
Transcriptome Analysis of Immune Response Against Edwardsiella tarda Infection in Spotted Sea Bass (Lateolabrax maculatus)

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Experimental Fish
2.2. Edwardsiella tarda Infection and Sample Collection
2.3. Transcriptome Sequencing
2.4. Transcriptome Data Analysis
2.5. QRT-PCR Validation
3. Results
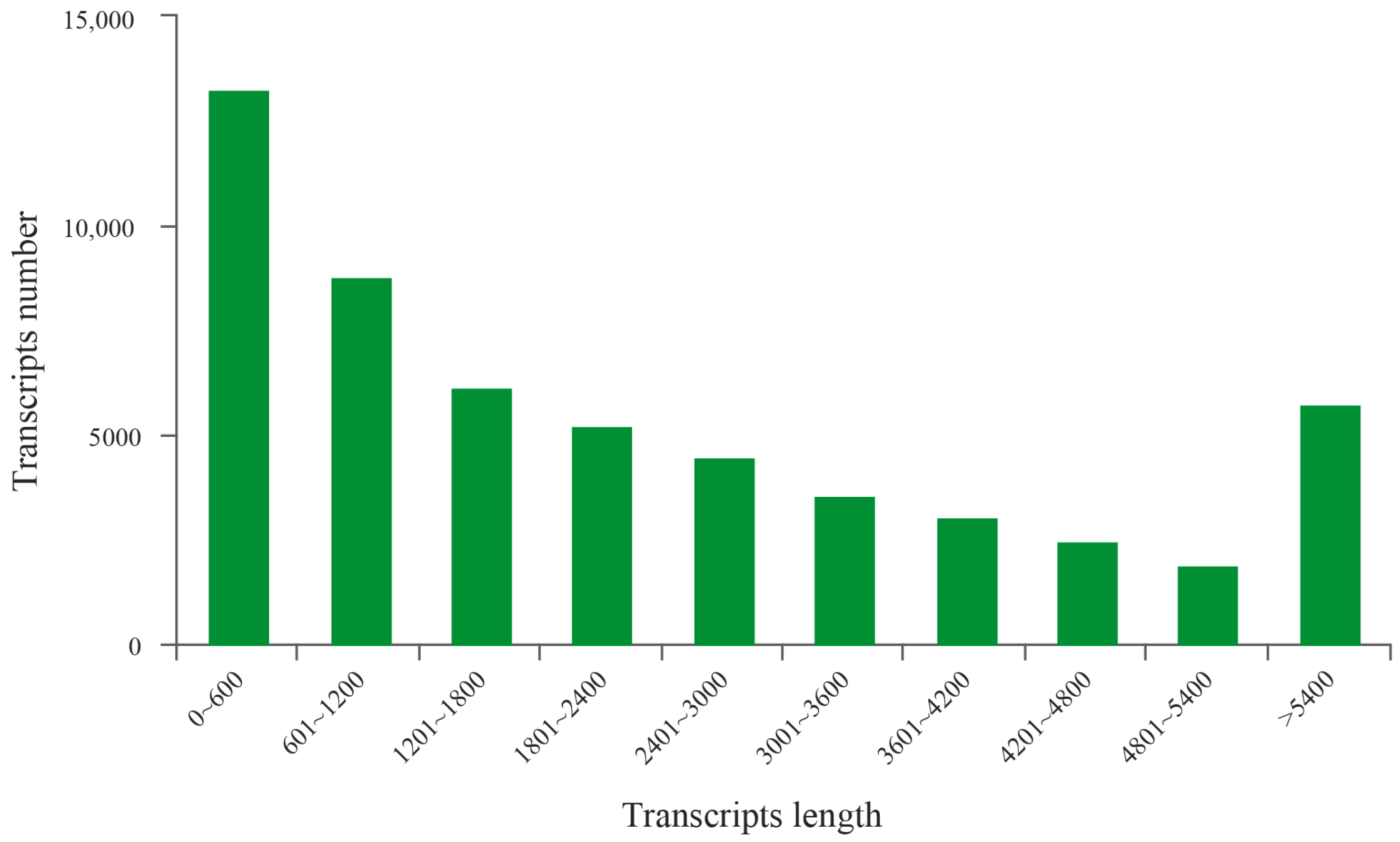
3.1. Transcriptome Assembly
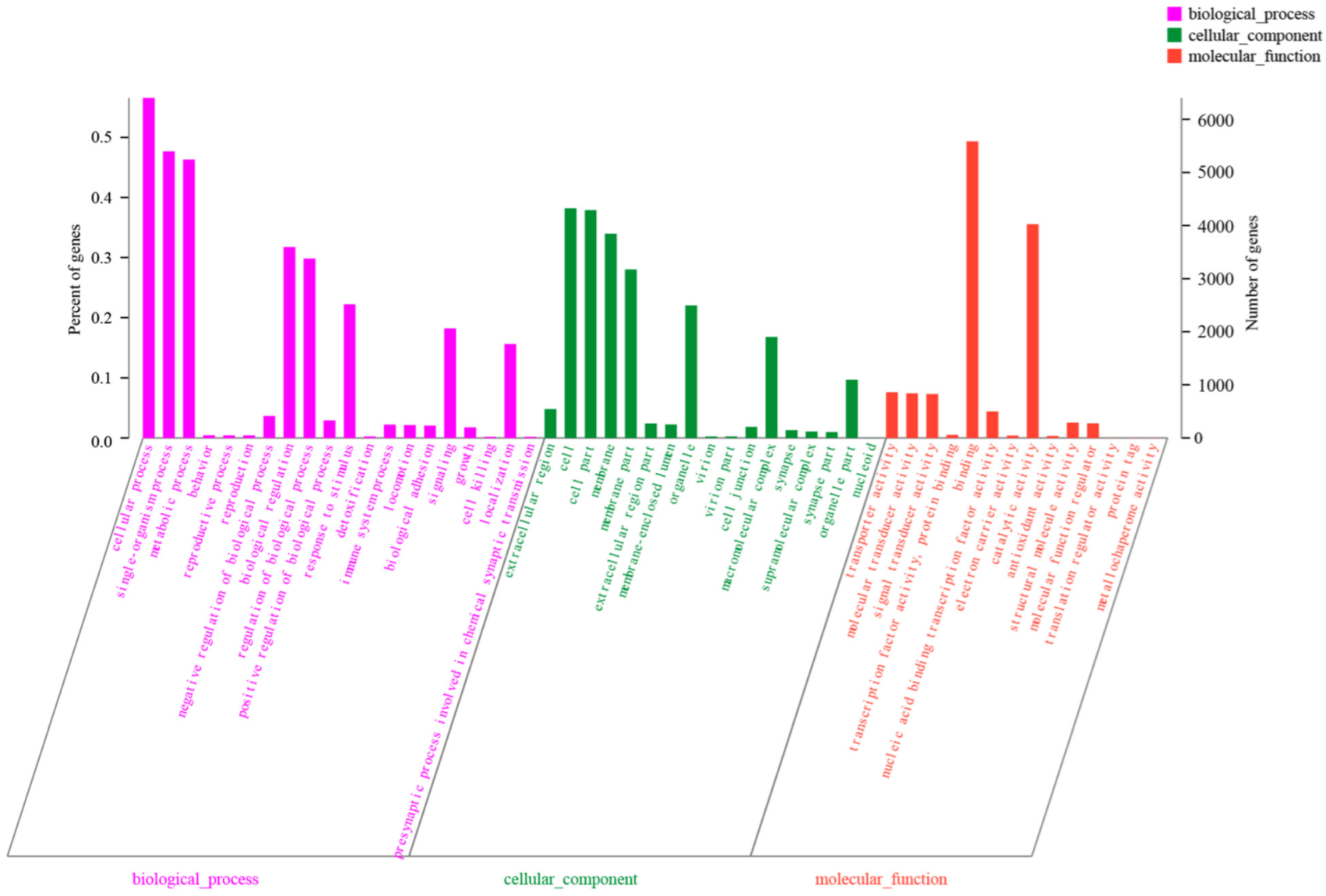
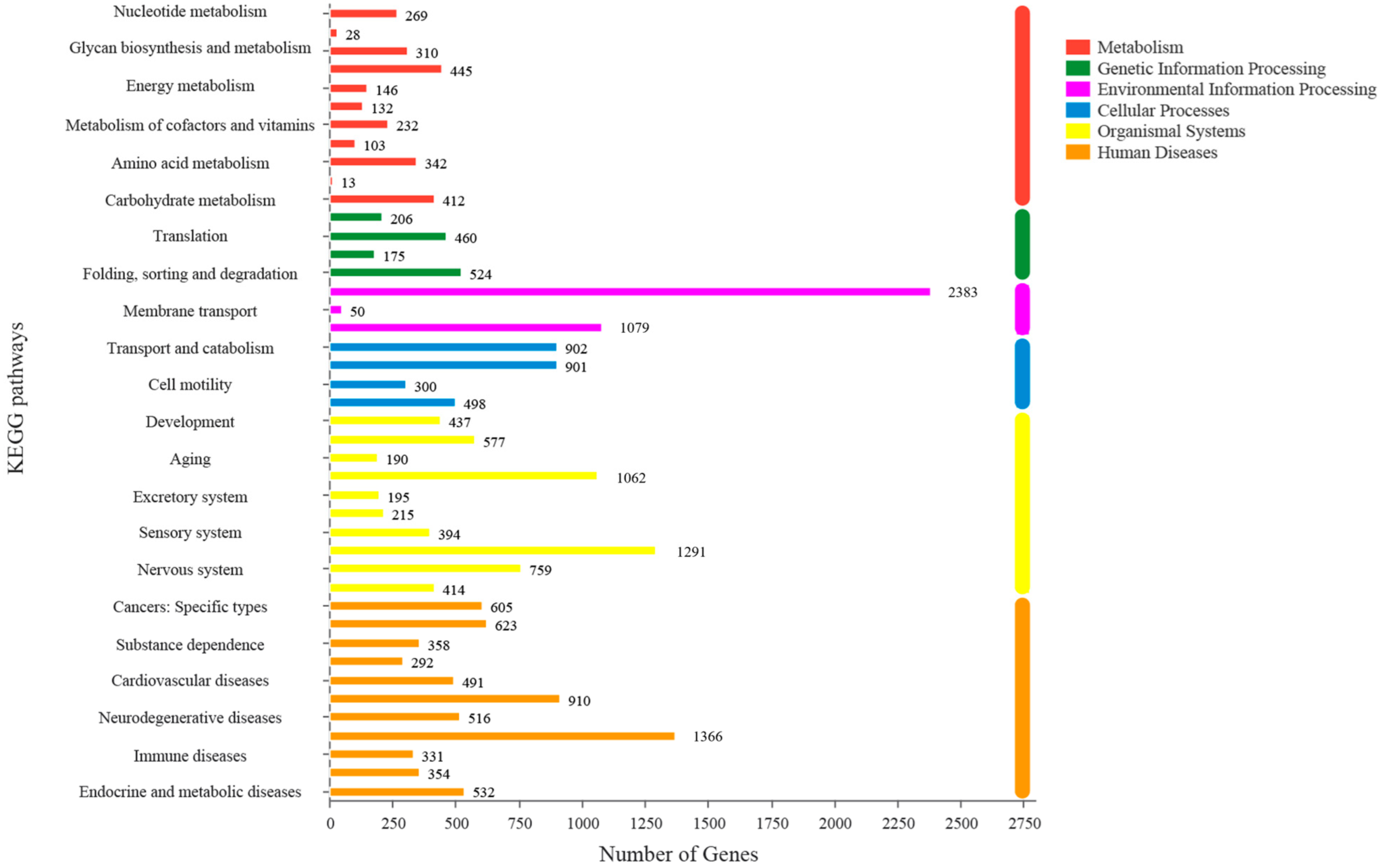
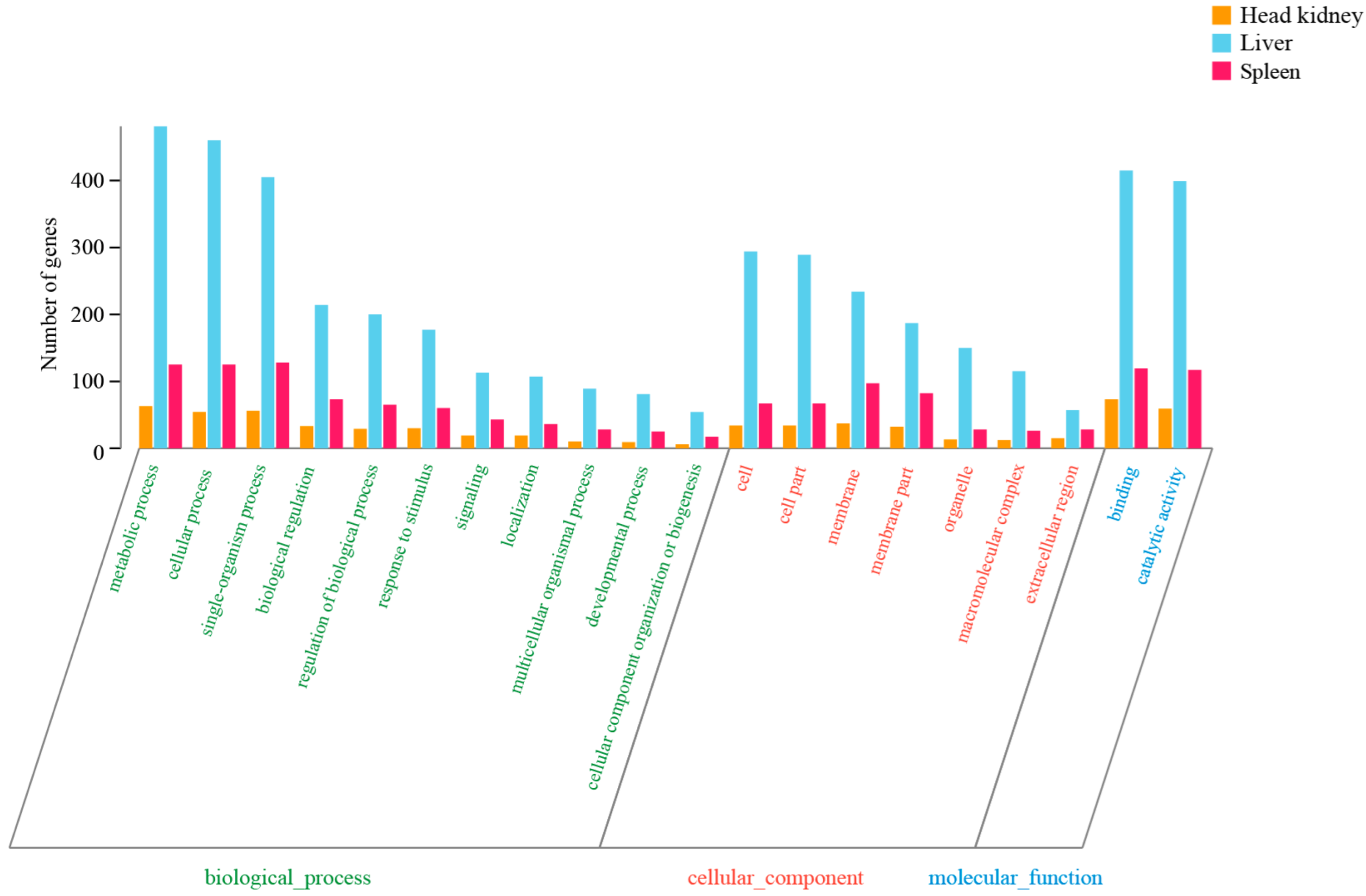
3.2. Annotation of Assembled Unigenes
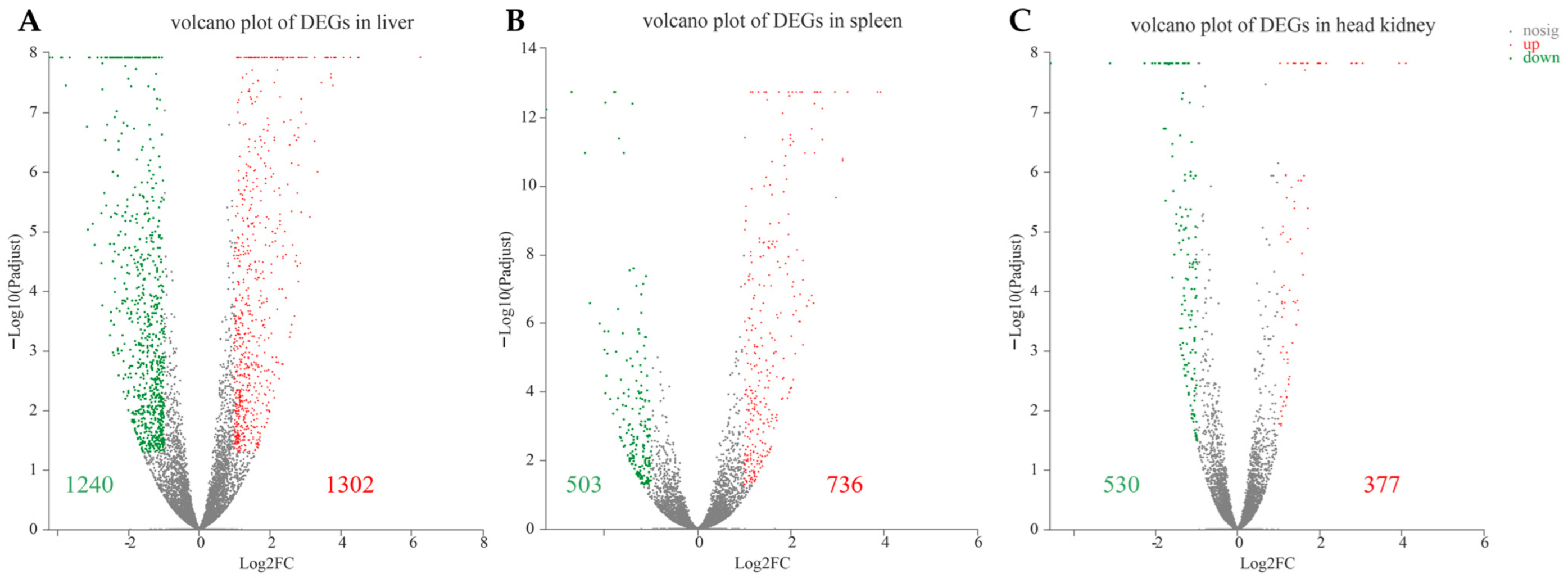
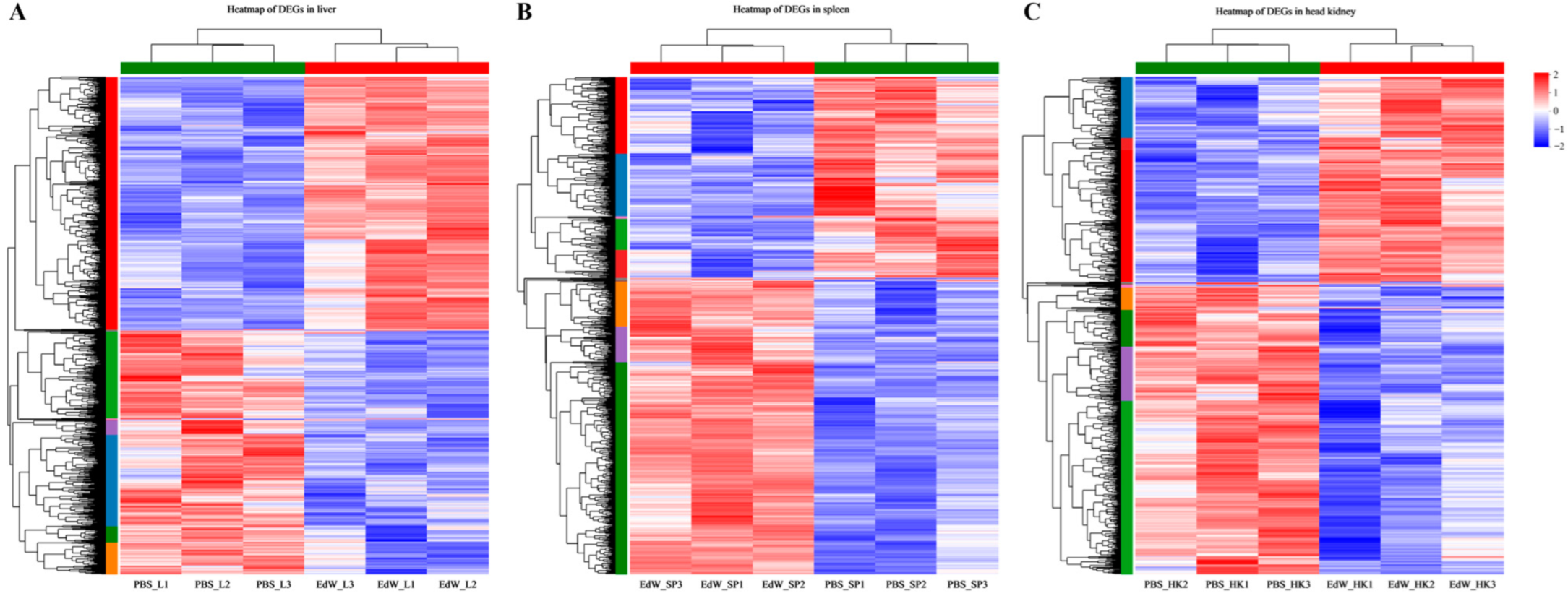
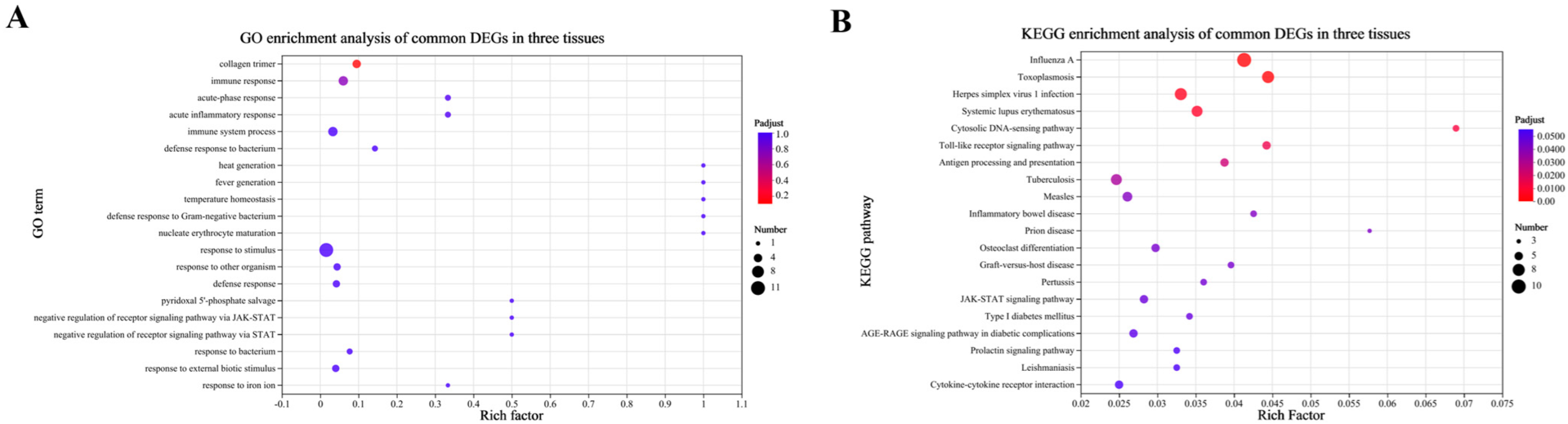
3.3. Identification of Differentially Expressed Genes
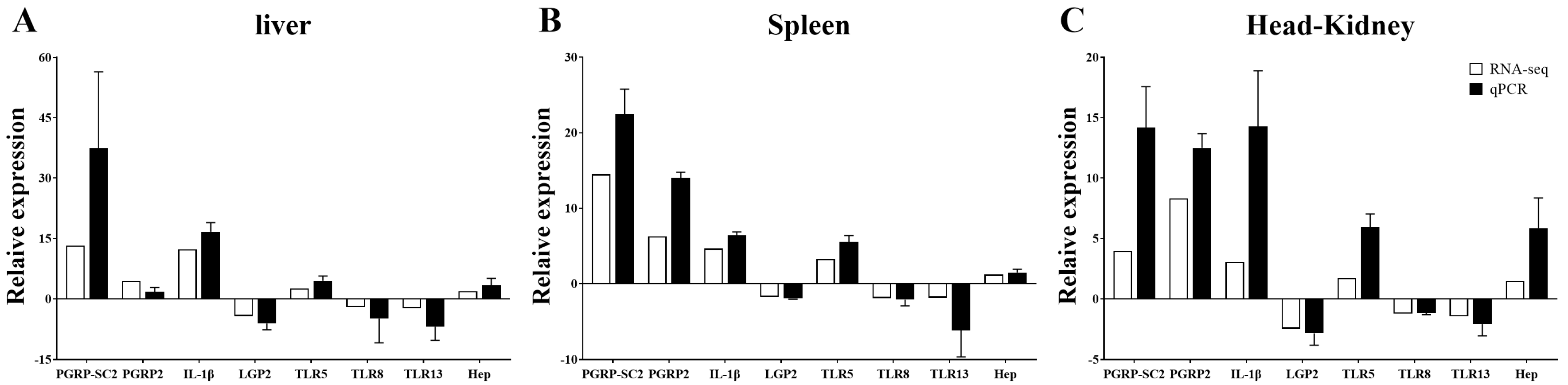
3.4. Validation of Differently Expressed Genes by qRT-PCR
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Okuda, J.; Arikawa, Y.; Takeuchi, Y.; Mahmoud, M.M.; Suzaki, E.; Kataoka, K.; Suzuki, T.; Okinaka, Y.; Nakai, T. Intracellular replication of Edwardsiella tarda in murine macrophage is dependent on the type III secretion system and induces an up-regulation of anti-apoptotic NF-kappaB target genes protecting the macrophage from staurosporine-induced apoptosis. Microb. Pathog. 2006, 41, 226–240. [Google Scholar] [CrossRef] [PubMed]
- Castro, N.; Toranzo, A.E.; Barja, J.L.; Nunez, S.; Magarinos, B. Characterization of Edwardsiella tarda strains isolated from turbot, Psetta maxima (L.). J. Fish. Dis. 2006, 29, 541–547. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.S.; Lee, K.W.; Jung, H.S.; Kim, J.; Yun, A.; Cho, S.H.; Kwon, M. Effects of dietary inclusion of yacon, ginger and blueberry on growth, body composition and challenge test of juvenile rockfish (Sebastes schlegeli) against Edwardsiella tarda. Aquacult Nutr. 2018, 24, 1048–1055. [Google Scholar] [CrossRef]
- Ling, S.H.; Wang, X.H.; Xie, L.; Lim, T.M.; Leung, K.Y. Use of green fluorescent protein (GFP) to study the invasion pathways of Edwardsiella tarda in in vivo and in vitro fish models. Microbiology 2000, 146 Pt 1, 7–19. [Google Scholar] [CrossRef]
- Mohanty, B.R.; Sahoo, P.K. Edwardsiellosis in fish: A brief review. J. Biosci. 2007, 32, 1331–1344. [Google Scholar] [CrossRef]
- Wu, J.; Liu, G.; Sun, Y.; Wang, X.; Fang, H.; Jiang, H.; Guo, Z.; Dong, J. The role of regulator FucP in Edwardsiella tarda pathogenesis and the inflammatory cytokine response in tilapia. Fish. Shellfish. Immunol. 2018, 80, 624–630. [Google Scholar] [CrossRef] [PubMed]
