–atom interactions if the direction of the magnetic moment of the metal–carbon structure in question is known.
We will determine this direction with the coordinates of the nanotorus pseudonode, which has zero mass. If this node in each torus has number
n + 1, and the node containing an iron atom has number
n, then, if we know the coordinates of the pseudonode, the direction cosines relative to the axes of the absolute basis will be determined as follows:
The procedure for determining the coordinates of a pseudonode is included in the general scheme for calculating the coordinates of nanotorus nodes. When the direction cosines are known α
i, β
i, γ
i (
i = 1, …,
N), the moment of force acting from the external magnetic field can be found as follows:
Here, μi = μ is the total magnetic moment of a toroidal molecule, and B is the magnetic induction. The value of the magnetic moment μ does not contain a subscript, since it is the same for all nanotori.
The velocity of the
kth atom during rotation of the
ith molecule is determined based on the classical theorem of the addition of velocities in the complex motion of an atom together with the center of mass of the molecule and rotation around it. Then, taking into account the fictitious force center with number
n + 1, we obtain
The initial conditions have the following form:
2.2. Numerical Scheme for Solving the Problem
Equations (1)–(12) can be considered as a Cauchy problem for a system of first-order evolution equations that serve to determine the coordinates and velocities of force centers, the coordinates and velocities of the centers of mass of carbon tori, and their angular velocities. All mentioned groups of equations are separated with respect to unknowns, except for the equations for angular velocities. In the last group of equations, by virtue of (4)–(6), the projections of the angular velocities of the tori on the axes of the absolute basis are associated with the components of the inertia tensor of an individual torus. The matrix of the system, which could be completely diagonal, becomes block-diagonal in its lower part. In this case, the blocks have a small size of 3 × 3. Because of this, it is possible to isolate the equations for the projections of angular velocities in an analytical way, using the direct method of solving systems of algebraic equations, i.e., Cramer’s rule. This can always be done since the determinant of the inertia tensor matrix of any molecular body is always different from zero.
If the system of initial first-order differential equations is separated, then it is suitable for applying explicit step-by-step methods of a significant order of accuracy. To improve the accuracy of calculations, these methods use the idea of recalculation at intermediate positions of a separate time step. The Runge–Kutta scheme of the standard fourth order of accuracy has this property [
62,
63]. This scheme was used to solve the problem. However, the procedure for separating the required functions was not implemented at the level of equations, but was synthesized into a high-precision time integration scheme. In other words, such division was carried out at each individual position of each time step. The result is a procedure that allows for a small relative error in calculating the total energy of the system, as well as the projections of the total angular momentum of all molecular bodies under consideration.
Next, we will analyze the application of the Runge–Kutta scheme of the fourth order of accuracy using the example of solving the equation for the angular moment, which is written in index-free form as
Vector
K can be a vector of sequentially written components of the angular moments of all molecular tori. If
r is also a column vector of the coordinates of all atoms of molecular tori, including the coordinates of the centers of mass of the tori, and
v is a column vector of the velocities of all these material and fictitious points, the kinematic relationship between coordinates and velocities, written in an absolute basis, looks as usual:
On the right side of Equation (13), the sums of the moments of van der Waals forces and magnetic forces are sequentially written. These moments ultimately depend only on the coordinates of the centers of force (including the point of zero mass). Therefore, according to the Runge–Kutta scheme, to find moments in intermediate positions of a separate time step (four positions correspond to a scheme of fourth order accuracy), we can write the following equations:
Here, v1 = v(t), v2 = v(t + Δt/2), v3 = v(t + Δt/2), v4 = v(t + Δt).
Then, the values of the angular moments at the next time step can be found as follows:
After a new value for the angular momentum of an individual torus is found, using Equations (4)–(6), we can determine the vector of the instantaneous angular velocity of the torus. However, this same vector can also be found using intermediate values of the angular moment:
Then, the idea of a recount will be fully realized. All calculations were carried out with a step of Δt = 10−6 ns. Software for simulating the translational and rotational dynamics of a nanotori column was developed using Visual Studio C++ 17.1.
