1. Introduction: Superconducting Infinite-Layered Nickelates Nd1−xSrxNiO2
The discovery of Bednorz and Muller [
1] started an intense search for novel superconductors with high values of the critical temperature
and gave very interesting open questions [
2]. However, in spite of a tremendous effort in the theory, the mechanism responsible for the pairing in cuprates is still unknown [
3]. Yet, this is one of the fundamental open problems in modern physics.
Perhaps less spectacular was the recent discovery of superconductivity in infinite-layered NdNiO
doped by Sr [
4] as the values of
are “only” close to 15 K [
5]. Nevertheless, it gave a new impulse to the theory of high-
superconductivity at large. To some extent, the nickelate superconductor family is rather similar to cuprate superconductors [
6], as once again two-dimensional (2D) planes of transition metal and oxygen ions play a central role here [
7]. With Ni
ions, one again has
electronic configuration and similar lattice structure, but the apical oxygens are absent [
8]. By following the same analysis for a NiO
layer as the one performed for a CuO
layer, we expect Ni
to exhibit antiferromagnetic (AFM) order. One finds that the charge-transfer energy and the gap in nickelate are larger than those of cuprates [
9,
10]. The doped holes reside on oxygen sites in cuprates forming the Zhang-Rice singlets [
11]. On the contrary, the doped holes will likely reside on Ni sites in doped Nd
Sr
NiO
.
Usually, nickelates are investigated by evaluating their electronic structures by density functional theory (DFT) and comparing them with those of cuprates [
6,
7,
8,
12,
13,
14]. This approach has its well known weak points—here we consider instead the tight-binding charge-transfer model [
10], similar to that for cuprates [
15], and shall concentrate on the electronic properties of nickelates. We shall also address the question of magnetic exchange and the absence of AFM order in nickelates [
16].
2. Charge-Transfer Model Revisited: Charge-Transfer Model for an NiO2 Plane
We introduced the multiband
Hamiltonian for a NiO
plane [
10] starting from a 2D Ni
O
cluster with periodic boundary conditions (PBCs). The basis set includes four orbitals per NiO
unit cell: two
orbitals
at each Ni
ion and one bonding
orbital (either
or
) at each oxygen ion in the 2D plane,
Here, the first two terms in the Hamiltonian (
1) stand for the kinetic energy:
includes the
hybridization
and
includes the inter-oxygen
hopping
,
where
(
) is the creation operator of an electron at nickel site
m (oxygen site
i) in an orbital
(
). Here,
z and
stand for
and
orbitals, while
stand for
and
orbitals. The elements
are accompanied by the phase factors, which follow from orbital phases [
15].
The one-particle (level) energies are included in
, where we introduce the charge-transfer energy between
d and
p orbitals,
The model is completed by the Coulomb interactions in the
d and
p orbitals. For the
d electrons,
where
stands for
and
orbitals.
(
) is electron creation (annihilation) operator at site
m in orbital
with spin
.
is a number operator.
stands for spin operator in
orbital at site
m. The two Kanamori parameters to describe the interactions between
electrons are
.
is Coulomb repulsion element for
orbital.
and
are Hund’s exchange and interorbital Coulomb interaction [
17]. Similar interactions with
are written for
p electrons.
