1. Introduction
The operating principle of a plasmonic nanolaser—the so-called spaser—is Surface Plasmon Amplification by Stimulated Emission of Radiation. The spaser concept was first proposed by Stockman and Bergman in 2003 [
1]. These authors developed a sophisticated quantum-plasmonics theory that models the spaser as a quantum generator for surface plasmon quanta through amplification by stimulated emission of a resonantly-coupled gain medium. A spaser consists of a noble-metal nanoparticle acting as nanocavity and a gain medium formed by resonantly coupled two-level emitters. The gain medium can either encapsulate the nanocavity or is incorporated in it. A prior condition for the emergence of the spasing state is maximum overlap between the emission spectrum of the gain medium and the surface plasmon resonance. Optical pumping of the gain medium stimulates resonance energy transfer between the two-level emitters and surface plasmons. In case of an efficient feedback mechanism, the resulting high local optical fields in turn excite the adjacent two-level systems, which again generates more identical surface plasmons in the same mode until the spasing state is accomplished [
2].
The first tentative experimental realization of a spaser utilized a core–shell nanostructure consisting of a gold nanosphere with a laser dye-doped silica shell [
3]. Unfortunately, the only evidence for spaser performance was the observation of non-exponential decay kinetics of the fluorescence emission monitored at 520 nm. Bi-exponential fitting resulted into two time constants: 4.3 ns and 1.6 ns. The 1.6 ns component was attributed to stimulated emission, which is questionable as being rather slow for stimulated-emission kinetics when excited with 90 ps-pulses. Gu et al. [
4] proposed the implementation of a gold-nanorod-based spaser with an encapsulating gain medium consisting of a dye-doped polystyrene shell. Instead of any indication of lasing action or stimulated emission, the authors observed a drastic reduction of the fluorescence intensity of the dye-doped polystyrene shell of the gold nanorod in comparison to the fluorescence emission of the equally dye-doped polystyrene spheres. The fluorescence quenching was attributed to excitation energy transfer from the initially excited dye molecules to the plasmonic gold nanorod. However, this demonstrates at least that the mean distance between the dye molecules—arbitrarily distributed in the polystyrene shell—and the gold nanorod surface is smaller than 5 nm [
5].
So far, a true lasing spaser composed of noble-metal nanostructures has not be experimentally realized, although remarkable research activities have been dedicated to the engineering of various promising nanoscale noble-metal core–shell structures [
6,
7,
8]. One major obstacle for the realization of the lasing spaser is the strong radiation damping of the noble metal nanocavity that could not be completely compensated in core–shell nanostructures with optical gain materials that comprised organic dye molecules, quantum dots, or rare-earth ions as two-level emitters. One reason might be the insufficient density of two-level emitters in the optical gain medium. In addition, inadequate arrangement of the two-level emitters may impede efficient dipole–dipole couplings with the surface plasmon modes in the nanocavity. Just as fundamental is the geometry of the spaser construct, which are mostly core–shell nanoparticles incorporating gold nanorods, nanospheres, or nanocubes.
In our opinion, the most promising spaser nanoconstructs are composed of gold nanorods (AuNR) with surface-attached excitonic systems. AuNRs are perfect optical antennas exhibiting two localized surface plasmon (LSP) modes due to electron oscillations along the nanorod axis (longitudinal LSP) and radial axis (transversal LSP), respectively. In contrast to the transversal LSP mode, the energy of the longitudinal mode crucially depends on the aspect ratio, and may therefore be tuned from the visible to the NIR just by following simple and reliable synthetic protocols. The spaser action requires an optical gain medium that amplifies the longitudinal LSP modes and compensates the losses of the plasmonic nanocavity. Effective gain media should consist of two-level quantum emitters that are placed in the near field of the dipolar LSPs, arranged with their transition dipole moments parallel to the polarization of the longitudinal LSPs. Equally essential is the spectral overlap between the longitudinal LSP resonance and the emission spectrum of the gain medium. This enables the LSPs to stimulate the emission in the gain medium, which in turn results in the excitation of more LSPs in the same mode, and so forth, providing a coherent feedback for the spaser. Thus, this nonradiative energy transfer to the spaser mode is the dominant process whose probability is by orders of magnitude greater than that of the free-space (far-field) emission.