- Zeng, C.; Tang, X.; Du, Y.; Sheng, X.; Xing, J.; Zhan, W. Dynamic distribution of formalin-inactivated Edwardsiella tarda in olive flounder (Paralichthys olivaceus) post intramuscular injection. Vet. Immunol. Immunopathol. 2018, 199, 53–60. [Google Scholar] [CrossRef]
- Morimoto, N.; Kondo, M.; Kono, T.; Sakai, M.; Hikima, J.I. Nonconservation of TLR5 activation site in Edwardsiella tarda flagellin decreases expression of interleukin-1beta and NF-kappaB genes in Japanese flounder, Paralichthys olivaceus. Fish. Shellfish. Immunol. 2019, 87, 765–771. [Google Scholar] [CrossRef]
- Tang, X.; Gong, J.; Zeng, C.; Sheng, X.; Xing, J.; Zhan, W. Dynamic distribution of formalin-inactivated Edwardsiella tarda in flounder (Paralichthys olivaceus) post intraperitoneal vaccination. Fish. Shellfish. Immunol. 2019, 89, 393–402. [Google Scholar] [CrossRef]
- Bujan, N.; Mohammed, H.; Balboa, S.; Romalde, J.L.; Toranzo, A.E.; Arias, C.R.; Magarinos, B. Genetic studies to re-affiliate Edwardsiella tarda fish isolates to Edwardsiella piscicida and Edwardsiella anguillarum species. Syst. Appl. Microbiol. 2018, 41, 30–37. [Google Scholar] [CrossRef]
- Noman Reza, M.A.; Mohapatra, S.; Shimizu, S.; Kitamura, S.I.; Harakawa, S.; Kawakami, H.; Nakayama, K.; Sawayama, E.; Matsubara, T.; Ohta, K.; et al. Molecular cloning, characterization and expression analysis of complement components in red sea bream (Pagrus major) after Edwardsiella tarda and red sea bream Iridovirus (RSIV) challenge. Fish. Shellfish. Immunol. 2018, 82, 286–295. [Google Scholar] [CrossRef]
- Reddacliff, G.L.; Hornitzky, M.; Whittington, R.J. Edwardsiella tarda septicaemia in rainbow trout (Oncorhynchus mykiss). Aust. Vet. J. 1996, 73, 30. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Wang, Q.L.; Lu, Y.; Chen, S.L.; Li, Q.; Sha, Z.X. Expression profiles of NODs in channel catfish (Ictalurus punctatus) after infection with Edwardsiella tarda, Aeromonas hydrophila, Streptococcus iniae and channel catfish hemorrhage reovirus. Fish. Shellfish. Immunol. 2012, 33, 1033–1041. [Google Scholar] [CrossRef]
- Janda, J.M.; Duman, M. Expanding the spectrum of diseases and disease associations caused by Edwardsiella tarda and related species. Microorganisms 2024, 12, 1031. [Google Scholar] [CrossRef] [PubMed]
- Yi, L.; Nie, P.; Yu, H.B.; Xie, H.X. Regulation of Type III Secretion of Translocon and Effector Proteins by the EsaB/EsaL/EsaM Complex in Edwardsiella tarda. Infect. Immun. 2017, 85, e00322-17. [Google Scholar] [CrossRef]
- Camp, K.L.; Wolters, W.R.; Rice, C.D. Survivability and immune responses after challenge with Edwardsiella ictaluri in susceptible and resistant families of channel catfish, Ictalurus punctatus. Fish. Shellfish. Immunol. 2000, 10, 475–487. [Google Scholar] [CrossRef]