3. Results and Discussion
We considered a microcanonical ensemble of
N interacting nanotori (see
Figure 1b), each of which has
n − 1 carbon atoms (C, 12 Da) and a single iron atom (Fe, 56 Da). The maximum total magnetic moment of each toroidal molecule
μmax was taken to be 3.23
μB, where
μB = 9.27∙10
−24 J/T is the Bohr magneton. At the initial moment of time, nanotori have zero translational and angular velocities:
,
. In this case, the planes of all tori are oriented parallel to the
xy-plane. We assumed that the total magnetic moment of the nanotorus is also collinear to the
xy-plane. It is assumed that doping of toroidal molecules with an iron atom can have different effects on the total magnetic moment of the molecule depending on the location, direction, and number of iron atoms; therefore, the maximum total magnetic moment of the molecule varies parametrically from 0 to
μmax. A cluster of
N molecules is subject to a magnetic field with a magnetic induction value
B varying from 0 to 5 T. Values of
B above 1 T are used mainly to understand the limiting values of the influence of the magnetic field, since this influence at
B less than 1 T has a large scatter. The Lennard-Jones potential parameters for iron and carbon atoms have the following values:
ε(Fe)/
kB = 27.7 K,
σ(Fe) = 0.454 nm;
ε(C)/
kB = 51.2 K,
σ(C) = 0.3653 nm where
kb = 1.38∙10
−23 J∙K
−1 is the Boltzmann constant; ε is the depth of the potential well; σ is the distance at which the potential energy is zero.
Before studying the group dynamics of a cluster of toroidal molecules (based on carbon nanotori C
120, C
192, C
252, and C
288) with one iron atom, it is necessary to determine the position of the minimum potential energy. For this purpose, the dependence of the total additive Lennard-Jones potential on the distance
s between the centers of mass of two toroidal molecules is used:
where
smk is the distance between the
mth and
kth atoms of the first and second nanotori.
Figure 2 shows the dimensionless dependences of the intermolecular potential energy of two molecules on the distance between them, divided by the number of interatomic interactions
n2. The position of the minimum energy
smin(C
n) for each
n = 120, 192, 252, 288 is shown by a vertical dashed line. As can be seen from the figure, the smallest value
smin is observed for the C
120 torus, and the largest is for C
252. At the initial moment of time, the centers of mass of the toroidal molecules are located at a distance
