Abstract
Duchenne muscular dystrophy (DMD) is an X-linked genetic disease in which the dystrophin gene is mutated, resulting in dysfunctional dystrophin protein. Without dystrophin, the dystrophin-glycoprotein complex (DGC) is unstable, leading to an increase in muscle damage. Moreover, the imbalance between muscle damage and repair leads to a chronic inflammatory response and an increase in the amount of fibrosis over time. The absence of dystrophin at the sarcolemma also delocalizes and downregulates nitric oxide synthase (nNOS) and alters enzymatic antioxidant responses, leading to an increase in oxidative stress. In this review, we analyze the pathogenic role of both inflammation and oxidative stress in muscular dystrophy.
1. Introduction
Duchenne muscular dystrophy (DMD) is a severe X-linked recessive disease affecting 1 of 5000 male births, being males hemizygous for chromosome X. Affected boys exhibit symptoms between 2 and 5 years of age, including motor developmental delay, abnormal gait, and muscle weakness [1]. The progressive muscle degeneration leads to loss of ambulation at 8–12 years with premature death due to cardio-respiratory failure [2]. The genetic causes of the disease are “out of frame” deletions (68%), duplications (11%), and small mutations (20%) in the dystrophin gene, the largest known gene with the highest spontaneous mutation rates in humans [3,4]. These alterations of the translational reading frame result in a dysfunctional or absent dystrophin protein with critical consequences for muscle tissue integrity.
Dystrophin is a cohesive cytoplasmic protein that interacts with a group of membrane proteins to form the dystrophin-associated protein complex (DAPC), which connects the myofiber cytoskeleton to the extracellular matrix. Critical components of DAPC are dystroglycans (DG) and sarcoglycans (SCG) forming the dystrophin-glycoprotein complex (DGC), and other associated proteins, such as α-dystrobrevin, syntrophins, caveolin-3, growth factor receptor-bound protein-2 (Grb2), and the neuronal nitric oxide synthase (nNOS) [5]. Since DAPC stability is determinant to support sarcolemma integrity, the absence of dystrophin protein alters the stability of membrane complexes, leading to aberrant intracellular signaling pathways and to a progressive increase of contraction-induced muscle damage [6,7].
Although the genetic basis of DMD pathology has been known for more than 30 years and a mouse model (mdx) has been developed, mechanisms leading to the extensive muscle wasting observed in DMD patients remain to be fully elucidated [8]. Indeed, while dystrophin deficiency is the primary genetic defect, secondary mechanisms are important features of the disease. The perturbation of calcium homeostasis along with chronic inflammation, mitochondrial dysfunction, and elevated oxidative stress characterize the dystrophic muscle and milieu. Moreover, repeated cycles of myofiber necrosis and ineffective regeneration contribute to the establishment of a chronic inflammatory response, which in turn amplifies tissue damage, leading to muscle degeneration and fatty tissue replacement [2,9]. Because no etiologic therapy is available for Duchenne muscular dystrophy, a better understanding of secondary pathogenic mechanisms downstream from the primary defect could be useful in developing new adjuvant treatments.
2. Animal Models of Muscular Dystrophy
Since muscular dystrophy emerged as a complex muscle disease, reliable animal models of the human disease are necessary to predict drug, gene, and cell therapy effectiveness in patients. The mdx mouse strain, lacking a functional dystrophin gene, has served as the animal model for human DMD and Becker muscular dystrophies [10]. However, while the skeletal muscles of mdx mice undergo extensive necrosis early in neonatal life, unlike the human disease, the affected muscle rapidly regenerates and regains structural and functional integrity [8,11,12,13]. The enhanced regenerative potential of mdx muscles and the upregulation of compensatory proteins, such as utrophin and integrins, are thought to be the basis of the reduced wasting of dystrophin-deficient muscles in mdx [14]. Moreover, among factors possibly explaining the difference between humans and mdx mice the extent of chronic inflammatory response has been suggested to be linked to the severity of dystropathology [15,16]. To compensate for these shortcomings of the mdx mouse, other mouse models of muscular dystrophy have been generated [17,18,19]. Nevertheless, we should take into consideration that double mutant mice like mdx mice deficient for MyoD [20], utrophin [18], parvalbumin [21], α7 integrin [22], or mTR (telomerase) [23] do not resemble the genetic background of DMD patients and are therefore less appropriate to predict therapeutic effects.
