
Genome-Wide Characterization and Analysis of Expression of the Histone Gene Family in Razor Clam, Sinonovacula constricta


Abstract
:1. Introduction
2. Materials and Methods
2.1. Genome Resource
2.2. Genome-Wide Identification of Histone Genes
2.3. Sequence Analysis and Chromosomal Localization of Histone Genes in S. constricta
2.4. Classification of Histone Genes
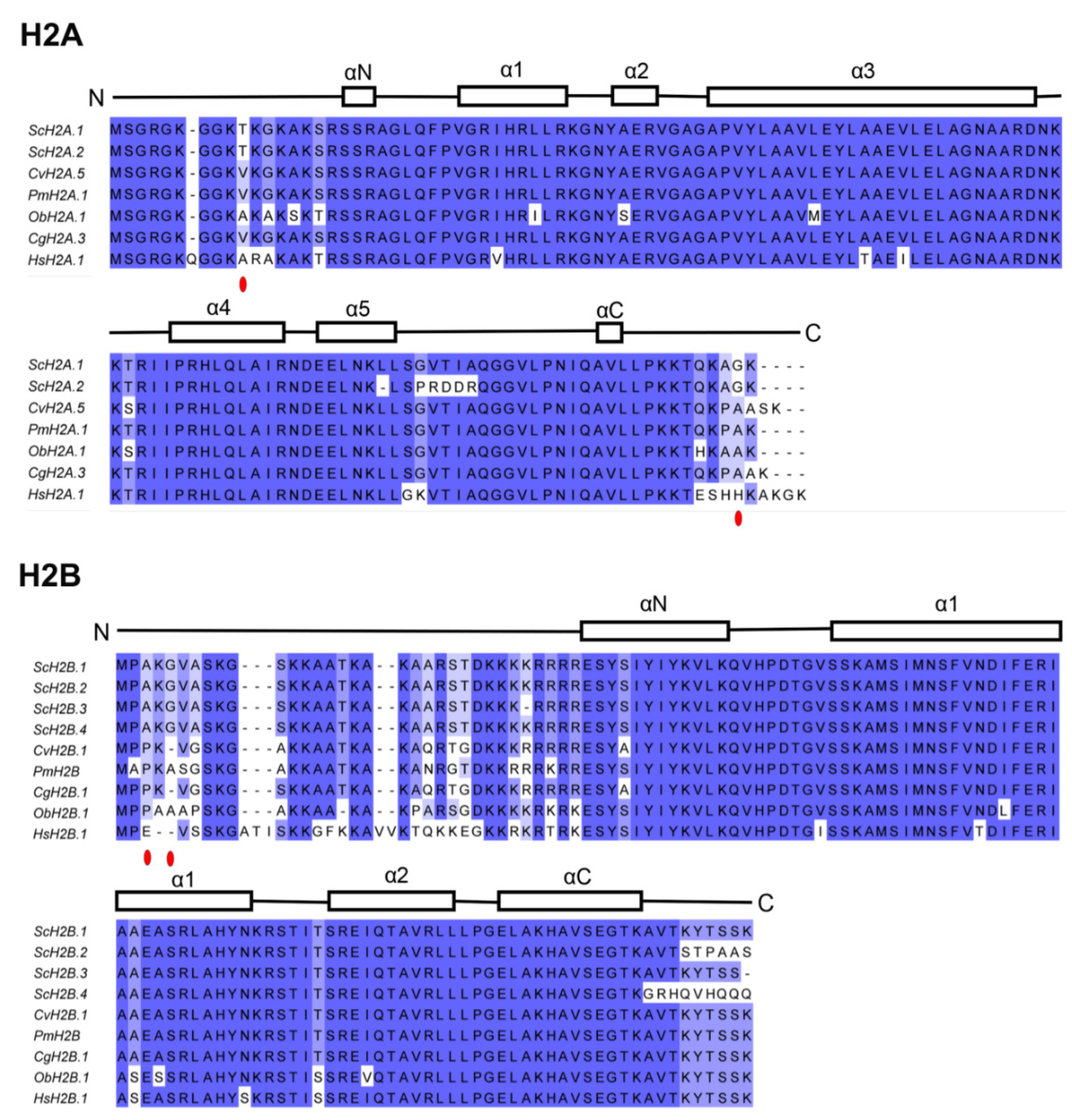
2.5. Sequence Alignment and Phylogenetic Analysis
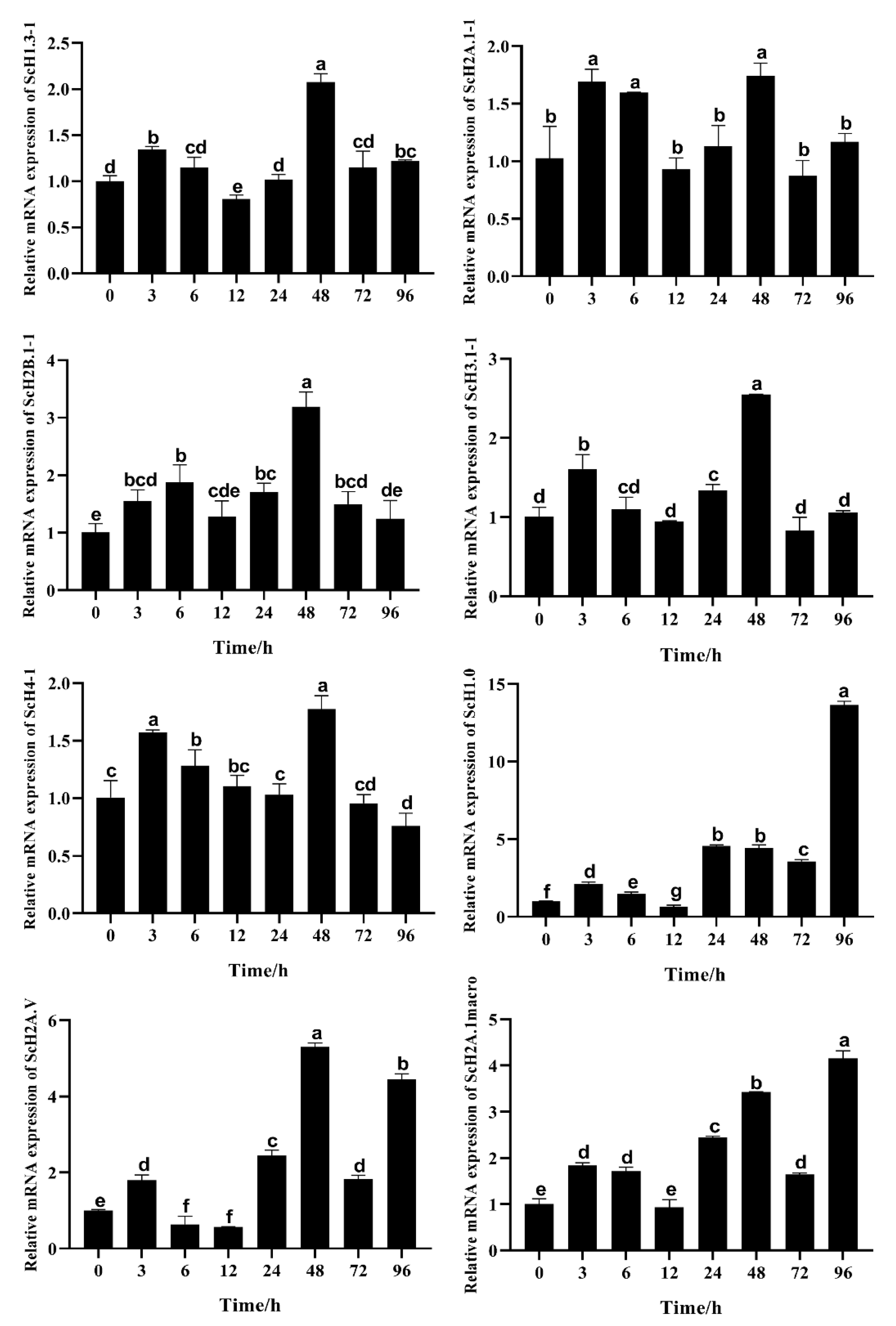
2.6. Expression Profiles Assessed by Transcriptomics and qRT-PCR
3. Results
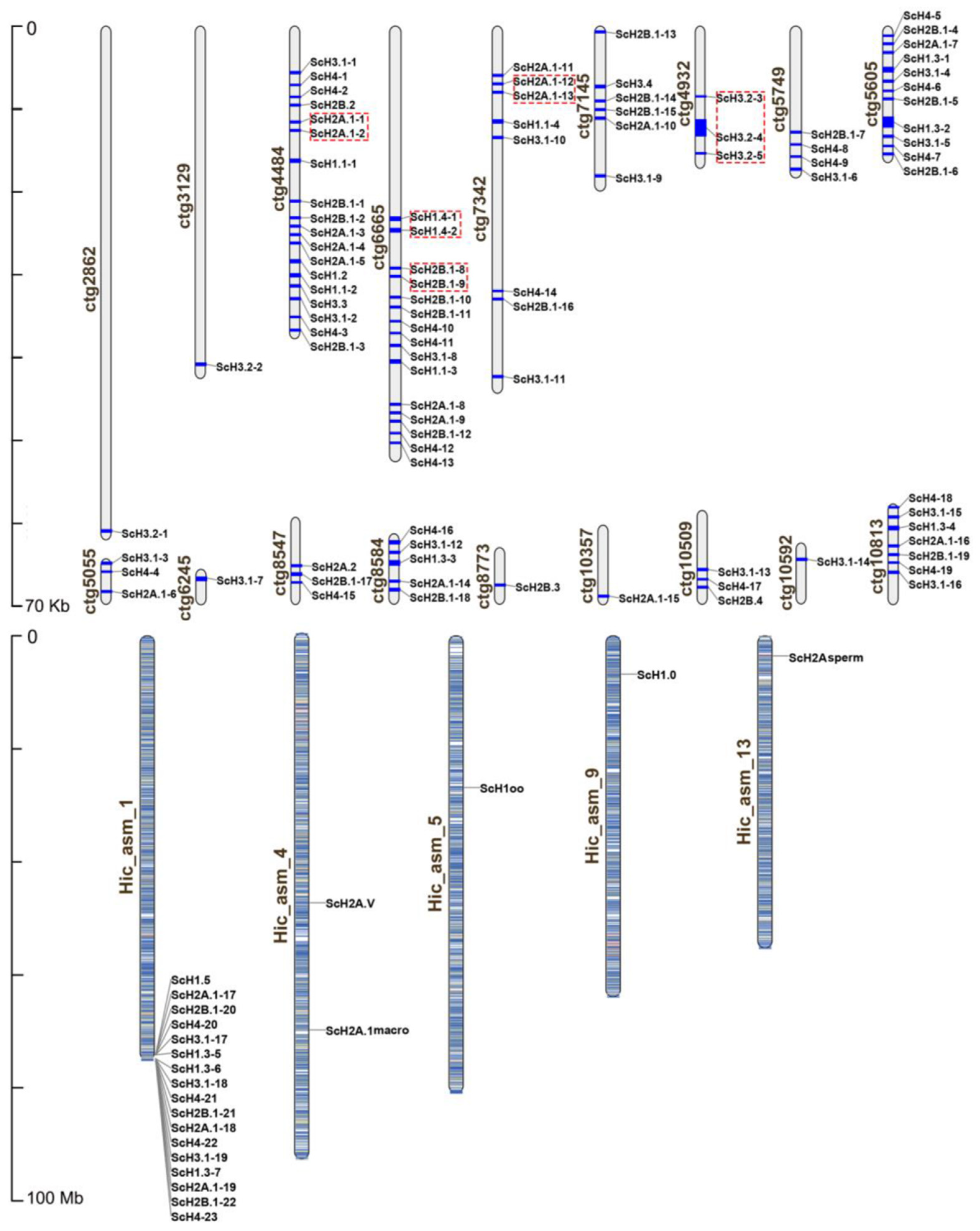
3.1. Genome-Wide Identification and Genomic Distribution of Histone Genes in S. constricta
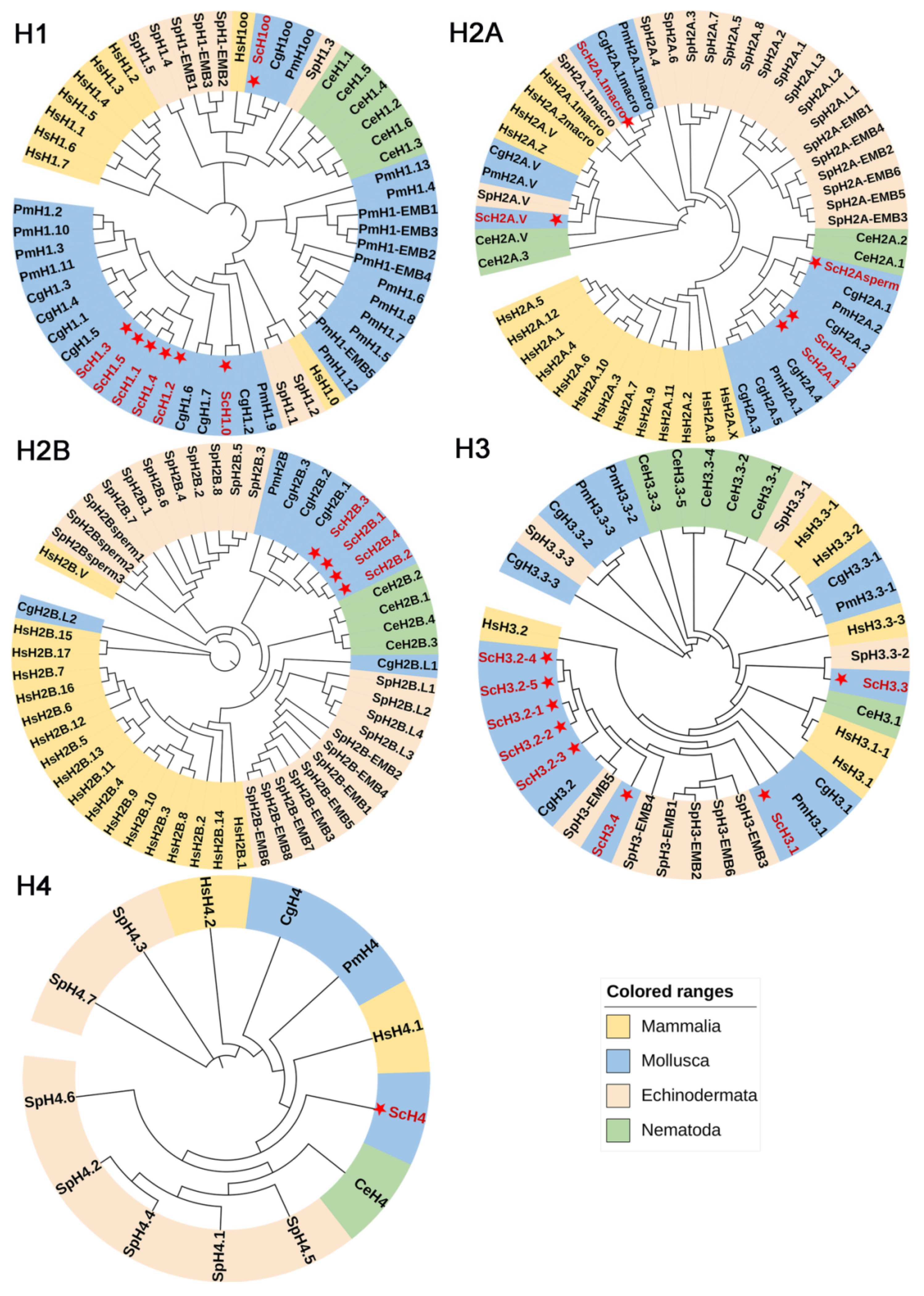
3.2. Phylogenetic Analysis of Histone Genes
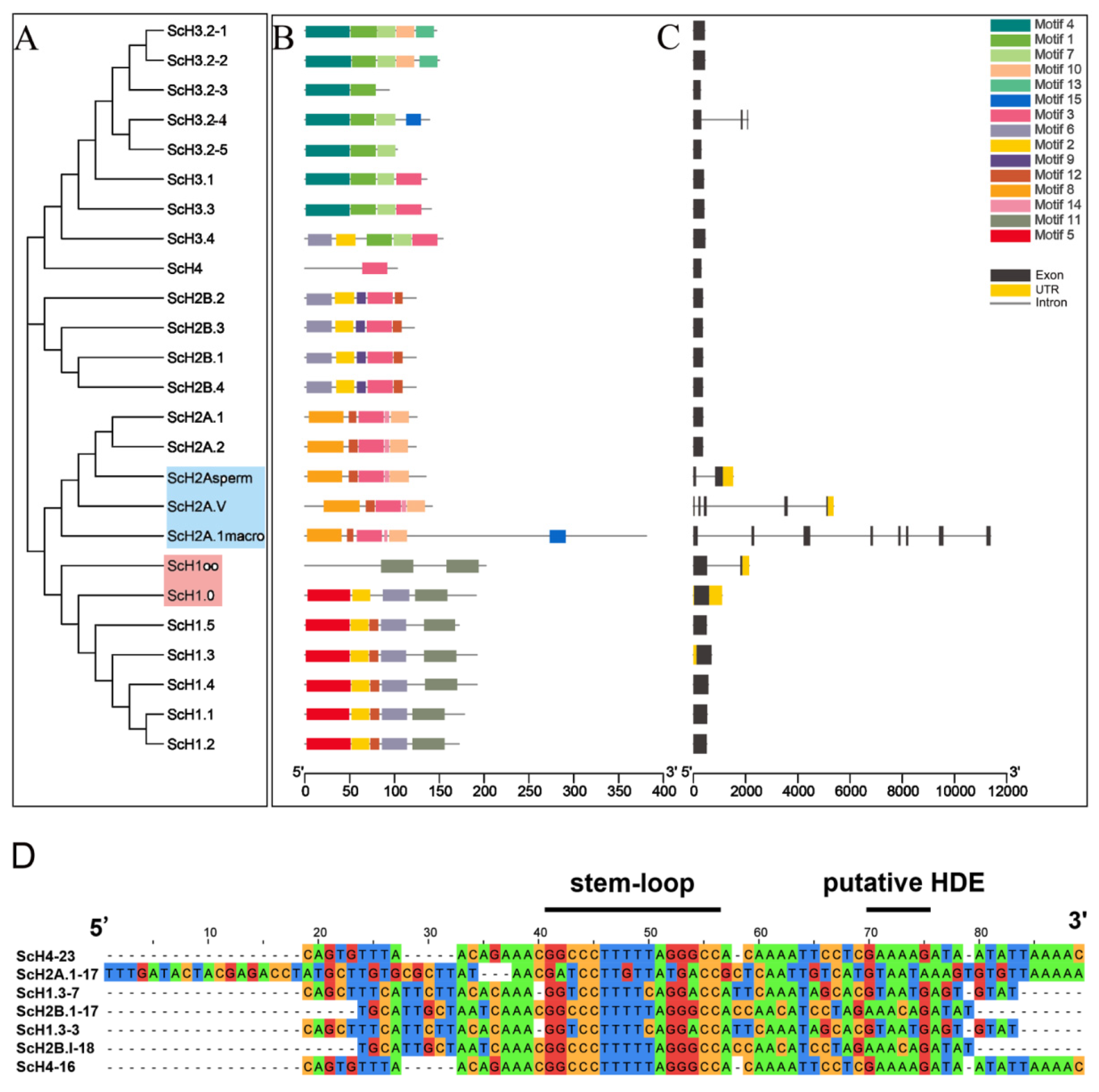
3.3. Gene Structure and Conserved Motif Characteristics of S. constricta Histone Genes
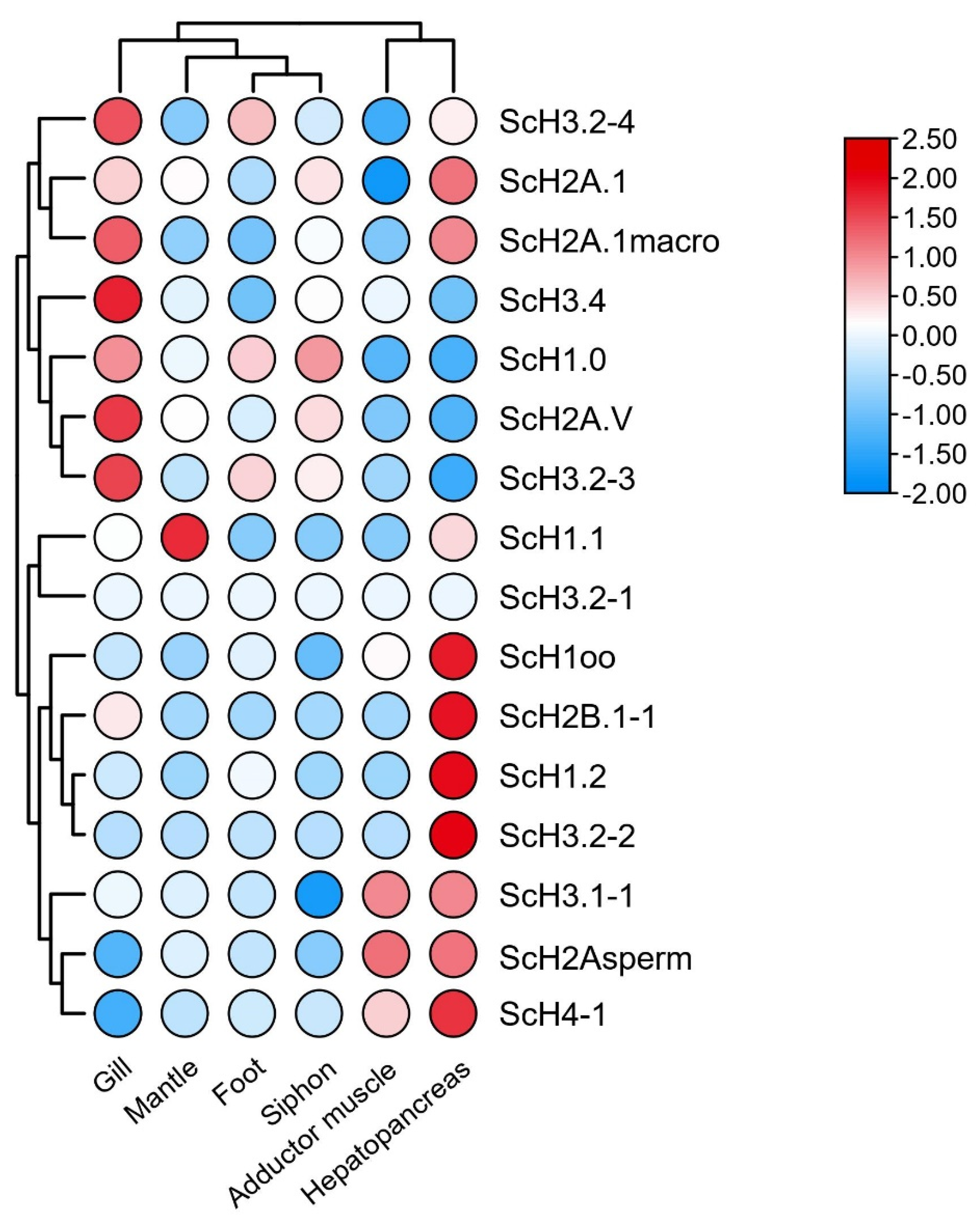
3.4. Gene Expression in S. constricta Assessed by Transcriptomics
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Gonzalez-Romero, R.; Rivera-Casas, C.; Frehlick, L.J.; Mendez, J.; Ausio, J.; Eirín-López, J.M. Histone H2A (H2A.X and H2A.Z) variants in molluscs: Molecular characterization and potential implications for chromatin dynamics. PLoS ONE 2012, 7, e30006. [Google Scholar]
- Li, C.; Song, L.; Zhao, J.; Zou, H.; Su, J.; Zhang, H. Genomic organization, nucleotide sequence analysis of the core histone genes cluster in Chlamys farreri and molecular evolution assessment of the H2A and H2B. DNA Seq. 2006, 17, 440–451. [Google Scholar] [CrossRef]
- Wolffe, A.P.; Kurumizaka, H. The nucleosome: A powerful regulator of transcription. Prog. Nucleic Acid Res. Mol. Biol. 1998, 61, 379–422. [Google Scholar]
- Kornberg, R.D. Chromatin structure: A repeating unit of histones and DNA. Science 1974, 184, 868–871. [Google Scholar] [CrossRef]