smin(C
n) from each other along the
z-axis, as shown in
Figure 1b. Thus, the
z-coordinate of the center of mass of the
ith nanotorus at the initial moment of time is set equal to (
i − 1)∙
smin(C
n), where
i = 1, …,
N.
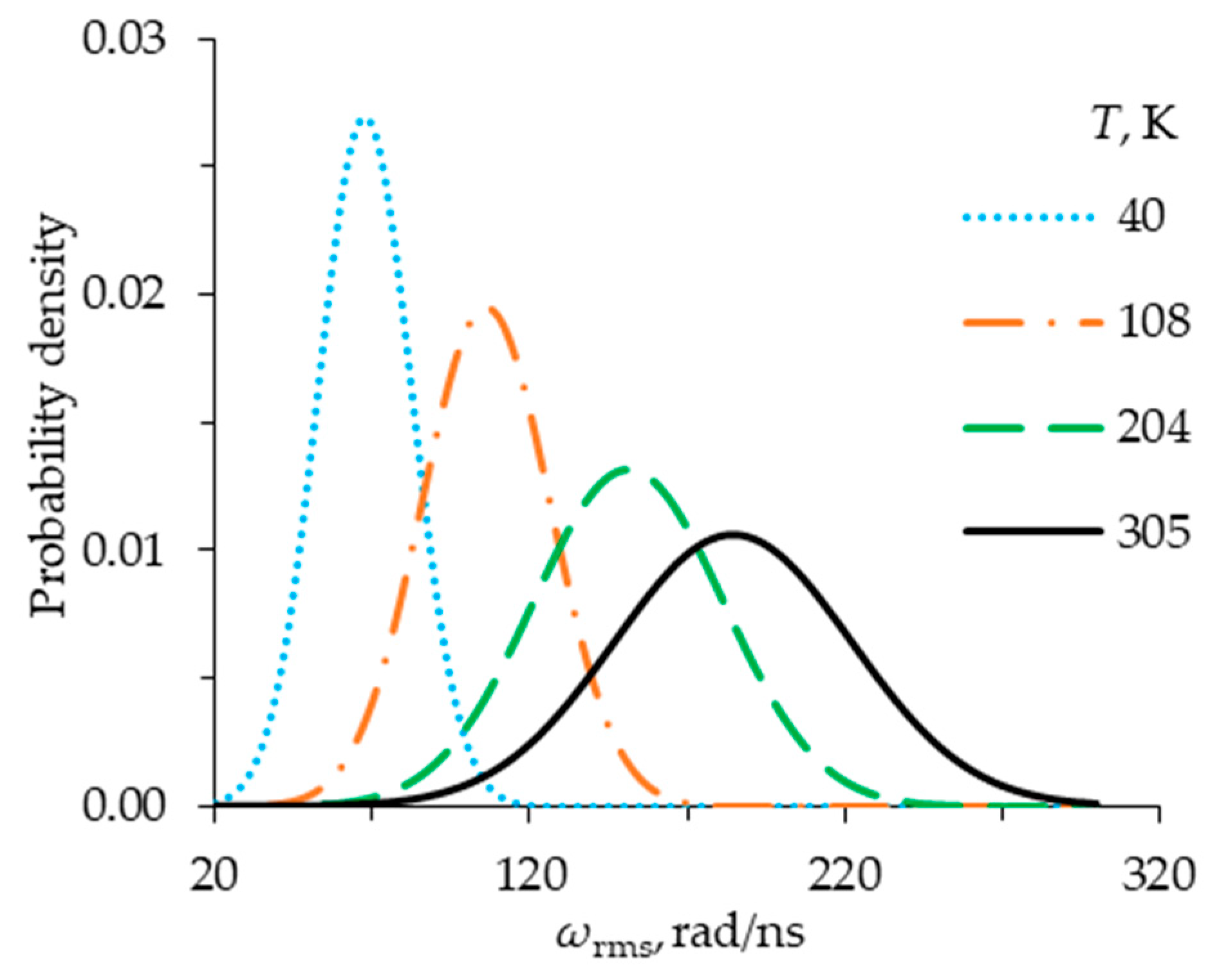
Figure 3 and
Figure 4 give a probabilistic description of the motion of molecules in a system consisting of three Fe@C
120 nanotori in the absence of an external magnetic field (
B = 0). By influencing the molecules with various small perturbations at the initial moments of time, we obtained a number of systems with different energy states and different velocities of thermal motion of the molecules. Molecules with average kinetic energy are characterized by the values of translational and rotational root-mean-square velocities
vrms(
t) and
ωrms(
t). According to the results obtained, the distributions of values
vrms(
t) and
ωrms(
t) obey the law of normal distribution (see
Figure 3 and
Figure 4). Numerical results showed that in the range from 40 to 350 K, Fe@C
120 nanotori arranged in a column retain their original columnar structure, while performing translational and rotational oscillations of varying intensity relative to each other, which directly depend on temperature. The average temperature value, characterizing the different thermal states of systems over a given period of time, was determined based on the assumption that molecules have 6 degrees of freedom, and the average value of the total kinetic energy in the system is 3
kBT.
We have chosen four cases that give different distributions of the root-mean-square velocity of translational and rotational motion of molecules in the system over a period of 10 ns (see
Figure 3 and
Figure 4). These cases correspond to the following average temperatures: 40, 108, 204, and 305 K. Note that since the system consists of a small number of molecules, the root-mean-square speeds
vrms and
ωrms have fairly large deviations at each moment of time. This suggests that, in reality, each nanotori system under consideration exists in a certain temperature range. In addition,
Figure 3 and
Figure 4 show that this range increases at higher temperatures. This means that systems with a higher average temperature and similar initial parameters will be characterized by a significantly greater scatter between the results. In this regard, it is convenient to use the case of
T = 40 K as a base case.