Thus, the mdx mouse is still the most widely used model to study DMD and also offers some advantages, considering that it is the most well-characterized and well-established murine model of DMD, in terms of pathogenic factors associated with the absence of dystrophin. There is an acute onset of pathology (increased myofiber necrosis and elevated blood creatine kinase) around 3 weeks of age, in which mdx mice display muscle weakness similar to DMD patients [8]. It has been reported that analysis of sera from 1 week to 7-month-old mdx mice revealed age-dependent changes in the level of biomarkers, with most biomarkers acutely elevated at 3 weeks of age [24], and these biomarkers were elevated at comparable levels in DMD patients. Although the use of mdx mice for providing insight regarding human DMD has generated considerable debate within the scientific community, it remains an invaluable experimental model. Indeed, over the last 30 years, many elegant papers have been published on the mdx mouse, including studies in which several therapeutic strategies have been analyzed [25,26,27,28,29,30,31,32].
3. Secondary Pathogenic Mechanisms Underlining DMD Pathology
3.1. Continuous Cycles of Myofiber Degeneration and Regeneration
The gradual loss of muscle tissue that occurs in patients affected by DMD has been attributed to an impaired regeneration process and to the progressive alteration of stem cell function in response to the chronic stimuli resulting from the constant repair of degenerating muscle fibers [33,34]. Indeed, repeated cycles of muscle degeneration and regeneration are key features of DMD pathology.
Recent findings demonstrated that the muscle resident stem (satellite) cells are directly involved in DMD pathology [35]. Satellite cells (SCs) are adult myogenic stem cells, residing in a specialized niche between the basal lamina and sarcolemma of individual myofibers, and their activation, proliferation, and differentiation are critical for muscle regeneration after injury [36,37]. However, it has been reported that lack of dystrophin affects SC polarity and asymmetric division as well as the ability to enter the myogenic program, thus contributing to the severity of disease [35,38]. In addition, prolonged muscle degeneration and chronic inflammation impair satellite cell activity and enhance fat and fibrotic tissue replacement [39,40].
Muscle regeneration is a highly coordinated program that requires a timely, controlled induction of muscle transcription factors (MRFs) and muscle specific genes [37,39]. Several studies reported how the complete differentiation program was impaired in muscles affected by DMD. Indeed, dystrophic satellite cells undergo extensive mitosis with a small population of progenitors contributing to the effective muscle repair [38,41]. In particular, accordingly with the elevated levels of paired box 7 (Pax7) expression in both DMD patients and mdx mice [42,43], cultured myoblasts from dystrophic muscles showed an increased number of Pax7-positive cells not expressing Myf5 (Myogenic factor 5) and a reduction of myogenin-differentiating cells [38,41]. Since dystrophin is expressed in activated satellite cells, dystrophin-deficient SCs display impaired polarity, abnormal division, and altered differentiation [41].
A functional dystrophin protein is also critical for the robustness of the muscle differentiation program, participating to the dystrophin/nNOS/HDAC2 (Histone deacetylase 2) pathway. It has been demonstrated that the absence of dystrophin disrupts the association between DAPC and nNOS (nitric oxide synthase), influencing the nitric oxide (NO) signaling pathway. An altered localization and expression of nNOS with a reduced production of NO has been observed in dystrophin-deficient muscles [44,45]. The impaired NO signaling mediates epigenetic changes through the direct modulation of HDAC2 nitrosylation state, thus affecting the expression of a specific subset of miRNAs and target genes that play an important role in muscle differentiation, homeostasis, and integrity [45].