In this contribution, we show that red spaser emission can be generated by amplifying longitudinal LSP modes in AuNRs via resonant excitation transfer from optically pumped laser dye molecules that are electrostatically surface-bound and operate thereupon as optical gain medium for the compensation of the plasmon losses. The laser dye is phthalocyanine tetrasulfonate (Pcts), which binds through its four negatively-charged sulfonate groups to the positively-charged cetyltrimethylammonium bromide (CTAB) bilayer surrounding the AuNR. Pcts exhibits fluorescence emission at 700 nm. Maximum spectral overlap was achieved by synthetically adjusting the longitudinal LSP resonance of the AuNRs around 700 nm. This spaser nanoconstruct was characterized using high-resolution transmission electron microscopy, UV/Vis absorption spectroscopy, and by performing fluorescence quenching experiments and femtosecond transient absorption spectroscopy experiments. Our work was presented at the 13th International Conference on “Atomically Controlled Surfaces Interfaces and Nanostructures” which was held in Frascati, Italy, October 9–15, 2016 [
9].
2. Results and Discussion
AuNRs with an aspect ratio of 2.4 (length × width = (55 ± 8) nm × (21 ± 5) nm) and a longitudinal LSP resonance at 690 nm were synthesized in aqueous solutions using the seed-mediated method developed by Ye et al. [
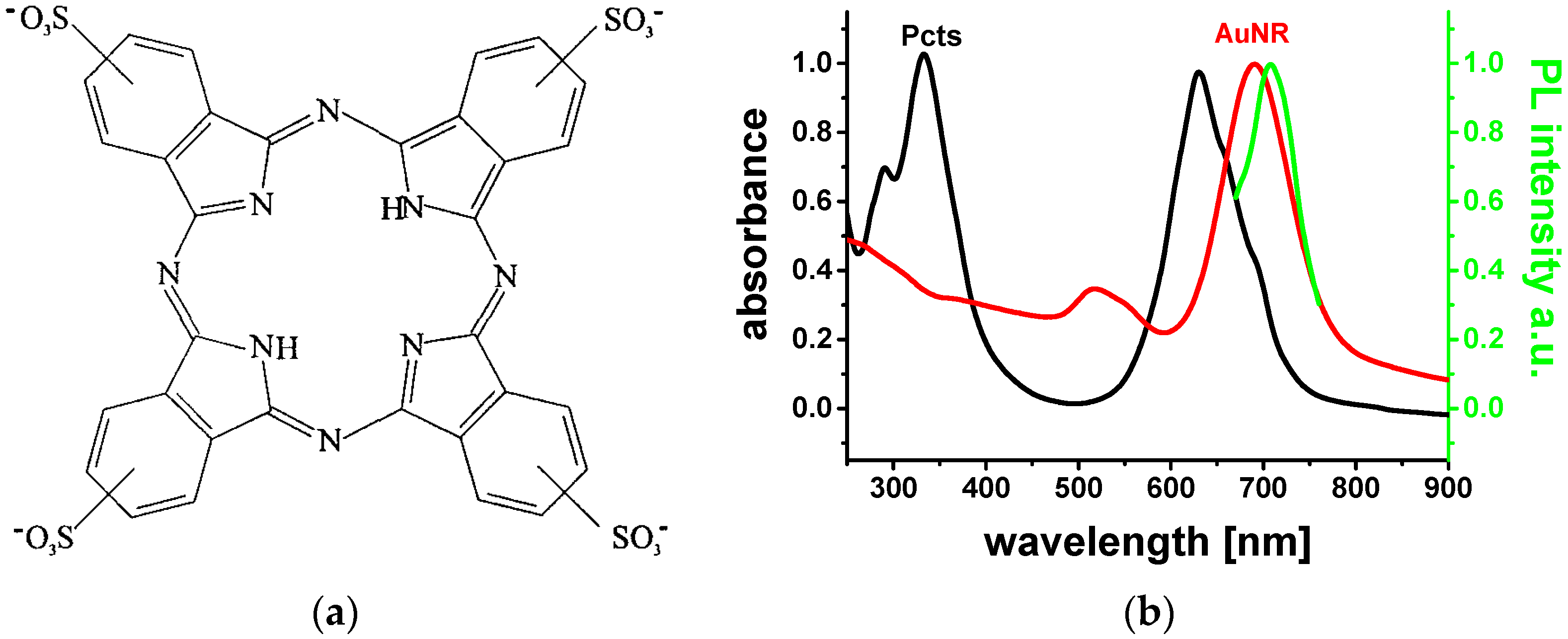
10]. The as-grown AuNRs were surrounded by a CTAB-bilayer shell, providing positive surface charges. The planar Pcts molecules were electrostatically bound at the CTAB bilayer through their four negatively-charged sulfonate anchor groups (
Figure 1a).