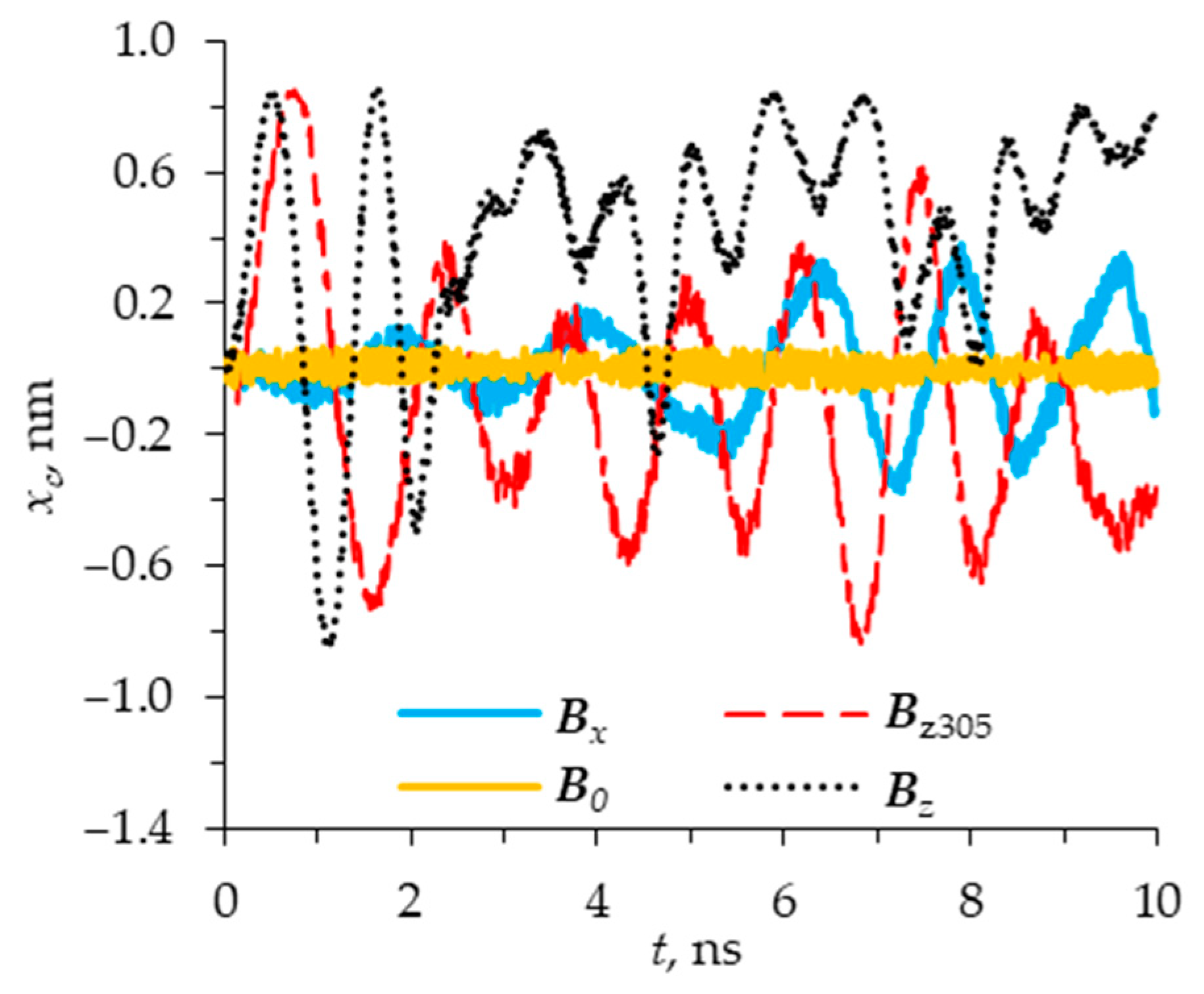
Figure 5,
Figure 6 and
Figure 7 show several special cases of the influence of a magnetic field on a column of 3 Fe@C
120 nanotori, each of which has 120 carbon atoms (C, 12 Da) and 1 iron atom (Fe, 56 Da). The total magnetic moment of the molecule
μ is taken to be equal to
μmax.
Figure 5,
Figure 6 and
Figure 7 show the
x-,
y-, and
z-coordinates of the centers of mass of the first nanotorus as a function of time
t. Curves
B0,
B1,
Bx,
Bz,
Bz305 correspond to the magnetic induction
B =
B0 = (0, 0, 0),
B =
Bx = (1, 0, 0),
B =
Bz = (0, 0, 1) at a temperature
T = 40 K, and
B =
Bz305 = (0, 0, 1) at
T = 305 K, respectively. The effect with the magnetic induction directed along the
y-axis is not considered since it is identical to the case
B =
Bx. In the absence of a magnetic field (B = B0), the centers of mass of the nanotori perform high-frequency oscillations with average linear deviations relative to the initial position equal to about 0.026 nm along the x- and y-axes and 0.004 nm along the z-axis. In general, the cluster maintains a stable position in space. Note that in the absence of a magnetic field, the motions of C120 and Fe@C120 molecules in the cluster differ slightly from each other. We also tried adding and concentrating different numbers of iron atoms (from one to five) at different locations on the nanotorus to break the symmetry of the molecular geometry and thus affect the stability of the system. However, we have not yet been able to do this, since the nanotorus rotates around its axis, taking such a position as to eliminate the appearance of instability caused by modification of the nanotori geometry. This suggests that the association of nanotori of this type is very stable.