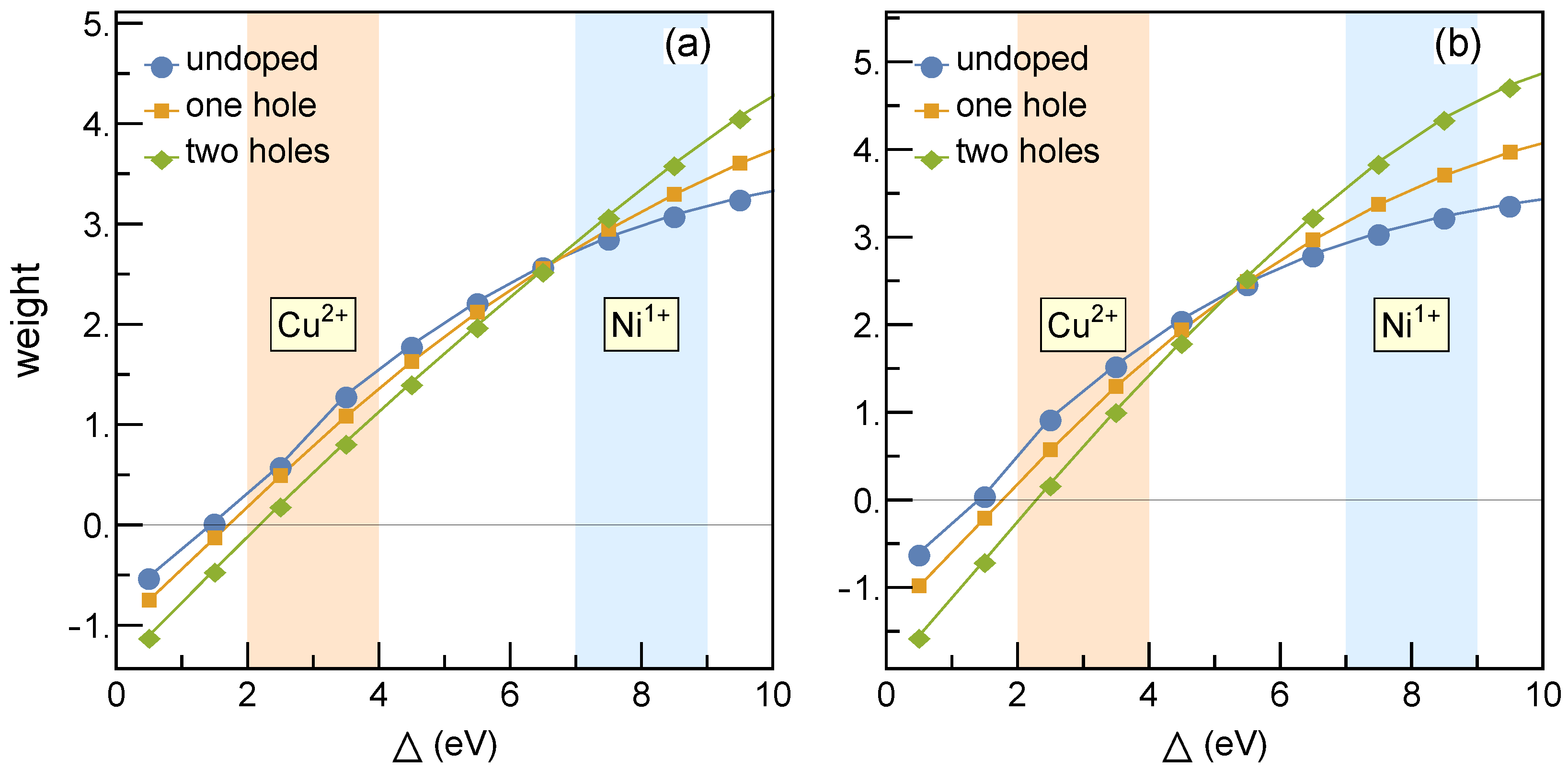
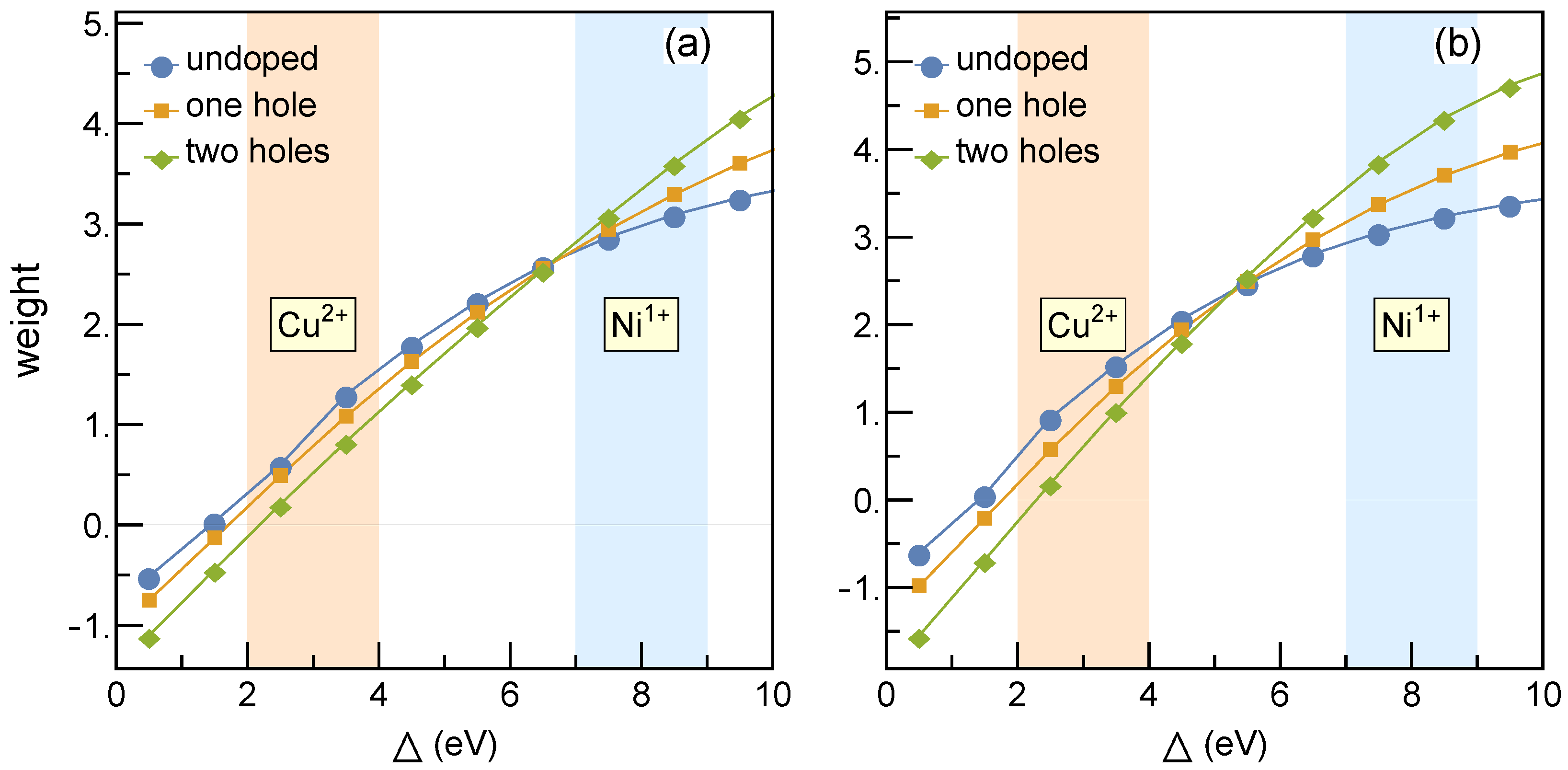
Figure 1 shows different weight distributions for hole occupations at Ni and O sites for the parameters of
Table 1. The Ni-O charge-transfer model has been studied via impurity [
9] as well as lattice approach [
10]. It has been established that the holes in undoped compounds remain within
orbitals. Thereby, we have assumed that Nd does not contribute to the electronic structure, and the system without Sr is a Mott insulator. Doping by Sr gives a doped hole, which tends to reside at nickel sites rather than at oxygen sites. The nickelate (cuprate) regime [
9] is highlighted in blue (orange) in
Figure 1. In the nickelate regime, the holes reside predominantly at Ni sites. This is the essential difference with cuprates where a doped hole (for hole doping) resides predominantly at oxygen and forms a Zhang-Rice singlet [
11].
Including intersite Coulomb repulsion
enhances the hole occupancy at Ni sites [
10] and shifts the doping crossover to lower values of the charge transfer energy
. All the on-site energies of the Ni(
) orbitals have been included in the Ni-O hybridization terms
[
9]. Similar results were obtained for finite
orbital splitting, where
eV should be considered the upper limit.
The NiO
compound is a Mott–Hubbard insulator. It is then possible to replace the charge-transfer model by the
d-only Hubbard model. The effective Ni–Ni hoppings can be derived from second-order perturbation theory [
18]. The next question is on which
d-orbitals the doped holes preferably reside?