- Zlatanova, J.; Bishop, T.C.; Victor, J.-M.; Jackson, V.; van Holde, K. The nucleosome family: Dynamic and growing. Structure 2009, 17, 160–171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allan, J.; Hartman, P.; Crane-Robinson, C.; Aviles, F. The structure of histone H1 and its location in chromatin. Nature 1980, 288, 675–679. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Reinberg, D. Chromatin higher-order structures and gene regulation. Curr. Opin. Genet. Dev. 2011, 21, 175–186. [Google Scholar] [CrossRef] [Green Version]
- Jiang, D.; Borg, M.; Lorkovi, Z.J.; Montgomery, S.A.; Osakabe, A.; Yelagandula, R.; Axelsson, E.; Berger, F. The evolution and functional divergence of the histone H2B family in plants. PLoS Genet. 2020, 16, e1008964. [Google Scholar] [CrossRef]
- Talbert, P.B.; Henikoff, S. Histone variants on the move: Substrates for chromatin dynamics. Nat. Rev. Mol. Cell Biol. 2017, 18, 115–126. [Google Scholar] [CrossRef] [PubMed]
- Gamble, M.J.; Kraus, W.L. Multiple facets of the unique histone variant macroH2A: From genomics to cell biology. Cell Cycle 2010, 9, 2568–2574. [Google Scholar] [CrossRef] [Green Version]
- Bönisch, C.; Hake, S.B. Histone H2A variants in nucleosomes and chromatin: More or less stable? Nucleic Acids Res. 2012, 40, 10719–10741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- López, M.D.; Samuelsson, T. Early evolution of histone mRNA 3′ end processing. RNA 2008, 14, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reddy, P.C.; Ubhe, S.; Sirwani, N.; Lohokare, R.; Galande, S. Rapid divergence of histones in Hydrozoa (Cnidaria) and evolution of a novel histone involved in DNA damage response in hydra. Zoology 2017, 123, 53–63. [Google Scholar] [CrossRef] [PubMed]
- Marzluff, W.F.; Wagner, E.J.; Duronio, R.J. Metabolism and regulation of canonical histone mRNAs: Life without a poly(A) tail. Nat. Rev. Genet. 2008, 9, 843–854. [Google Scholar] [CrossRef] [Green Version]
- Talbert, P.B.; Henikoff, S. Histone variants—Ancient wrap artists of the epigenome. Nat. Rev. Mol. Cell Biol. 2010, 11, 264–275. [Google Scholar] [CrossRef] [PubMed]