In the presence of a magnetic field with a magnetic induction directed along the x-axis (B = Bx), an additional oscillation mode appears in the xy-plane. The average linear deviations in all x-, y-, and z-directions increase to 0.12, 0.26, and 0.13 nm, respectively. The influence of an external magnetic field does not lead to the destruction of the column into molecules moving independently in space. However, it leads to a fairly strong change in the position of the nanotorus relative to the initial state, although μ and Bx are parallel at the initial moment of time and therefore the torque should be equal to 0 according to Equation (1). However, a magnetic field can affect nanotori due to the thermal movement of molecules, as a result of which the orientation of the molecules changes and conditions for non-zero arise.
In the cases of
B =
Bz and
B =
Bz305, the picture is radically different from the previous two cases,
B =
B0 and
B =
Bx. A dramatic increase in the average linear deviation of the center of mass of the nanotorus is observed in all directions and most strongly in the
z-direction up to 0.45 and 0.54 nm at
B =
Bz and
B =
Bz305, respectively (see
Figure 5).
This shows that the direction of the magnetic field relative to the magnetic moment mu plays a large role in the dynamics of the molecules located in the cluster. The cases
B =
Bz305 and
B =
Bz have similar features. It can be seen that the nanotorus periodically returns to its original position (see
Figure 5), but with different frequencies. In general, the results presented in
Figure 3,
Figure 4 and
Figure 5 show that intense movements of a toroidal molecule in the presence of a magnetic field occur in the case when the magnetic induction is directed perpendicular to the column of nanotori and, therefore, to the magnetic moment
μ.
Figure 6 illustrates in more detail the case of exposure to a magnetic field at
B =
Bz305 in order to understand how the magnetic field affects the molecular cluster in general. In
Figure 6, curves 1, 2, and 3 correspond to the number of the nanotorus in the column, consisting of three molecules. As can be seen from
Figure 8, the centers of mass of the first and third nanotori change places under the influence of a magnetic field
Bz305 = 1 T. The center of mass of the average molecule maintains its position. It can be seen that during the time interval under consideration, stable group rotations of the nanotori system are maintained relative to the position at which the common center of mass was located at the initial moment of time.
The mutually perpendicular external magnetic field (B = Bz305) and the magnetic moment of the molecule μ create conditions for rotating nanoobjects by 90 degrees. However, the system acquires a sufficiently large mechanical impulse to neutralize the orientational effect of the external magnetic field at the moment when B and μ become parallel. In addition, each molecule has different orientations of the magnetic moment μi, the central molecule performs predominantly its own rotation around the axis, and the remaining molecules move in orbit around a common center of mass. This is due to the relative position and geometric shape of the nanotori, as well as the van der Waals attractive forces that maintain the coordinated movement of the nanotori. Thus, the magnetic field directed along the column (along the z-axis) generates periodic collective orbital motion of this column around a common center of mass and relative to the xy-plane. In this case, the collective periodic motion occurs on a nanosecond time scale. At the same time, the structure of the system itself is preserved despite the complex interaction between the nanotori, which arises during collective motion due to the influence of an external magnetic field.
Until now, we have considered cases in which the external magnetic field at the initial moment of time is strictly perpendicular or parallel to the total magnetic moment of the molecule. However, if the magnetic moment of the molecule μ is not directed perpendicular to the direction of the external magnetic field, then the results will be similar to those for the magnetic moment of the molecule, which has a smaller value. In addition, nanotori can contain different numbers of iron atoms, which can either reduce the total magnetic moment of the molecule μ due to the different directions of individual moments of the iron atoms, or increase it. The total magnetic moment of a molecule also depends on the position of the Fe atom in the nanotorus structure, and therefore can have values significantly lower than μmax.
As the curves
Bx in
Figure 5,
Figure 6 and
Figure 7 show, if the directions of the external magnetic field and the magnetic moment coincide, a π-rotation of the system (by 180 degrees) is not observed. To perform a rotation, a deviation of the magnetic moment of the molecule μ from the direction of the external magnetic field is required such that at the initial moment of time the projection onto the vector
B =
Bz is less than the projection onto the plane perpendicular to
B =
Bz (i.e.,
μz <
μxy). It is worth noting that some additional deviation is always present due to the thermal movement of molecules in the system, even if it was absent at the initial moment.