Figure 1b depicts the UV/Vis absorption spectra of the aqueous Pcts (black solid line) and AuNRs solution (red solid line), and the photoluminescence (PL) spectrum obtained upon excitation of the AuNR colloid at 530 nm (green solid line).
The aqueous AuNR solution under study exhibits transversal LSP resonance and longitudinal LSP resonance as a small broad bump around 520 nm and a prominent band at 690 nm in the UV/Vis absorption spectrum, respectively. The PL emission peaking at 708 nm is assigned to the longitudinal plasmonic emission of the AuNRs. Pcts dissolved in water shows two strong broad absorption bands: the Soret band appears in the near-UV (332 nm) and the Q-band in the red (630 nm) [
11]. The Q-band arises from in-plane electronic (
π → π*) transitions. This implies that the transition dipole moments of photo-excited Pcts molecules at the AuNR surface are predominantly parallel to the dipole moment of the longitudinal LSP modes. Pcts is anchored in a planar configuration at the CTAB bilayer, which has a thickness of ca. 3.5 nm. In addition, the longitudinal LSP resonance spectrally overlaps with the fluorescence emission spectrum of Pcts (
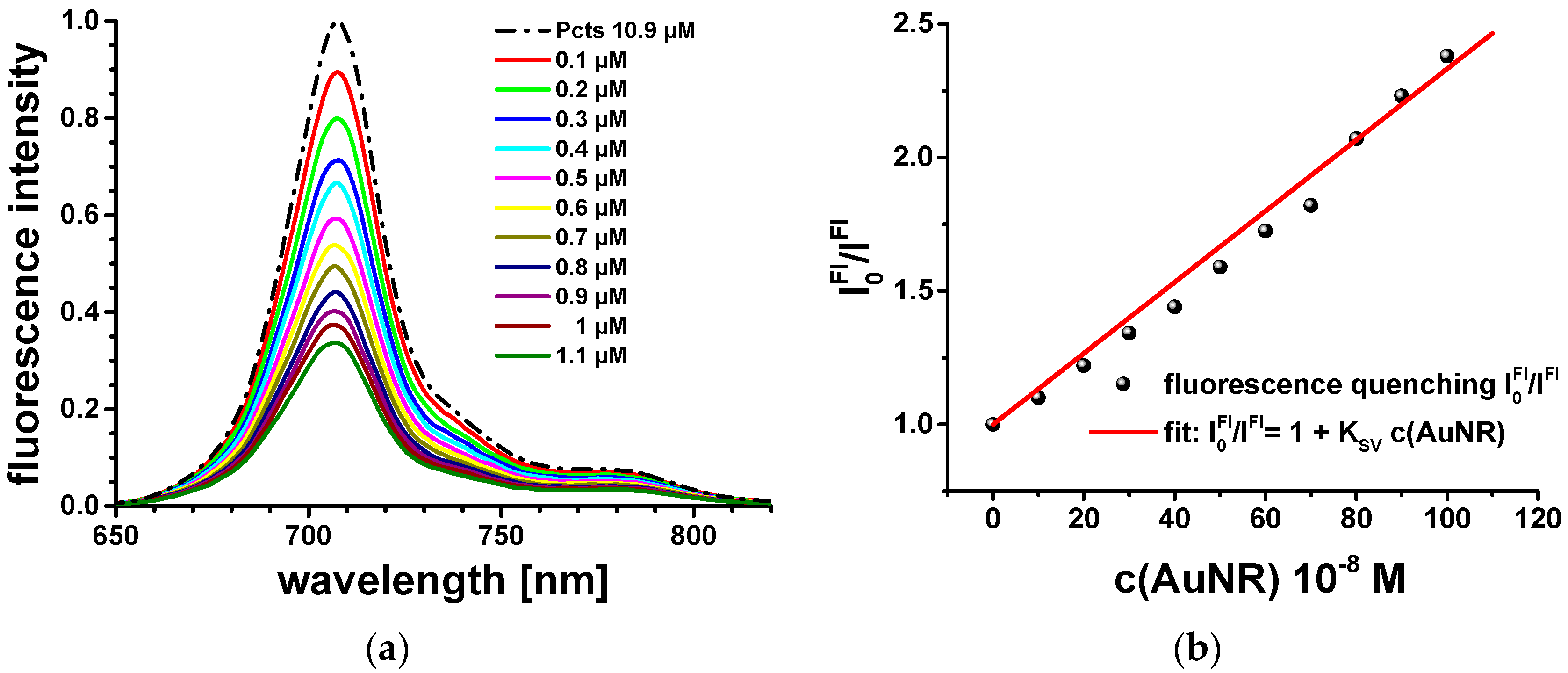
Figure 2a). Therewith, the prior conditions for strong dipole–dipole couplings and resonance excitation energy transfer between electronically excited Pcts at the AuNR surface and the longitudinal LSP modes of the AuNRs should be fulfilled. Fluorescence quenching