3.2. Inflammation
Muscles of patients affected by Duchenne muscular dystrophy are known to exhibit consistent inflammatory changes, with these changes often observed soon after birth [15,46]. Factors related to chronic inflammation dominate the molecular signature of skeletal muscle in both DMD patients and mdx mice [15,16,46]. Indeed, gene profiling studies have provided evidence for coordinated activity of different mediators of the inflammatory response in dystrophic muscle, including cytokine and chemokine signaling, leukocyte adhesion and diapedesis, several invasive cell type-specific markers, and complement system activation [11].
Despite the complexity of the DMD-associated muscle inflammation, macrophages are reported as the primary inflammatory cell type involved in dystrophinopathy, and the balance between M1 and M2 phenotypic populations is known to affect the progression of pathology in mdx mice [47]. In particular, M1 macrophages represent the classically activated population able to participate in Th1 immune response and capable of tissue damage involved in the early acute phase of pathology in mdx (4 weeks of age) [47,48]. A later-invading population of M2 macrophages characterizes the mdx dystrophic muscle during the regenerative stage of disease (12 weeks of age) by promoting muscle growth and regeneration, an event that is not observed in human patients [47,48,49]. Indeed, alternatively activated macrophages expressing markers as CD163 are known to produce anti-inflammatory factors (IL-10) and to enhance satellite cell proliferation and muscle repair [47].
In light of the diversified role of immune cells in dystrophic muscle, specific released cytokines and chemokines and other inflammatory mediators are likely to play an important role in the pathogenesis of DMD. The pro-inflammatory M1 polarized cells, characterized by the expression of CD68 and iNOS (inducible nitric oxide synthase), upon activation, actively secrete chemoattractant molecules such as transforming growth factor- β (TGF-β), interleukin-6 (IL-6), tumor necrosis factor α (TNFα), and interleukin-1 β (IL-1β).
IL-6 is a pleiotropic cytokine that is involved in several intracellular signaling pathways under both physiological and pathological conditions. In particular, this cytokine exerts its biological activities through the activation of the canonical pathway, principally mediated by the membrane-bound receptor gp130, and by the induction of an alternative signaling mediated by the soluble IL-6 receptor α (IL-6r) [50]. Based on the activation of either classic or trans-signaling, IL-6 can respectively exert regenerative/homeostatic activities or promote pro-inflammatory pathways [51].
Indeed, IL-6 is locally and transiently produced in response to exercise and injury, and it plays an important role in satellite cell proliferation and muscle growth [52,53]. In contrast, the administration of high doses or long-term exposure of animal models to IL-6 increased muscle proteolysis [54,55,56].
IL-6, which is able to promote the switch from acute to chronic inflammation, is over-produced in both DMD and mdx muscles, whereas its circulating levels are significantly reduced in human patients after steroid treatment, and in mdx mice during the stabilization stage of disease (24 weeks of age) [8,28,57,58]. In light of this evidence, IL-6 is considered a key factor contributing to DMD progression and thus a potential target for the development of therapeutic approaches.
We recently demonstrated that a blockade of endogenous IL-6 receptor α, which mediates the IL-6 trans-signaling, conferred on dystrophic muscles resistance to degeneration and alleviated both morphological and functional consequences of the primary genetic defect. Pharmacological inhibition of IL-6r led to changes in the dystrophic muscle environment, favoring anti-inflammatory responses, improvement in muscle repair, and functional homeostatic maintenance of dystrophic muscle [28]. These data support different conclusions if compared to the study reported by Kostek and co-workers [59]. The two studies present several differences in the methodologies employed, including dosing regimen, and in the analysis and outcomes examined. Although improvements were observed in the Kostek et al. study, most of them did not reach statistical significance. Despite this, interestingly—and in agreement with molecular results of our study—they found increased levels of IL-10 in anti-IL6r-treated mdx mice. This piece of data is in evident contradiction with their conclusion that inflammation is increased. It is reasonable to speculate that by inhibiting IL-6 trans-signaling activity we reduced the inflammatory response in dystrophic mice and created a qualitative environment that makes the dystrophic muscle more resistant to the damage exerted by mechanical contraction [28].