- Mutz, K.O.; Heilkenbrinker, A.; Lonne, M.; Walter, J.G.; Stahl, F. Transcriptome analysis using next-generation sequencing. Curr. Opin. Biotechnol. 2013, 24, 22–30. [Google Scholar] [CrossRef]
- Lu, A.J.; Hu, X.C.; Wang, Y.; Zhu, A.H.; Shen, L.L.; Tian, J.; Feng, Z.Z.; Feng, Z.J. Skin immune response in the zebrafish, Danio rerio (Hamilton), to Aeromonas hydrophila infection: A transcriptional profiling approach. J. Fish. Dis. 2015, 38, 137–150. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, S.; Chen, S.; Chen, Y.; Liu, Y.; Shao, C.; Wang, Q.; Lu, Y.; Gong, G.; Ding, S.; et al. Transcriptome analysis revealed changes of multiple genes involved in immunity in Cynoglossus semilaevis during Vibrio anguillarum infection. Fish. Shellfish. Immunol. 2015, 43, 209–218. [Google Scholar] [CrossRef]
- Huang, Y.; Huang, X.; Yan, Y.; Cai, J.; Ouyang, Z.; Cui, H.; Wang, P.; Qin, Q. Transcriptome analysis of orange-spotted grouper (Epinephelus coioides) spleen in response to Singapore grouper iridovirus. BMC Genom. 2011, 12, 556. [Google Scholar] [CrossRef]
- Chen, J.; Li, C.; Huang, R.; Du, F.; Liao, L.; Zhu, Z.; Wang, Y. Transcriptome analysis of head kidney in grass carp and discovery of immune-related genes. BMC Vet. Res. 2012, 8, 108. [Google Scholar] [CrossRef]
- Zhang, X.; Mu, Y.; Mu, P.; Ao, J.; Chen, X. Transcriptome analysis reveals comprehensive insights into the early immune response of large yellow croaker (Larimichthys crocea) Induced by Trivalent Bacterial Vaccine. PLoS ONE 2017, 12, e0170958. [Google Scholar] [CrossRef]
- Wu, R.; Sheng, X.; Tang, X.; Xing, J.; Zhan, W. Transcriptome analysis of flounder (Paralichthys olivaceus) gill in response to lymphocystis disease virus (LCDV) infection: Novel insights into fish defense mechanisms. Int. J. Mol. Sci. 2018, 19, 160. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, B.; Liu, M.; Jiang, K.; Wang, M.; Wang, L. Comparative transcriptome analysis reveals the different roles between hepatopancreas and intestine of Litopenaeus vannamei in immune response to aflatoxin B1 (AFB1) challenge. Comp. Biochem. Physiol. C Toxicol. Pharmacol. 2019, 222, 1–10. [Google Scholar] [CrossRef]
- Cai, Y.; Wang, S.; Guo, W.; Xie, Z.; Zheng, Y.; Cao, Z.; Zhou, Y. Transcriptome analysis provides insights into the immune responsive pathways and genes in the head kidney of tiger grouper (Epinephelus fuscoguttatus) fed with Spatholobus suberectus, Phellodendron amurense, or Eclipta prostrata. Fish. Shellfish. Immunol. 2018, 73, 100–111. [Google Scholar] [CrossRef] [PubMed]