experiments were performed to provide evidence for excitation energy transfer. Therefore, the fluorescence spectrum of a 2 mL aqueous 10.9 µM Pcts solution was recorded, and the fluorescence intensity was quenched by successively adding a 10 µL aqueous 200 µM AuNR solution. The fluorescence intensity ratio
was obtained by measuring the fluorescence intensity of Pcts (
) and that of Pcts in the presence of AuNRs (
), respectively.
follows the Stern–Volmer kinetics, and the Stern–Volmer coefficient was determined as
KSV = 1.22 × 10
6 M
−1 (
Figure 2b). The Stern–Volmer coefficient demonstrates efficient fluorescence quenching, which is taken as evidence for strong dipole–dipole couplings and the resulting ultrafast resonant excitation energy transfer between surface-bound Pcts molecules and longitudinal surface plasmons of the AuNRs. Stationary optical excitation conditions—as applied for steady-state fluorescence spectroscopy experiments—are not sufficient to overcome the threshold of spaser performance. Further proofs for ultrafast resonant excitation energy transfer and the inherent feedback mechanism resulting in spasing emission were obtained from the analysis of the ultrafast excitation and relaxation dynamics of Pcts, AuNRs, and Pcts-capped AuNRs (Pcts@AuNRs).
Therefore, the optical response of the Pcts, AuNRs, and Pcts@AuNRs to ultrashort (150 fs) laser pulses (387 nm) was examined by conducting fs transient absorption (TA) spectroscopy experiments.