- Lim, C.; Knowles, B.; Solter, D.; Messerschmidt, D. Epigenetic control of early mouse development. Curr. Top. Dev. Biol. 2016, 120, 311–360. [Google Scholar]
- Rooney, A.P.; Piontkivska, H.; Nei, M. Molecular evolution of the nontandemly repeated genes of the histone 3 multigene family. Mol. Biol. Evol. 2002, 19, 68–75. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.M.; Cao, L.; Nie, P.; Chang, M.X. Histone H2A cooperates with RIP2 to induce the expression of antibacterial genes and MHC related genes. Dev. Comp. Immunol. 2019, 101, 103455. [Google Scholar] [CrossRef]
- Richards, R.C.; O’Neil, D.B.; Thibault, P.; Ewart, K.V. Histone H1: An antimicrobial protein of Atlantic salmon (Salmo salar). Biochem. Biophys. Res. Commun. 2001, 284, 549–555. [Google Scholar] [CrossRef]
- Arockiaraj, J.; Gnanam, A.J.; Kumaresan, V.; Palanisamy, R.; Bhatt, P.; Thirumalai, M.K.; Roy, A.; Pasupuleti, M.; Kasi, M. An unconventional antimicrobial protein histone from freshwater prawn Macrobrachium rosenbergii: Analysis of immune properties. Fish Shellfish Immunol. 2013, 35, 1511–1522. [Google Scholar] [CrossRef]
- Patat, S.A.; Carnegie, R.B.; Kingsbury, C.; Gross, P.S.; Chapman, R.; Schey, K.L. Antimicrobial activity of histones from hemocytes of the Pacific white shrimp. Eur. J. Biochem. 2004, 271, 4825–4833. [Google Scholar] [CrossRef] [PubMed]
- De Zoysa, M.; Nikapitiya, C.; Whang, I.; Lee, J.-S.; Lee, J. Abhisin: A potential antimicrobial peptide derived from histone H2A of disk abalone (Haliotis discus discus). Fish Shellfish Immunol. 2009, 27, 639–646. [Google Scholar] [CrossRef]
- Li, C.; Song, L.; Zhao, J.; Zhu, L.; Zou, H.; Zhang, H.; Wang, H.; Cai, Z. Preliminary study on a potential antibacterial peptide derived from histone H2A in hemocytes of scallop Chlamys farreri. Fish Shellfish Immunol. 2007, 22, 663–672. [Google Scholar] [CrossRef]