- Ahn, D.H.; Kang, S.; Park, H. Transcriptome analysis of immune response genes induced by pathogen agonists in the Antarctic bullhead notothen Notothenia coriiceps. Fish. Shellfish. Immunol. 2016, 55, 315–322. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.H.; Miyata, M.; Chan, C.; Ngoh, S.Y.; Liew, W.C.; Saju, J.M.; Ng, K.S.; Wong, F.S.; Lee, Y.S.; Chang, S.F.; et al. Differential Transcriptomic Response in the Spleen and Head Kidney Following Vaccination and Infection of Asian Seabass with Streptococcus iniae. PLoS ONE 2014, 9, e99128. [Google Scholar] [CrossRef]
- Zhu, J.; Li, C.; Ao, Q.; Tan, Y.; Luo, Y.; Guo, Y.; Lan, G.; Jiang, H.; Gan, X. Trancriptomic profiling revealed the signatures of acute immune response in tilapia (Oreochromis niloticus) following Streptococcus iniae challenge. Fish. Shellfish. Immunol. 2015, 46, 346–353. [Google Scholar] [CrossRef]
- Zhang, R.; Zhang, L.L.; Ye, X.; Tian, Y.Y.; Sun, C.F.; Lu, M.X.; Bai, J.J. Transcriptome profiling and digital gene expression analysis of Nile tilapia (Oreochromis niloticus) infected by Streptococcus agalactiae. Mol. Biol. Rep. 2013, 40, 5657–5668. [Google Scholar] [CrossRef]
- Pan, C.; Zhu, Y.R.; Cao, K.X.; Li, J.X.; Wang, S.Y.; Zhu, J.H.; Zeng, X.M.; Zhang, H.Q.; Qin, Z.W. Transcriptome, intestinal microbiome and histomorphology profiling of differences in the response of Chinese sea bass (Lateolabrax maculatus) to Aeromonas hydrophila infection. Front. Microbiol. 2023, 14, 1103412. [Google Scholar] [CrossRef]
- Wang, B.T.; Hu, J.M.; Feng, J.; Zhang, Y.Q.; Sun, Y.X.; Jiang, B.; Li, W.; Liu, C.; Huang, Y.H.; Su, Y.L. Acute septicemia and immune response of spotted sea bass (Lateolabrax maculatus) to Aeromonas veronii infection. Fish. Shellfish. Immun. 2022, 124, 47–55. [Google Scholar] [CrossRef]
- Liu, H.; Wang, M.L.; Du, J.Y.; Wang, S.; Zhang, Z.; He, T.T.; Wang, Y.; Chen, Y.; Wang, W.; Li, X.J. Skin transcriptome of lenok trout (Brachymystax lenok) provides new insight on lectin genes and immune response mechanisms to Aeromonas salmonicida infection. Comp. Biochem. Phys. D 2025, 54, 101439. [Google Scholar] [CrossRef]
- Huang, R.S.; Tao, Y.F.; Jiang, B.J.; Badran, M.F.; Zhu, J.; Hua, J.X.; Wang, Q.C.; Lu, S.Q.; Saleh, M.H.L.; Aboueleila, R.H.M.; et al. Integrated gill transcriptome and biochemical indices analyses reveal that acute salinity stress induces oxidative stress and immune and metabolic disorders in Red Tilapia (Oreochromis spp.). Aquaculture 2025, 599, 742108. [Google Scholar] [CrossRef]