Figure 9 shows the parametric dependence of the time of the first π-rotation
tz1 on the relative value of the magnetic moment of the molecule for systems of Fe@C
120 nanotori with different average temperatures in the presence of a magnetic field
Bz = 1 T. The value
tz1 shows the response time of the nanotori system to the influence of an external magnetic field and its transition to the oscillations on a nanosecond time scale. Parametric analysis (see
Figure 8) shows that the same external magnetic field has a weaker effect on systems with higher temperatures. The average value of the function
tz1(
μ) increased by 81%, 96%, and 165% compared to the cases of
T = 40 K for
T = 108, 204, and 305 K, respectively. It is also clear that with a decrease in the total magnetic moment of the molecule μ, the rotation time of the system
tz1 increases. It follows that when nanotori are saturated with iron atoms and at a higher temperature, most likely, the influence of the external magnetic field will decrease.
It is of interest to study the influence on collective rotations of such factors as the size of the nanotori cluster and the magnitude of magnetic induction.
Figure 10 shows the dependence of the half-period
Tz (i.e., the time the cluster rotates through an angle of 180 degrees) on the cluster size at
μ =
μmax,
Bz = 1, and 5 T, calculated as the average value over a time interval from 0 to 10 ns. The blue square and orange circle correspond to calculations with
Bz = 1 and
Bz = 5 T, respectively. As can be seen, there is a linear dependence of the half-cycle of rotation on the number of nanotori in the cluster at
Bz = 5 T. The dotted line is obtained by the least squares method from data for
Bz = 5 T. It shows that increasing the column size by one nanotorus increases the rotation time by Δ
Tz = 0.176 ns. Nanotori in a larger cluster need to travel longer orbital distances, so
Tz increases with cluster size in proportion to the orbital radius. However, there is no obvious linear dependence of the average rotation time on the number of nanotori in the cluster at
Bz = 1 T. As can be seen from
Figure 10, the half-period of rotation of systems with three, four, and five nanotori increase with the number of nanotori, but a strong decrease in the half-period
Tz is observed for systems with five and eight nanotori. This is due to the process of chaotic motion of molecules, which can have the same strong effect as the magnetic field at Bz = 1 T, but unlike the latter, thermal motion introduces a random character into the process of rotation of the nanotori cluster. However, obtaining a relationship based on linear regression for
Bz = 1 T (dashed curve) can be useful for comparing the effectiveness of increasing the magnitude of magnetic induction. The results of the numerical data give Δ
Tz = 0.362 ns for
Bz = 1 T. Thus, an increase in the magnitude of magnetic induction by 5 times increases Δ
Tz by only 2.06 times. Note that numerical calculations have shown that even one molecule with an iron atom (Fe@C
120) is enough to cause a system of two to eight C
120 nanotori to rotate.
Figure 11 presents the results of a parametric study of the influence of the magnitude of magnetic induction and the type of molecule (Fe@C
120, Fe@C
192, Fe@C
252, and Fe@C
288) on the cluster rotation frequency ω
z.
Figure 11 shows the dependence of the average rotation frequency
ωz of a column consisting of three nanotori on the value of magnetic induction
Bz. The frequency lies in the mega- and gigahertz ranges. It can be seen that the greater the magnetic induction, the greater the rotation frequency of the column. The magnetic field has a greater effect on Fe@C
120 and less on Fe@C
288 because these nanotori have the lowest and highest masses compared to others. Judging by the trends observed from
Figure 11, groups of nanotori with a large total number of atoms will rotate in a magnetic field mainly with frequencies in the megahertz range. The dependence of the cluster rotation frequency on the magnetic induction value for
Bz ≥ 1 has a form close to a linear dependence (hence
Tz (
Bz) has a hyperbolic dependence). However, it can be seen that, in general, the frequency at small values of
Bz < 1 is lower than it could be with a linear dependence. This is due to the thermal movement of molecules, which causes scatter and generally reduces the frequency at low magnetic field values (
Bz < 1), although it can increase, as in the case of a Fe@C
120 molecule at
Bz = 1 T.
Note that we have presented results concerning continuous exposure to an external magnetic field, but even short-term exposure lasting less than a nanosecond makes it possible to create conditions for long-term collective motion of nanotori, since the main task that an external magnetic field solves is to unbalance a cluster of several nanotori by creating a directed mechanical impulse of sufficient magnitude. As the calculation results show, a magnetic field up to B = 5 T does not lead to the destruction of the columnar structure into individual independently moving molecules. To make a group of two to eight C120 nanotori rotate using a magnetic field, it is enough that among them there is at least one molecule with a single iron atom (Fe@C120). In this case, it does not really matter where in the molecular structure the Fe atom is located and where the molecule is located in the cluster, but the total magnetic moment of the molecule must be quite large and not equal to zero.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}