Moreover, despite the implication of IL-6 signaling pathway in myoblast differentiation, the IL6r blockade resulted in an improved stabilization of dystrophic muscle phenotype with an enhanced expression of factors related to muscle differentiation and maturation [28]. It has also been reported that inhibition of the IL-6 intracellular mediator, namely the Jak/Stat pathway, stimulates muscle regeneration in both aged and dystrophic mice [60,61].
The opposite effects exerted by IL-6 can be justified considering the concept of hormesis, in which a low dose of a substance/molecule is stimulatory/homeostatic and a high dose is inhibitory/catabolic. Moreover, the positive effects of IL-6 are normally associated with its transient production and short-term action. In contrast, persistent inflammatory conditions, including muscular dystrophy, are associated with long-lasting elevated systemic IL-6 levels. Thus, muscle tissue benefits from low and transient increases in IL-6, whereas it is damaged by exposure to persistently high levels of this cytokine [28,58,62].
Along with IL-6, other pro-inflammatory cytokines, such as TNFα and IL-1β, are able to induce skeletal muscle proteolysis and to promote leukocytes influx inside the muscle [63]. Moreover, secreted cytokines can also activate peripheral neutrophils invading tissue and producing ROS (reactive oxygen species) [64,65]. Thus, the persistent activation and maintenance of the M1 pro-inflammatory population in dystrophic muscle contributes to the establishment of chronic inflammation, contributing to the severity of disease.
3.3. Oxidative Stress
Oxidative stress derives from an increased production of reactive oxygen/nitrogen species (ROS and RNS) and a decreased, insufficient, or imbalanced antioxidant response, leading to cellular dysfunction, damage, and tissue degeneration [66,67]. In skeletal muscle the impact of ROS is physiologically well contained through the activation of an endogenous program of antioxidant response, including the expression of redox-sensitive transcription factors [68,69,70,71]. However, elevated levels of free radicals generated under pathologic conditions can overwhelm the antioxidant defense leading to protein carbonylation and DNA damage. A role of oxidative stress in DMD pathology has been proposed [66,72].
The master regulator of the endogenous antioxidant defense is the NF-E2-related factor 2 (Nrf2), a transcription factor that is able to modulate the expression of phase II enzyme, counteracting ROS detrimental effects. Under physiological conditions, Nrf2 is known to be sequestered in the cytoplasm in a complex with its modulator KEAP1 (Kelch like-ECH-associated protein 1), which leads to Nrf2 ubiquitination and degradation [69,73]. Upon pro-oxidant conditions, Nrf2 translocates into the nucleus where it induces the expression of genes involved in glutathione synthesis, such as glutamate cysteine ligase (GCLC and GCLM), in the detoxification of xenobiotics (i.e., NAD(P)H quinone dehydrogenase (NQO1) and heme oxygenase (HO-1)), and in ROS neutralization such as superoxide dismutase (SOD1, SOD2) and catalase (CAT-1). Thus, Nrf2 represents a potential therapeutic target for DMD. Indeed, the induction of Nrf2, by the Nrf2 activator sulforaphane (SFN), was shown to reduce muscle inflammation and fibrosis in dystrophic mice, thereby improving muscle function [74,75,76]. The central role of the Nrf2 pathway in DMD pathophysiology is further supported by clinical studies in which the expression of Nrf2 was found to be upregulated in very young DMD patients early before the appearance of symptoms but not during the progression of disease, when the severity of muscular dystrophy is clearly observed [77].
Although the impact of ROS can be limited by antioxidant systems and enzymes, the excess of ROS production in a dystrophic context might impair the cellular antioxidant defense, leading to irreversible oxidative damage. A complete transcriptome analysis on biopsies from DMD patients revealed the reduced expression of several antioxidant enzymes including CAT-1, SOD1/2, and thioredoxin, indicating a link between an impaired antioxidant response and DMD progression [78]. Our recent study supports this evidence. We recently reported the existence of an antioxidant compensatory mechanism in muscles of young mdx mice and human patients before the onset of DMD symptoms (0–2 years). In particular, we observed a significant upregulation of several Nrf2-dependent genes and enzymes to counteract the strong induction of NOX2 (NADPH oxidase 2) expression, the major source of ROS production in dystrophic muscles [77,79].