- Seo, J.-K.; Stephenson, J.; Crawford, J.M.; Stone, K.L.; Noga, E.J. American oyster, Crassostrea virginica, expresses a potent antibacterial histone H2B protein. Mar. Biotechnol. 2010, 12, 543–551. [Google Scholar] [CrossRef]
- Li, A.; Eirín-López, J.M.; Ausió, J. H2AX: Tailoring histone H2A for chromatin-dependent genomic integrity. Biochem. Cell Biol. 2005, 83, 505–515. [Google Scholar] [CrossRef] [PubMed]
- González-Romero, R.; Ausió, J.; Méndez, J.; Eirín-López, J.M. Histone genes of the razor clam Solen marginatus unveil new aspects of linker histone evolution in protostomes. Genome 2009, 52, 597–607. [Google Scholar] [CrossRef] [Green Version]
- Shen, H.; Qi, Z.; Wei, H. Molecular evolution of the histone gene family in Schistosoma japonicum. Int. J. Med. Parasit. Dis. 2015, 42, 67–73. [Google Scholar]
- Montes-Rodríguez, I.M.; Rodríguez-Pou, Y.; González-Méndez, R.R.; Lopez-Garriga, J.; Ropelewski, A.J.; Cadilla, C.L. Characterization of histone genes from the bivalve Lucina pectinata. Int. J. Environ. Res. Public Health 2018, 15, 2170. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Zhang, Y.-P.; Qian, Y.-H.; Zeng, Q.-T. Phylogenetic relationships of Drosophila melanogaster species group deduced from spacer regions of histone gene H2A-H2B. Mol. Phylogenetics Evol. 2004, 30, 336–343. [Google Scholar] [CrossRef]
- Dong, Y.; Zeng, Q.; Ren, J.; Yao, H.; Lv, L.; He, L.; Ruan, W.; Xue, Q.; Bao, Z.; Wang, S.; et al. The chromosome-level genome assembly and comprehensive transcriptomes of the razor clam (Sinonovacula constricta). Front. Genet. 2020, 11, 664. [Google Scholar] [CrossRef]
- Morton, B. The functional morphology of Sinonovacula constricta with a discussion on the taxonomic status of the Novaculininae (Bivalvia). J. Zool. 1984, 202, 299–325. [Google Scholar] [CrossRef]
- Wang, W.X.; Xu, Z.Z. Larval swimming and postlarval drifting behavior in the infaunal bivalve Sinonovacula constricta. Mar. Ecol. Prog. Ser. 1997, 148, 71–81. [Google Scholar] [CrossRef] [Green Version]