- Xing, M.C.; Rong, Z.; Zhao, X.; Gao, X.W.; Hou, Z.G.; Zhang, L.H.; Khor, W.; Xu, Y.H.; Chen, L.; Wu, C.B. Transcriptome analysis reveals hypoxic response key genes and modules as well as adaptive mechanism of crucian carp (Carassius auratus) gill under hypoxic stress. Front. Immunol. 2025, 16, 1543605. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Wen, H.; Wang, H.; Ren, Y.; Zhao, J.; Li, Y. RNA-Seq analysis of salinity stress-responsive transcriptome in the liver of spotted sea bass (Lateolabrax maculatus). PLoS ONE 2017, 12, e0173238. [Google Scholar] [CrossRef]
- Cock, P.J.; Fields, C.J.; Goto, N.; Heuer, M.L.; Rice, P.M. The Sanger FASTQ file format for sequences with quality scores, and the Solexa/Illumina FASTQ variants. Nucleic Acids Res. 2010, 38, 1767–1771. [Google Scholar] [CrossRef]
- Conesa, A.; Gotz, S.; Garcia-Gomez, J.M.; Terol, J.; Talon, M.; Robles, M. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef]
- Finn, R.D.; Clements, J.; Eddy, S.R. HMMER web server: Interactive sequence similarity searching. Nucleic Acids Res. 2011, 39, W29–W37. [Google Scholar] [CrossRef]
- Xie, C.; Mao, X.Z.; Huang, J.J.; Ding, Y.; Wu, J.M.; Dong, S.; Kong, L.; Gao, G.; Li, C.Y.; Wei, L.P. KOBAS 2.0: A web server for annotation and identification of enriched pathways and diseases. Nucleic Acids Res. 2011, 39, W316–W322. [Google Scholar] [CrossRef]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T.; et al. Gene ontology: Tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef]
- Tatusov, R.L.; Koonin, E.V.; Lipman, D.J. A genomic perspective on protein families. Science 1997, 278, 631–637. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M.; Sato, Y.; Kawashima, M.; Furumichi, M.; Tanabe, M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 2016, 44, D457–D462. [Google Scholar] [CrossRef]
- Medzhitov, R.; Janeway, C.A., Jr. Decoding the patterns of self and nonself by the innate immune system. Science 2002, 296, 298–300. [Google Scholar] [CrossRef]
- Hoffmann, J.A. The immune response of Drosophila. Nature 2003, 426, 33–38. [Google Scholar] [CrossRef]
- Janeway, C.A., Jr.; Medzhitov, R. Innate immune recognition. Annu. Rev. Immunol. 2002, 20, 197–216. [Google Scholar] [CrossRef] [PubMed]
- Gulati, A.; Kaur, D.; Krishna Prasad, G.V.R.; Mukhopadhaya, A. PRR function of innate immune receptors in recognition of bacteria or bacterial ligands. Adv. Exp. Med. Biol. 2018, 1112, 255–280. [Google Scholar] [CrossRef]
- Farre, E.M.; Liu, T. The PRR family of transcriptional regulators reflects the complexity and evolution of plant circadian clocks. Curr. Opin. Plant Biol. 2013, 16, 621–629. [Google Scholar] [CrossRef] [PubMed]