The asymmetric distribution of holes suggests that one could replace the Ni-O model (
1) with a Ni
d-only model as oxygen
p-orbitals become unimportant. The DFT calculation shows the band structure of NdNiO
that two bands are crossing Fermi level. In the orbital-resolved band structure, the lower band has
character and the upper band contains both Nd and Ni contributions. The large charge transfer energy as well as the presence of electron pocket at
are the two striking features of the nickelate compound. The empty 5d states of Nd are responsible for providing electron pocket. The empty
states are below the Fermi level, in other words, these states provide the hole states into Ni band by the so-called ’self-doped’ effect [
19]. Furthermore, the
states was shown to be hybridized with Ni apical orbitals, i.e.,
and 4
s orbitals [
20] lead us to construct the effective two-band model consisting of Ni in-plane orbital,
, and Ni off-plane orbital, the modified
s orbital. In this work, we present the character of doped holes in the realistic two-band model of NdNiO
compound.
3. Electronic Structure Calculations
The ab initio electronic structure calculations using DFT were performed with the Quantum Espresso code [
21,
22,
23] using a plane-wave pseudopotential method, combining a projector augmented wave method [
24] and a specific choice of pseudopotentials [
25]. Within this work, we chose an energy cutoff of 600 eV and a
point centered Brillouin zone mesh of size
. For all calculations we used the same crystal structure published in [
12].
The two-band model is derived by performing a Wannier projection onto the DFT band structure as implemented within the Wannier90 interface [
26], including onsite energies and hopping parameters of each Wannier orbital. The projection was performed onto a Ni(
) and an apical Ni(
s) Wannier orbital within an energy window ranging from
to
eV. The DFT band structure, represented here by the projected Wannier bands and its density of states (DOS), are shown in
Figure 2. Panel
Figure 2a shows the DFT band structure (black lines) and Wannier bands (red dashed lines). The right panel
Figure 2b contains the resulting Wannier DOS around the Fermi energy for the bands of two symmetries:
and
s. It is clear that the fit cannot be perfect and the electron/hole estimation is wrong, but this plays a minor role fr the questions asked here and our result qualitatively matches previous studies [
12,
20].
The Wannier Hamiltonian is given within the real space notation and is of the form,
The basis is ordered following the convention
. Explicit numerical values for the hopping parameters are given in
Table 2 for the terms with leading contributions, i.e., terms larger than
(the other terms were neglected). Note that the relation
holds for Wannier models. As a consequence, both terms (for distances
and
) need to be included in further calculations. From
, a tight-binding Hamiltonian can be constructed by applying a Fourier transformation of the form,
where
describes the distances of the Wannier orbitals
and is typically represented in terms of the lattice vectors.
4. Effective Two-Band Hamiltonian
Following the idea of neglecting oxygen orbitals, we construct an effective model containing only Ni() orbitals. The band structure calculation shows that only two bands contribute at Fermi level. Therefore, the two-band model of nickelates is capable of reproducing the physics of nickelates. It consists of orbital and the s orbital, which includes rare-earth states and Ni apical orbitals.
We consider the two-band Hamiltonian
. The kinetic part is
while the interactions are given in a similar way to Equation (
5). The orbitals
and
s have the diagonal energies
, being 0 and
. The bands are constructed following Ref. [
20]. The oxygen
orbitals are included implicitly in
ones. Note that here, one should not confuse
symmetry with Ni(
) orbital. The former is an effective orbital including in-plane oxygen, while the latter is solely a Ni(
) orbital. The
s orbital contains contributions from Nd(
), Ni(
) and Ni(
) orbitals. The symmetry of this hybrid orbital is the same as that of atomic
s orbital.
Instead of Nd(
) electronic states, the empty Nd(
) orbitals are responsible for the striking electron pocket at the
point in the band structure [
6]. The Nd atoms are originally located in the off-plane direction of Ni-O plane. In this effective model, the rare-earth atoms are included into Ni atoms via
s orbital, indicating the model is three-dimensional and the Coulomb interaction as well as Hund’s coupling need to be screened. For instance, the two electrons sitting within
s orbital could be located either at Nd or at Ni atom. Therefore, the Coulomb repulsion between these two electrons within the
s orbital is reduced. We introduce a parameter
to represent the reduced Coulomb interaction and Hund’s coupling, i.e.,
and
.
stands for weak (strong) screening effect from rare-earth atoms, and we consider two parameter sets
A and
B, given in
Table 3. We study the effective two-band model via Lanczos algorithm [
27] on a
unit cell.