- Sousa, H.; Hinzmann, M. Review: Antibacterial components of the bivalve’s immune system and the potential of freshwater bivalves as a source of new antibacterial compounds. Fish Shellfish Immunol. 2020, 98, 971–980. [Google Scholar] [CrossRef] [PubMed]
- Lv, L.-Y.; Liang, X.-F.; He, S. Genome-wide identification and characterization of olfactory receptor genes in Chinese perch, Siniperca chuatsi. Genes 2019, 10, 178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.; Xia, R. TBtools: An integrative toolkit developed for interactive analyses of big biological data. Mol. Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef]
- Bailey, T.L.; Boden, M.; Buske, F.A.; Frith, M.; Grant, C.E.; Clementi, L.; Ren, J.; Li, W.W.; Noble, W.S. MEME SUITE: Tools for motif discovery and searching. Nucleic Acids Res. 2009, 37, W202–W208. [Google Scholar] [CrossRef] [PubMed]
- Ronquist, F.; Huelsenbeck, J.P. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 2003, 19, 1572–1574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Gerard, T.; Jose, C. Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst. Biol. 2007, 56, 564–577. [Google Scholar]
- Ronquist, F.; Teslenko, M.; van der Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef] [Green Version]
- Waterhouse, A.M.; Procter, J.B.; Martin, D.M.; Clamp, M.; Barton, G.J. Jalview Version 2—A multiple sequence alignment editor and analysis workbench. Bioinformatics 2009, 25, 1189–1191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ponte, I.; Romero, D.; Yero, D.; Suau, P.; Roque, A. Complex evolutionary history of the mammalian histone H1.1-H1.5 gene family. Mol. Biol. Evol. 2017, 34, 545–558. [Google Scholar] [CrossRef] [Green Version]
- Eirin-Lopez, J.M.; Ishibashi, T.; Ausio, J. H2A.Bbd: A quickly evolving hypervariable mammalian histone that destabilizes nucleosomes in an acetylation-independent way. FASEB J. 2008, 22, 316–326. [Google Scholar] [CrossRef] [PubMed]