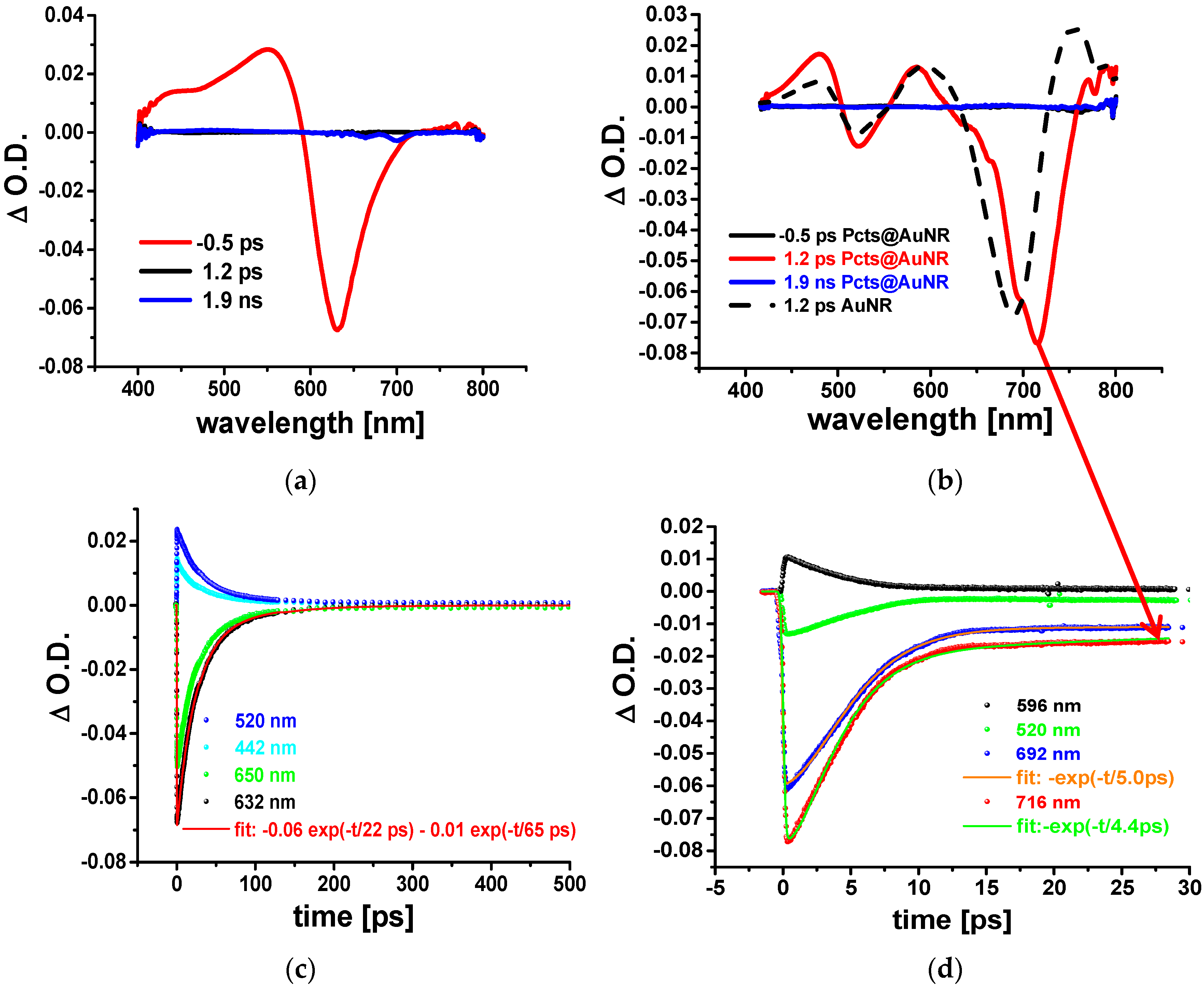
Figure 3 displays the time evolution of the change in the optical density (ΔOD) of aqueous solutions of Pcts (a) and AuNRs and Pcts@AuNRs (b). The TA spectra of Pcts showed a negative ΔOD signature around 630 nm that arises from photo-induced bleaching of the ground-state absorption at 632 nm (
Figure 1b). The positive ΔOD signature at shorter wavelengths was ascribed to photo-induced absorption transitions emerging from the S
1 state of Pcts. The kinetic traces detected at four different wavelengths: 442 nm, 520 nm, 632 nm, and 650 nm were fitted to a bi-exponential decay function. The resulting time constants with
t1 = 22 ps and
t2 = 65 ps reflect ultrashort decay or rise dynamics. In case of a laser dye such as Pcts picosecond-resolved dynamics are characteristic for stimulated emission, providing the deactivation of the S
1 state and the repopulation of the S
0 ground state [
11].