In contrast, the progressive increase of NOX2 expression in symptomatic DMD patients (2–9 years of age) failed to activate the Nrf2-mediated antioxidant response [77]. These data strongly indicate that the imbalance between oxidant and antioxidant systems is involved in DMD progression [78,80,81,82].
In skeletal muscle, ROS production is strictly dependent on mitochondrial calcium overload and NOX2 activation [83]. Calcium is known to mediate several regulatory mechanisms in skeletal muscle; however, dystrophic myofibers showed deregulated Ca2+ entry leading to the alteration of muscle homeostasis. Potential sources of Ca2+ inside dystrophin-deficient muscle fibers are membrane tears and the activation of stretch activated ion channels (SACs) [84,85,86,87]. Indeed, the SACs blockade induced by using streptomycin has been shown to reduce intracellular calcium levels in mdx mice [86,88]. Moreover, it has been reported that membrane stressors, such as isometric and eccentric contraction, induce increased ROS production in mdx mice, leading to Ca2+ signaling dysfunction through the oxidation of Ca2+ channels [89,90]. Elevated intracellular concentration of Ca2+ can alter mitochondria permeability and function, thereby inducing degenerative pathways and ROS production [86,91,92,93,94]. Reactive oxygen molecules, in turn, are able to react with fatty acids inducing lipid peroxidation, resulting in membrane insult [95].
The other critical source of ROS in dystrophic muscles is the activation of NOX2, which can be directly linked to the DMD primary defect since it derives from a mechanotransduction-dependent pathway. Indeed, recent studies demonstrated that dystrophin is a microtubule-associated protein (MAP) [78,96] that is able to link the actin and intermediate filaments of cytoskeleton to the transmembrane dystroglycan complex [96,97]. Thus, the lack of dystrophin can influence microtubule (MT) stability and organization; mdx mice exhibited MT disorganization in skeletal muscle, showing an increased expression of α and β tubulin subunits [96]. Of note, Khairallah and colleagues indicated that the MT-network is responsible for stretch-dependent NOX2 activation, a process called X-ROS, also confirming NOX2 complex as the source of ROS production in adult mdx muscles [78]. It has been demonstrated that the X-ROS signaling is altered in adult mdx hindlimb muscles but not in young dystrophic muscles, suggesting a relation between X-ROS alteration and DMD progression. However, in diaphragm muscle, which is the most severely affected muscle in mdx, an altered X-ROS signaling early before the onset of the necrotic phase of pathology (2 weeks of age) has been observed [79]. In particular, we recently reported that pre-necrotic mdx diaphragms showed an enhanced expression of both MT network components (α-, β-, and detyrosinated-tubulin) and gp91phox, the catalytic subunit of NOX2 complex, with an increased production of ROS compared to age-matched wild type mice [79]. The importance of the MT-network deregulation in dystrophin-deficient mice was also revealed in young mdx and wild type muscles in which the taxol-induced enhancement of MT density resulted in altered X-ROS signaling [78].
Another important player in the production of ROS in dystrophin-deficient muscles is the NADPH (nicotinamide adenine dinucleotide phosphate) content. Under physiologic conditions, the cytosolic enzyme glucose-6-phosphate dehydrogenase (G6PD) is able to maintain elevated levels of NADPH, an electron carrier molecule critical for the activity of the glutathione system. However, in dystrophic muscles most of the NADPH produced is used by the NOX complex to convert molecular oxygen to the reactive radical O2−. Of note, G6PD mRNA was found deregulated in mdx muscles and identified as a target of miR-1 family [45]. In particular, Cacchiarelli and coworkers demonstrated that elevated levels of G6PD mRNA in mdx mice correlate with the reduced expression of miR-1 due to the deregulated dystrophin/nNOS/HDAC2 pathway [45].
Altogether, these findings support the hypothesis that ROS production is a direct mechanism downstream of the primary defect of DMD and are consistent with a model in which the absence of dystrophin protein leads to a dysregulated nNOS pathway and MT network (Figure 1). The increased density of microtubules, explained by a possible compensatory upregulation of tubulins, might induce NOX2 assembly/activation, probably through the microtubule-associated NOX2 subunit Rac1 (Ras-related C3 botulinum toxin substrate 1), leading to a consequent production of ROS. Moreover, the enhanced NADPH production by G6PD, due to the reduced expression of miR-1, can continuously provide substrate to the NOX2 activity.