- Kowalski, A. Nuclear and nucleolar activity of linker histone variant H1.0. Cell. Mol. Biol. Lett. 2016, 21, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rutowicz, K.; Puzio, M.; Halibart-Puzio, J.; Lirski, M.; Kotliński, M.; Kroteń, M.A.; Knizewski, L.; Lange, B.; Muszewska, A.; Śniegowska-Świerk, K.; et al. A specialized histone H1 variant is required for adaptive responses to complex abiotic stress and related DNA methylation in Arabidopsis. Plant Physiol. 2015, 169, 2080–2101. [Google Scholar] [CrossRef] [Green Version]
- Teng, L.; Fan, X.; Nelson, D.R.; Han, W.; Zhang, X.; Xu, D.; Renault, H.; Markov, G.V.; Ye, N. Diversity and evolution of cytochromes P450 in stramenopiles. Planta 2019, 249, 647–661. [Google Scholar] [CrossRef] [Green Version]
- Hentschel, C.C.; Birnstiel, M.L. The organization and expression of histone gene families. Cell 1981, 25, 301–313. [Google Scholar] [CrossRef]
- Miller, D.; Harrison, P.; Mahony, T.; McMillan, J.; Miles, A.; Odorico, D.; Ten Lohuis, M. Feature of histone gene structure and organization are common to diploblastic and tripoblastic metazoans. J. Mol. Evol. 1993, 37, 245–253. [Google Scholar] [CrossRef]
- Sanicola, M.; Ward, S.; Childs, G.; Emmons, S.W. Identification of a Caenorhabditis elegans histone H1 gene family: Characterization of a family member containing an intron and encoding a poly (A)+ mRNA. J. Mol. Biol. 1990, 212, 259–268. [Google Scholar] [CrossRef]
- Eirín-López, J.M.; Ruiz, M.F.; Gonzalez-Tizon, A.M.; Martínez, A.; Sanchez, L.; Mendez, J. Molecular evolutionary characterization of the mussel Mytilus histone multigene family: First record of a tandemly repeated unit of five histone genes containing an H1 subtype with “orphon” features. J. Mol. Evol. 2004, 58, 131–144. [Google Scholar] [CrossRef]
- Eirín-López, J.M.; González-Romero, R.; Dryhurst, D.; Méndez, J.; Ausió, J. Long-Term Evolution of Histone Families: Old Notions and New Insights into Their Mechanisms of Diversification across Eukaryotes. 2009. Available online: https://link.springer.com/chapter/10.1007/978-3-642-00952-5_8 (accessed on 12 October 2020).