- West, A.P.; Koblansky, A.A.; Ghosh, S. Recognition and signaling by toll-like receptors. Annu. Rev. Cell Dev. Biol. 2006, 22, 409–437. [Google Scholar] [CrossRef]
- O’Neill, L.A.; Golenbock, D.; Bowie, A.G. The history of Toll-like receptors—Redefining innate immunity. Nat. Rev. Immunol. 2013, 13, 453–460. [Google Scholar] [CrossRef]
- Hopkins, P.A.; Sriskandan, S. Mammalian Toll-like receptors: To immunity and beyond. Clin. Exp. Immunol. 2005, 140, 395–407. [Google Scholar] [CrossRef]
- Stein, C.; Caccamo, M.; Laird, G.; Leptin, M. Conservation and divergence of gene families encoding components of innate immune response systems in zebrafish. Genome Biol. 2007, 8, R251. [Google Scholar] [CrossRef]
- Roach, J.C.; Glusman, G.; Rowen, L.; Kaur, A.; Purcell, M.K.; Smith, K.D.; Hood, L.E.; Aderem, A. The evolution of vertebrate Toll-like receptors. Proc. Natl. Acad. Sci. USA 2005, 102, 9577–9582. [Google Scholar] [CrossRef] [PubMed]
- Baoprasertkul, P.; Peatman, E.; Abernathy, J.; Liu, Z. Structural characterisation and expression analysis of toll-like receptor 2 gene from catfish. Fish. Shellfish. Immunol. 2007, 22, 418–426. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.N.; Wang, Z.Y.; Yao, C.L. Characterization of Toll-like receptor 3 gene in large yellow croaker, Pseudosciaena crocea. Fish. Shellfish. Immunol. 2011, 31, 98–106. [Google Scholar] [CrossRef]
- Meijer, A.H.; Gabby Krens, S.F.; Medina Rodriguez, I.A.; He, S.; Bitter, W.; Ewa Snaar-Jagalska, B.; Spaink, H.P. Expression analysis of the Toll-like receptor and TIR domain adaptor families of zebrafish. Mol. Immunol. 2004, 40, 773–783. [Google Scholar] [CrossRef]
- Palti, Y.; Rodriguez, M.F.; Vallejo, R.L.; Rexroad, C.E., 3rd. Mapping of Toll-like receptor genes in rainbow trout. Anim. Genet. 2006, 37, 597–598. [Google Scholar] [CrossRef] [PubMed]
- Baoprasertkul, P.; Peatman, E.; Somridhivej, B.; Liu, Z. Toll-like receptor 3 and TICAM genes in catfish: Species-specific expression profiles following infection with Edwardsiella ictaluri. Immunogenetics 2006, 58, 817–830. [Google Scholar] [CrossRef]
- Chen, X.; Wang, Q.; Yang, C.; Rao, Y.; Li, Q.; Wan, Q.; Peng, L.; Wu, S.; Su, J. Identification, expression profiling of a grass carp TLR8 and its inhibition leading to the resistance to reovirus in CIK cells. Dev. Comp. Immunol. 2013, 41, 82–93. [Google Scholar] [CrossRef]
- Wang, R.H.; Li, W.; Fan, Y.D.; Liu, Q.L.; Zeng, L.B.; Xiao, T.Y. TLR22 structure and expression characteristic of barbel chub, Squaliobarbus curriculus provides insights into antiviral immunity against infection with grass carp reovirus. Fish. Shellfish. Immunol. 2017, 66, 120–128. [Google Scholar] [CrossRef]