The undoped nickelate corresponds to quarter-filling, i.e., 8 electrons. The stoichimetric compound has
configuration, where Ni(
) orbital is half-filled, and the weak hybridization of Ni and Nd causes Ni(
) orbital to be away from half-filling and creates the self-doping effect [
19,
28]. The Ni–Ni hopping integrals are obtained via fitting Wannier functions on DFT band calculation. We aim to address the question: where are the doped holes located in the two-band model?
In the absence of an electron hoppings Hamiltonian, the ground state is a trivial antiferromagnet where
orbital is half-filled and the two canonical AFM phases,
C-AFM and
G-AFM, are degenerate. Including hopping elements to further neighbors leads to metallic behavior. In one-band Hubbard model, the metal–insulator transition occurs when
, where
is the number of neighbors in the 2D plane. Similarly, the two-band description can lead to partial orbital-selective Mott transition, where one band is insulating and the other one remains metallic. In the one-band version, the two relevant configurations are singly occupied or form double occupation within a single site. The quarter filling two-band version, however, contains several possible configurations where the lowest energy is still a single occupancy followed by a local triplet state with energy
[
17].
5. Results and Discussion
We begin with electronic density distribution on
clusters obtained by exact diagonalization with twisted boundary condition (TBC). The hopping parameters used here are given in
Table 2. With PBC, one requires hopping integrals
to be scaled by the factor of 1/2 due to double-counting at the boundary. To avoid this additional factor in the hopping terms, we replace the PBC with TBC. Instead of having a constant phase
as in PBCs, the hopping terms at the boundary are modified by twisted angles
, giving
; for more details, see Refs. [
29,
30]. The PBC corresponds to
, while
is obtained for the anti-periodic boundary condition. In what follows, the observables are obtained by averaging over several twisted angles.
In
Figure 3a,b, we show the undoped orbital-resolved densities, i.e., the electron occupancy within each orbital:
(predominantly occupied) and
s (it usually has decent amount of electrons). Finite occupancy of
s orbital is expected due to self-doping effect. We further see that increasing of the parameter
causes the occupation on
orbital to approach half-filling. Once the
orbitals are almost half-filled, we show below that the
G-AFM ground state is the only dominant magnetic ground state. Including screening effect into
orbital by lowering its Coulomb interaction gives this orbital as slightly more favorable, see also
Figure 3a,b.
By adding one hole, we see that both orbitals can be occupied by holes depending on how strong the screening effects of
s orbital are, see
Figure 3c,d. The screening effect on
orbitals makes little changes in the one-hole case. An interesting scenario arises when two holes are added into the undoped Ni-O plane. The unscreened
remains roughly the same, as
s orbital is effectively favorable for holes at small
. When taking the screening effect into account, holes are effectively occupying the
orbital regardless of the parameter
.
Theoretical studies based on Ni(
) bands [
10,
19,
28] have suggested that high-spin
state Ni
is favorable for hole doping. In contrast, RIXS measurements [
31] show that two holes are residing mainly within Ni(
). Due to the limitation of the measurement, we cannot determine the occupation of the rare-earth
states. The difference between the predictions of the theoretical model and the experiment arises from the rare-earth atoms. In the Ni(
) with
configuration, the
is half-filled and forming a high-spin state together with another
orbital with energy
[
10]. This energy is significantly smaller than the energy
U for a low-spin
state.
On the contrary, the
s orbital, in the hole picture, has lower energy than
orbital and the quarter-filling of electrons corresponds to
-filling by holes via particle-hole transformation. In this case, the
s orbital is filled by holes and leaves half-filled
orbital—then holes reside mainly on
orbital. This finding suggests that for hole doping, the one-band Hubbard model may be sufficient [
32]. This intuitive picture of hole configuration requires the
to be nearly half-filled, in other words, it suggests that the ground state is a strong antiferromagnet as in cuprates [
11,
33]. However, NMR experiments [
34,
35] report no observation of long-range AFM order in the
RNiO
(
R = Nd,La) down to 2 K.