- Müller, K.; Schmitt, R. Histone genes of Volvox carteri: DNA sequence and organization of two H3-H4 gene loci. Nucleic Acids Res. 1988, 16, 4121–4136. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doyle, J.J.; Kanazin, V.; Shoemaker, R.C. Phylogenetic utility of histone H3 intron sequences in the perennial relatives of soybean (Glycine: Leguminosae). Mol. Phylogenet. Evol. 1996, 6, 438–447. [Google Scholar] [CrossRef]
- Dominski, Z.; Marzluff, W.F. Formation of the 3′ end of histone mRNA: Getting closer to the end. Gene 2007, 396, 373–390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cong, M.; Wu, H.; Cao, T.; Ji, C.; Lv, J. Effects of ammonia nitrogen on gill mitochondria in clam Ruditapes philippinarum. Environ. Toxicol. Pharmacol. 2018, 65, 46–52. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, B.; Koner, D.; Bhuyan, G.; Saha, N. Differential expression of multiple glutamine synthetase genes in air-breathing magur catfish, Clarias magur and their induction under hyper-ammonia stress. Gene 2018, 671, 85–95. [Google Scholar] [CrossRef]
- Tanaka, M.; Hennebold, J.D.; Macfarlane, J.; Adashi, E.Y. A mammalian oocyte-specific linker histone gene H1oo: Homology with the genes for the oocyte-specific cleavage stage histone (cs-H1) of sea urchin and the B4/H1M histone of the frog. Development 2001, 128, 655–664. [Google Scholar] [CrossRef] [PubMed]
- Furuya, M.; Tanaka, M.; Teranishi, T.; Matsumoto, K.; Hosoi, Y.; Saeki, K.; Ishimoto, H.; Minegishi, K.; Iritani, A.; Yoshimura, Y. H1foo is indispensable for meiotic maturation of the mouse oocyte. J. Reprod. Dev. 2007, 53, 895–902. [Google Scholar] [CrossRef] [Green Version]
- Eirin-Lopez, J.M.; Ausio, J. H2A. Z-mediated genome-wide chromatin specialization. Curr. Genom. 2007, 8, 59–66. [Google Scholar] [CrossRef]
- Subramanian, V.; Fields, P.A.; Boyer, L.A. H2A. Z: A molecular rheostat for transcriptional control. F1000Prime Rep. 2015. Available online: https://pubmed.ncbi.nlm.nih.gov/25705384/ (accessed on 22 October 2020).
- Mattiroli, F.; D’Arcy, S.; Luger, K. The right place at the right time: Chaperoning core histone variants. EMBO Rep. 2015, 16, 1454–1466. [Google Scholar] [CrossRef] [Green Version]
- Kawasaki, H.; Iwamuro, S. Potential roles of histones in host defense as antimicrobial agents. Infect. Disord. Drug Targets 2008, 8, 195–205. [Google Scholar] [CrossRef]
- Alkhalil, A.; Hammamieh, R.; Hardick, J.; Ichou, M.A.; Jett, M.; Ibrahim, S. Gene expression profiling of monkeypox virus-infected cells reveals novel interfaces for host-virus interactions. Virol. J. 2010, 7, 1–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lü, A.; Hu, X.; Xue, J.; Zhu, J.; Wang, Y.; Zhou, G. Gene expression profiling in the skin of zebrafish infected with Citrobacter freundii. Fish Shellfish Immunol. 2012, 32, 273–283. [Google Scholar] [CrossRef] [PubMed]
- Dorrington, T.; Villamil, L.; Gómez-chiarri, M. Upregulation in response to infection and antibacterial activity of oyster histone H4. Fish Shellfish Immunol. 2011, 30, 94–101. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | Primer Sequence (5′-3′) |
|---|---|
| ScH1.3-1F | GCTCGTCCAGACAGGCAAT |
| ScH1.3-1R | TTTGGCGTCTTGGCTTTC |
| ScH2A.1-1F | CTGGACGAGGAAAAGGAG |
| ScH2A.1-1R | CGGAGAGGAGTTTGTTCA |
| ScH2B.1-1F | ATGCCGGCTAAAGGAGTTG |
| ScH2B.1-1R | TACTTGGTGACGGCTTTGG |
| ScH3.1-1F | TGAAGAAGCCCCACAGGT |
| ScH3.1-1R | TTAAGCACGCTCTCCACGG |
| ScH4-1F | CGGTAAAGGAGGAAAAGG |
| ScH4-1R | CTTCAGGGCGTAGACAAC |
| ScH2A.VF | CAGGCAACGCCAGTAAGGAT |
| ScH2A.VR | AATGACACCACCGCCAGCAA |
| ScH2A.1macroF | CTGATGCCATAGTTCACCCAA |
| ScH2A.1macroR | GAGCATTTTGAGAGCCCCAT |
| ScH1.0F | CCCACCCGAAATACAGCGAGA |
| ScH1.0R | CTTCCTCAGAGCCATCTTCAGC |
| 18SF | TCGGTTCTATTGCGTTGGTTTT |
| 18SR | CAGTTGGCATCGTTTATGGTCA |
| Histone Type | Top Blast Hit | e-Value | Identity | AA | No. of Gene Copies | Exon | PI | MW | GRAVY |
|---|---|---|---|---|---|---|---|---|---|
| H1.1 | XP_022314758.1: histone H1-delta-like [Crassostrea virginica] | 6 × 10−39 | 74% | 178 | 4 | 1 | 10.77 | 18.35 | −0.794 |
| H1.2 | XP_022314758.1: histone H1-delta-like [Crassostrea virginica] | 8 × 10−39 | 74% | 172 | 1 | 1 | 10.74 | 17.74 | −0.794 |
| H1.3 | XP_022313748.1: histone H1-delta-like [Crassostrea virginica] | 1 × 10−38 | 72% | 192 | 7 | 1 | 10.82 | 19.77 | −0.822 |
| H1.4 | XP_022314758.1: histone H1-delta-like [Crassostrea virginica] | 8 × 1039 | 74% | 192 | 2 | 1 | 10.82 | 19.80 | −0.844 |