- Ng, C.S.; Kato, H.; Fujita, T. Recognition of viruses in the cytoplasm by RLRs and other helicases--how conformational changes, mitochondrial dynamics and ubiquitination control innate immune responses. Int. Immunol. 2012, 24, 739–749. [Google Scholar] [CrossRef]
- Gu, T.; Lu, L.; An, C.; Chen, B.; Wei, W.; Wu, X.; Xu, Q.; Chen, G. MDA5 and LGP2 acts as a key regulator though activating NF-kappaB and IRF3 in RLRs signaling of mandarinfish. Fish. Shellfish. Immunol. 2019, 86, 1114–1122. [Google Scholar] [CrossRef]
- Pollpeter, D.; Komuro, A.; Barber, G.N.; Horvath, C.M. Impaired cellular responses to cytosolic DNA or infection with Listeria monocytogenes and vaccinia virus in the absence of the murine LGP2 protein. PLoS ONE 2011, 6, e18842. [Google Scholar] [CrossRef]
- Paria, A.; Deepika, A.; Sreedharan, K.; Makesh, M.; Chaudhari, A.; Purushothaman, C.S.; Rajendran, K.V. Identification, ontogeny and expression analysis of a novel laboratory of genetics and physiology 2 (LGP2) transcript in Asian seabass, Lates calcarifer. Fish. Shellfish. Immunol. 2017, 62, 265–275. [Google Scholar] [CrossRef]
- Kim, M.Y.; Jang, J.H.; Lee, J.W.; Cho, J.H. Molecular cloning and characterization of peptidoglycan recognition proteins from the rockfish, Sebastes schlegeli. Fish. Shellfish. Immunol. 2010, 28, 632–639. [Google Scholar] [CrossRef] [PubMed]
- Jang, J.H.; Kim, H.; Cho, J.H. Molecular cloning and functional characterization of peptidoglycan recognition protein OmPGRP-L2 from the rainbow trout, Oncorhynchus mykiss. Vet. Immunol. Immunopathol. 2017, 192, 28–32. [Google Scholar] [CrossRef] [PubMed]
- Li, J.H.; Yu, Z.L.; Xue, N.N.; Zou, P.F.; Hu, J.Y.; Nie, P.; Chang, M.X. Molecular cloning and functional characterization of peptidoglycan recognition protein 6 in grass carp Ctenopharyngodon idella. Dev. Comp. Immunol. 2014, 42, 244–255. [Google Scholar] [CrossRef]
- Banyer, J.L.; Hamilton, N.H.; Ramshaw, I.A.; Ramsay, A.J. Cytokines in innate and adaptive immunity. Rev. Immunogenet. 2000, 2, 359–373. [Google Scholar] [PubMed]
- Floege, J.; Luscher, B.; Muller-Newen, G. Cytokines and inflammation. Eur. J. Cell Biol. 2012, 91, 427. [Google Scholar] [CrossRef]
- Hong, S.; Zou, J.; Crampe, M.; Peddie, S.; Scapigliati, G.; Bols, N.; Cunningham, C.; Secombes, C.J. The production and bioactivity of rainbow trout (Oncorhynchus mykiss) recombinant IL-1 beta. Vet. Immunol. Immunopathol. 2001, 81, 1–14. [Google Scholar] [CrossRef]