In case of plasmonic nanostructures such as AuNRs, pump laser pulses with energies much higher than that corresponding to the LSPs may induce instantaneous interband transitions, which create a nonthermal distribution of “hot” electrons. The dephasing of the LSPs occurs as the hot electrons rapidly equilibrate via intrinsic damping pathways (bulk or electron-surface scattering), leading to a Fermi distribution at a higher electron temperature. Such a thermalization process generates a photo-induced bleaching of the ground-state population density and photo-induced absorption due to transitions of excited species. These processes should then result in negative ΔOD signatures in proximity to the LSP resonances and positive ΔOD signatures in between. The positive and negative ΔOD signatures decay on the time scale of a few picoseconds as the hot-electron distribution relaxes via phonon emission (with the electron–phonon scattering time constant increasing with the laser pump fluence) and subsequent phonon–phonon scattering, ultimately dissipating heat into the solvent.
The TA spectra of the AuNRs (broken black line) and Pcts@AuNRs (red solid line) contain two negative ΔOD signatures which are ascribed to photo-induced bleaching through the transversal LSP resonance (516 nm) and longitudinal LSP resonance (690 nm and 705 nm, respectively) (
Figure 3b). In contrast to pristine AuNRs [
5], an additional narrow band at 716 nm emerges from the maximum of the longitudinal LSP bleach band that is red-shifted to 705 nm due to the changed environmental refractive index. Such a sharp intensive peak at wavelengths, with negative ΔOD values and clearly red-shifted from the longitudinal LSP resonance and fluorescence emission of Pcts (700 nm) is typical for spaser emission. In contrast to fluorescence, spaser emission performs as a focused coherent radiation, and thus has a much higher intensity; this facilitates its observation in fs TA spectra. Another typical feature of spaser emission persists in its ultrafast decay dynamics. The analysis of the mono-exponential kinetic traces at 692 nm (lLSP: longitudinal LSPs) and 716 nm (se: spaser emission) yielded the time constants:
tlLSP = 5 ps and
tse = 4.4 ps. The exponential decay behavior of the longitudinal LSP bleach with
tlLSP = 5 ps is attributed to electron–phonon scattering events. The spasing emission displays a distinctly faster decay dynamics, with
tse = 4.4 ps. Unfortunately pump pulses with fluencies larger than those applied here (300 nJ/cm
2) were observed to degrade the aqueous samples of AuNRs and Pcts@AuNRs. In future, we will improve the performance of AuNR-based spaser by engineering heat-resistant surface coatings as optical gain media.
3. Materials and Methods
3.1. Materials
The following chemicals were purchased from different suppliers and were used as received. Hexadecyltrimethyl-ammonium bromide (CTAB, >98.0%), 5-bromosalicylic acid (>98%), sodium salicylate (99%), phthalocyanine tetrasulfonate hydrate (>98%), L-ascorbic acid (99.5%), silver nitrate (AgNO3, >99%), and sodium borohydride (NaBH4, 99%) were purchased from Sigma Aldrich (Steinheim, Germany). Hydrogen tetrachloroaurate (III) trihydrate (≥99.9% p.a.) were purchased from Carl Roth (Karlsruhe, Germany) and sodium borohydride (>97%) from Fluka (Buchs, Switzerland). Milli-Q deionised water (18 MΩ) was used throughout the experiments.