| H1.5 | XP_022313748.1: histone H1-delta-like [Crassostrea virginica] | 2 × 10−38 | 72% | 172 | 1 | 1 | 10.61 | 17.78 | −0.684 |
| H1oo | XP_002733520.1: PREDICTED: sperm-specific protein PHI-2B/PHI-3-like [Saccoglossus kowalevskii] | 8 × 10−15 | 42% | 202 | 1 | 2 | 10.32 | 21.62 | −0.951 |
| H1.0 | AYV75054.1: histone 1.B [Phacoides pectinatus] | 4 × 10−39 | 83% | 191 | 1 | 1 | 11.11 | 20.07 | −1.023 |
| H2A.1 | XP_033730396.1: histone H2A [Pecten maximus] | 1 × 10−79 | 98% | 125 | 19 | 1 | 10.9 | 13.38 | −0.343 |
| H2A.2 | XP_011450417.1: histone H2A [Crassostrea gigas] | 2 × 10−71 | 92% | 124 | 1 | 1 | 10.87 | 13.46 | −0.594 |
| H2A.V | NP_001116980.1: histone H2A.V [Strongylocentrotus purpuratus] | 2 × 10−28 | 99% | 142 | 1 | 5 | 10.31 | 15.26 | −0.406 |
| H2A.1macro | XP_033754346.1: core histone macro-H2A.1-like isoform X2 [Pecten maximus] | 2 × 10−169 | 67% | 381 | 1 | 8 | 9.95 | 40.62 | −0.159 |
| H2Asperm | XP_022326311.1: histone H2A-like [Crassostrea virginica] | 3 × 10−76 | 88% | 135 | 1 | 2 | 10.72 | 14.44 | −0.450 |
| H2B.1 | XP_022318982.1: histone H2B-like [Crassostrea virginica] | 3 × 10−67 | 100% | 124 | 22 | 1 | 10.52 | 13.71 | −0.624 |
| H2B.2 | XP_009053399.1: hypothetical protein LOTGIDRAFT_147373, partial [Lottia gigantea] | 1 × 10−66 | 95% | 124 | 1 | 1 | 10.43 | 16.91 | −0.535 |
| H2B.3 | AYV75055.1: histone 2B [Phacoides pectinatus] | 8 × 10−77 | 97% | 122 | 1 | 1 | 10.47 | 13.46 | −0.570 |
| H2B.4 | XP_009053399.1: hypothetical protein LOTGIDRAFT_147373, partial [Lottia gigantea] | 3 × 10−64 | 95% | 124 | 1 | 1 | 10.67 | 13.85 | −0.745 |
| H3.1 | XP_001862696.1: Histone H3c [Culex quinquefasciatus] | 2 × 10−93 | 100% | 136 | 19 | 1 | 11.27 | 15.39 | −0.604 |
| H3.2-1 | ROT69816.1: histone H3 [Penaeus vannamei] | 3 × 10−55 | 64% | 147 | 1 | 1 | 10.78 | 16.73 | −0.448 |
| H3.2-2 | ROT69816.1: histone H3 [Penaeus vannamei] | 4 × 10−54 | 99% | 150 | 1 | 1 | 10.5 | 16.96 | −0.439 |
| H3.2-3 | XP_001618210.2: histone H3 [Nematostella vectensis] | 9 × 10−53 | 90% | 94 | 1 | 1 | 11.45 | 10.61 | −0.615 |
| H3.2-4 | XP_018951417.1: PREDICTED: histone H3-like [Cyprinus carpio] | 4 × 10−54 | 78% | 139 | 1 | 3 | 10.85 | 15.75 | −0.717 |
| H3.2-5 | XP_022206407.1: histone H3-like [Nilaparvata lugens] | 5 × 10−53 | 89% | 103 | 1 | 1 | 11.26 | 11.60 | −0.737 |
| H3.3 | XP_002422632.1: histone H3, putative [Pediculus humanus corporis] | 1 × 10−72 | 79% | 154 | 1 | 1 | 10.73 | 17.68 | −0.684 |
| H3.4 | XP_025913950.1: histone H3-like [Apteryx rowi] | 3 × 10−88 | 95% | 141 | 1 | 1 | 11.26 | 23.51 | −0.494 |
| H4 | XP_018963044.1: PREDICTED: histone H4-like [Cyprinus carpio] | 2 × 10−65 | 100% | 103 | 23 | 1 | 11.36 | 11.37 | −0.521 |
| Species Name | H1 | H2A | H2B | H3 | H4 | Total |
|---|---|---|---|---|---|---|
| Homo sapiens | 9 | 26 | 23 | 17 | 15 | 90 |
| Xenopus laevis | 10 | 31 | 21 | 19 | 14 | 95 |
| Drosophila melanogaster | 23 | 21 | 23 | 25 | 23 | 115 |
| Branchiostoma belcheri | 2 | 25 | 25 | 15 | 8 | 75 |
| Caenorhabditis elegans | 6 | 19 | 16 | 20 | 16 | 77 |
| Strongylocentrotus purpuratus | 8 | 28 | 31 | 19 | 20 | 106 |
| Octopus bimaculoides | 9 | 14 | 7 | 12 | 5 | 47 |
| Sinonovaula constricta | 17 | 23 | 25 | 26 | 23 | 114 |
| Crassostrea virginica | 16 | 33 | 27 | 10 | 15 | 100 |
| Crassostrea gigas | 11 | 13 | 8 | 8 | 1 | 39 |
| Pecten maximus | 30 | 17 | 12 | 6 | 11 | 74 |
| Tegillarca granosa | 7 | 4 | 1 | 3 | 0 | 18 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lian, J.; Lv, L.; Yao, H.; Lin, Z.; Dong, Y. Genome-Wide Characterization and Analysis of Expression of the Histone Gene Family in Razor Clam, Sinonovacula constricta. Fishes 2022, 7, 5. https://doi.org/10.3390/fishes7010005
Lian J, Lv L, Yao H, Lin Z, Dong Y. Genome-Wide Characterization and Analysis of Expression of the Histone Gene Family in Razor Clam, Sinonovacula constricta. Fishes. 2022; 7(1):5. https://doi.org/10.3390/fishes7010005
Chicago/Turabian StyleLian, Jiaying, Liyuan Lv, Hanhan Yao, Zhihua Lin, and Yinghui Dong. 2022. "Genome-Wide Characterization and Analysis of Expression of the Histone Gene Family in Razor Clam, Sinonovacula constricta" Fishes 7, no. 1: 5. https://doi.org/10.3390/fishes7010005
APA StyleLian, J., Lv, L., Yao, H., Lin, Z., & Dong, Y. (2022). Genome-Wide Characterization and Analysis of Expression of the Histone Gene Family in Razor Clam, Sinonovacula constricta. Fishes, 7(1), 5. https://doi.org/10.3390/fishes7010005