- Bae, J.S.; Shim, S.H.; Hwang, S.D.; Kim, J.W.; Park, D.W.; Park, C.I. Molecular cloning and expression analysis of interleukin (IL)-15 and IL-15 receptor alpha from rock bream, Oplegnathus fasciatus. Fish. Shellfish. Immunol. 2013, 35, 1209–1215. [Google Scholar] [CrossRef]
- Guo, M.; Tang, X.; Sheng, X.; Xing, J.; Zhan, W. The effects of IL-1beta, IL-8, G-CSF and TNF-alpha as molecular adjuvant on the immune response to an E. tarda subunit vaccine in flounder (Paralichthys olivaceus). Fish. Shellfish. Immunol. 2018, 77, 374–384. [Google Scholar] [CrossRef] [PubMed]
- Andersen, L.K.; Abernathy, J.W.; Farmer, B.D.; Lange, M.D.; Sankappa, N.M.; Mcentire, M.E.; Rawles, S.D. Analysis of striped bass (Morone saxatilis) and white bass (M. chrysops) splenic transcriptome following Streptococcus iniae infection. Mar. Biotechnol. 2025, 27, 51. [Google Scholar] [CrossRef]
- Zhao, Y.; He, Z.; Liu, Y.; Ren, Y.; Ren, J.; Zhang, Y.; Wang, Y.; Wang, G.; San, L.; Hou, J. Isolation, identification and the pathogenicity characterization of Pseudomonas putida 1C3 and its activation on immune responses in Japanese flounder (Paralichthys olivaceus). Fish. Shellfish. Immun. 2025, 160, 110208. [Google Scholar] [CrossRef]
- Su, L.; Guo, B.S.; Jiang, L.R.; Lin, Y.P.; Xu, Q.Y.; Zheng, D.; Xiu, Y.J. Intestinal epithelial cells of Japanese flounder (Paralichthys olivaceus) as an in vitro model for studying intestine immune function based on transcriptome analysis. Fish. Shellfish. Immun. 2024, 148, 109473. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, Z.; Li, X.; Zhang, Q.; Wang, W.; Wang, H.; Pan, T.; Gao, Q. Transcriptome Analysis of Immune Response Against Edwardsiella tarda Infection in Spotted Sea Bass (Lateolabrax maculatus). Fishes 2025, 10, 153. https://doi.org/10.3390/fishes10040153
Sun Z, Li X, Zhang Q, Wang W, Wang H, Pan T, Gao Q. Transcriptome Analysis of Immune Response Against Edwardsiella tarda Infection in Spotted Sea Bass (Lateolabrax maculatus). Fishes. 2025; 10(4):153. https://doi.org/10.3390/fishes10040153
Chicago/Turabian StyleSun, Zhaosheng, Xia Li, Qingling Zhang, Wei Wang, Huan Wang, Tingshuang Pan, and Qian Gao. 2025. "Transcriptome Analysis of Immune Response Against Edwardsiella tarda Infection in Spotted Sea Bass (Lateolabrax maculatus)" Fishes 10, no. 4: 153. https://doi.org/10.3390/fishes10040153
APA StyleSun, Z., Li, X., Zhang, Q., Wang, W., Wang, H., Pan, T., & Gao, Q. (2025). Transcriptome Analysis of Immune Response Against Edwardsiella tarda Infection in Spotted Sea Bass (Lateolabrax maculatus). Fishes, 10(4), 153. https://doi.org/10.3390/fishes10040153