3.2. Gold Nanorod Synthesis
The AuNR synthesis procedures used here were developed by Ye et al. [
10]. The AuNR seed solution was prepared by dissolving CTAB (1 mmol, 365 mg) in 5 mL water and heating the solution until it became clear. HAuCl
4 (5 mL of 0.5 mM) was added to the CTAB solution under gentle stirring. A freshly-prepared solution of NaBH
4 (0.6 mL of 10 mM, diluted to 1 mL) was injected into the Au(III)-CTAB solution under vigorous stirring (1200 rpm). The color of the solution changed from yellow to reddish-brown. The stirring was stopped after 2 min. The seed solution aged at room temperature for 30 to 60 min before use. The growth solution was prepared by dissolving 1.8 g CTAB and 110 mg 5-bromosalcylic acid in 50 mL of water at 70 °C under gentle stirring, until all CTAB was dissolved and a transparent solution was obtained. The solution cooled down to 30 °C. Then, 2.4 mL of a 4 mM solution of AgNO
3 was added. The mixture was kept undisturbed at 30 °C for 15 min. Afterwards, 50 mL of a 1 mM HAuCl
4 solution was added and gently stirred (300–400 rpm) for 15 min at 30 °C. A 64 mM solution of ascorbic acid was added, and the solution was vigorously stirred for 30 s until it became colorless. The AuNR solution was used immediately after preparation. The Pcts@AuNR samples were prepared by isolating the AuNRs from the reaction mixture and mixing them with Pcts. The as-synthesized AuNR solution was centrifuged at 8500 rpm for 25 min, the supernatant was removed, and deionized water was added. This washing procedure was repeated twice. At the end, the AuNRs were redispersed in 5 mL of water and mixed with 2 mL of 10
−3 M Pcts solution. The Pcts@AuNRs were separated from free Pcts molecules by performing the washing procedure as described above and resdispersing the Pcts@AuNRs in 5 mL water.
3.3. Steady-State Spectroscopy
The UV/Vis absorption spectra were measured using a Perkin Elmer Lambda2 spectrometer (Bodenseewerk Perkin-Elmer GmbH, Überlingen, Germany). The fluorescence spectra were recorded on a Horiba Jobin-Yvon FluoroMax-3 spectrofluorometer (Horiba, Bensheim, Germany) using the magic-angle polarization configuration. All experiments were performed at room temperature employing 10 mm quartz cuvettes.
3.4. Femtosecond Transient Absorption Spectroscopy Measurements
The experiments were conducted on a Clark MXR CPA 2101 laser system (Horiba, Bensheim, Germany) in conjunction with an Ultrafast TAPPS HELIOS detection system (Ultrafast systems, Sarasota, USA), consisting primarily of an Er3+-doped glass-fiber-based spectrometer. The output pulses at 387 nm with a 150 fs pulse duration and a 1 kHz repetition rate were used as pump pulses. Aqueous AuNR, Pcts, and Pcts@AuNR solutions were transferred into quartz cuvettes with 2 mm optical path and pumped at pump fluencies of 120 to 250 μJ/cm2. Transient absorption spectra were recorded in the visible region between 400 and 800 nm in the magic angle configuration with respect to the polarization vectors of the pump and probe pulse (the chirp of this spectral range was approximately 350 fs). TA spectra were obtained as the temporal evolution of the changes in the optical density (ΔOD) of the sample. Therefore, a chopper wheel provided the blocking of each second pump pulse so that the probe pulse was alternately transmitted through a pump-pulse-excited and a ground-state sample. The intensity of the transmitted probe pulse after the pump pulse-excited sample, I*(λ,τ), and that without pump-pulse excitation, I0(λ), were measured as a function of the delay time, τ. The ΔOD values were determined as ΔOD(λ,τ) = log[I0(λ)/I*(λ,τ)]. For I0(λ) > I*(λ,τ), the ΔOD signal attains positive values, and is assigned to absorption transitions of excited species (the so-called photoinduced absorption), whereas negative values of ΔOD result from I0(λ) < I*(λ,τ) and are ascribed to photoinduced bleaching of the ground state population density or stimulated emission.



{kind=link}
{kind=link}
{kind=link}