According to the DFT+sicDMFT approach [
28], the paramagnetic ground state has the lowest energy followed by
C–AFM with energy difference about 20 meV/atom and
G-AFM with energy 105 meV/atom. The
C–AFM state has parallel spins along the
c axis, while
G–AFM phase has antiparallel spin alignment to its nearest neighbor in all directions. To address the magnetic order in this two-band model, the spin structure factor of undoped profile,
is shown in
Figure 4. With this small cluster size we can observe only six relevant
-points in the Brillouin zone and only simple types of AFM order can be investigated. As a universal feature, the spin structure factor at
is enhanced as the
orbital is close to forming a half-filled band; it indicates that
G–AFM order is favored for electron doping [
13], see
Figure 4.
At small
, the spin structure factor (
9) shows a competition between several magnetic ordered states, dominated by
and
spin correlations, which stand for
C–AFM and
G–AFM order. Other types of order, in particular
A–AFM order, are not favored. In the recent
x-ray scattering experiment [
36], the existence of AFM correlations was confirmed, but the small cluster size prevents us from concluding whether AFM long-range order could be stable or not in this parameter regime.
The screening effect from rare-earth atoms [
19,
37] could be responsible for the competition of magnetic orders. At small
where the screening effect from rare-earth is strong, the electrons tend to form on-site triplets with energy
[
10], coexisting with singly occupied
orbitals. Then, increasing
enhances the interactions on
s orbitals and the on-site triplet energy surpasses the bandwidth of
s orbital, i.e.,
(
). The largest hopping elements are the in-plane hoppings along
x and
y direction of
orbital. Another transition then occurs when the on-site triplet energy overcome the bandwidth of
orbital, becoming a Mott insulator.
Figure 5 shows the competition between low-spin and high-spin states in NiO
planes of the novel Ni-layered superconductors for decreasing number of holes, i.e., for electron doping. Here we use
and
, where
N is the number of Ni sites. We show here that the double occupancy
is enhanced with increasing number of holes in the NiO
plane. Increasing
increases the double occupancy
, while the double occupancies in
s orbitals are almost absent. Thus, the model reduces in this regime to the one-band Hubbard model [
32].
Simultaneously, the amplitude of high-spin states
T at Ni sites is reduced in the regime of large
. Therefore, we conclude that electron doped materials have preferably low-spin configuration. On the contrary, hole doping may favor locally high-spin
states [
10] instead of singlets
for the double occupancies of
orbitals. Whether such local triplets could play a role in nickelate superconductivity is still an open question.
6. Summary and Conclusions
In summary, we have replaced the half-filled charge-transfer model by the effective two-band model at quarter-filling. The symmetries of each band are given by and s symmetry. The contributions of Nd and Ni atoms to s orbital lead to the reduction of Hund’s exchange and Coulomb repulsion, with its strength being scaled by the parameter . The model shows the three competing phases: metal, orbital-selective, and Mott insulator. The Mott insulator is realized when the splitting between onsite triplet and singly occupied state is larger than the size of the bandwidth, similar to one-band Hubbard model. It is followed by the orbital-selective Mott insulator where the orbital splitting separates the and s bands from each other.
The nonmagnetic state is unlikely within this small unit cell and only AFM configuration can be realized. While the G–AFM phase is clearly dominating at large , the competition between C–AFM and G–AFM is found at low where the on-site triplet competes with AFM ground state, indicating the tendency toward AFM ordering. The holes are doped differently among the three phases. In metallic phase, the itinerant s orbital is favorable for holes. On the other hand, in the orbital-selective phase, the situation is slightly complicated while adding one hole favors s orbital, but adding two holes can effectively lead again to occupation, as in the Mott insulating phase.
When the screening effect on orbital is included, the stability of the orbital-selective phase is enhanced and the metallic phase vanishes. The two-band model at quarter-filling, therefore, connects the two controversial scenarios of which orbitals preferred by doped-holes via the parameter . The parameter somehow represents the screening effect from rare-earth orbital, showing the importance of rare-earth atoms for the electronic structure of superconducting nickelate.
Finally, we remark that the present approach has several limitations, which could be seen as open questions at present, to be resolved in the future. First of all, electronic phase separation could occur in nickelates as in other superconductors, here we give the data for iron superconductors [
38]. A very interesting open problem is how many electronic components contribute to the superconducting state and whether orbital selective superconductivity may occur in nickelates [
20]. One could address these questions in a more satisfactory way only by extending the two-band model by spin-orbit coupling.


 ,
, 





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}