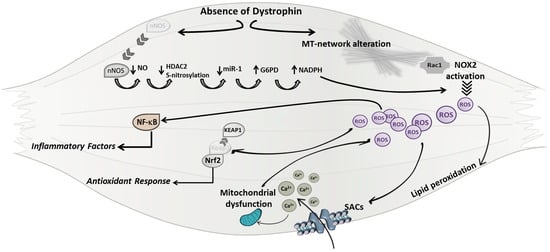
Figure 1.
A schematic model of mechanisms underlining Duchenne muscular dystrophy (DMD) genetic defect leading to oxidative stress. In dystrophic muscles the lack of dystrophin protein can directly induce reactive oxygen species (ROS) production leading to the alteration of sarcolemma integrity, mitochondrial calcium (Ca2+) overload, and to the activation of the NF-κB (Nuclear factor-kappa B) pathway. MT (microtubule); Rac1 (Ras-related C3 botulinum toxin substrate 1); NOX2 (NADPH oxidase 2); SACs (stretch activated ion channels); Nrf2 (NF-E2-related factor 2); KEAP1 (Kelch like-ECH-associated protein 1); nNOS (nitric oxide synthase); NO (nitric oxide); HDAC2 (Histone deacetylase 2); miR (micro RNA); Glucose-6-phosphate dehydrogenase (G6PD); NADPH (nicotinamide adenine dinucleotide phosphate).
Besides a direct insult to membrane lipids, elevated levels of ROS can in turn induce the over-activation of Ca2+ channels, such as SACs (stretch activated ion channels), leading to a deregulated and excessive calcium influx inside skeletal muscle fibers and thus to the activation of degenerative pathways.
4. Crosstalk between Oxidative Stress and Inflammatory Response
In light of recent reports indicating a pivotal role for elevated levels of ROS in the induction of degenerative pathways in dystrophic muscle [67], it is possible that oxidative stress is the first pathogenic mover of muscle damage, stimulating the activation of the inflammatory response that can act as an amplifier mechanism, enhancing muscle degeneration. It has been frequently indicated that the important inflammatory response established in dystrophic muscle is a direct consequence of myonecrosis and strongly contributes to muscle damage (Figure 2). In addition, elevated levels of ROS production have been related to the sprawling invasion of inflammatory cells into dystrophin-deficient muscle tissue (Figure 2). Indeed, phagocytic inflammatory cells, such as macrophages and neutrophils, are known to be an important source of ROS through the expression of NADPH oxidase (NOX2).
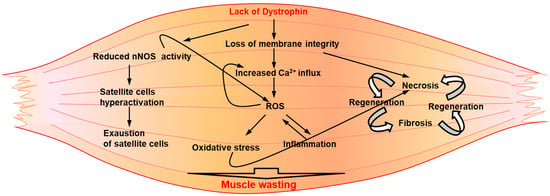
Figure 2.
A schematic representation of secondary pathogenic mechanisms underlining DMD pathology. Downstream of the genetic defect, a pathologic interplay between several degenerative circuitries might contribute to exacerbate the disease.
A crosstalk between ROS and inflammation has been well demonstrated, based also on the evidence that inflammatory signaling, such as that mediated by NF-κB and TNFα activity, is responsive to oxidative stress [66,98].
NF-κB (Nuclear factor-kappa B) is a ubiquitous transcription factor implicated in the promotion of muscle wasting in dystrophic context. In fact, NF-κB activation is induced by several inflammatory mediators, such as IL-1β, TNFα, and metalloproteinases, all molecules found elevated in DMD [57,98,99,100,101,102]. Active NF-κB is able to translocate to the nucleus, inducing the expression of genes encoding for cytokines and chemokines, cell adhesion molecules, and matrix metalloproteinases [103,104,105,106,107].
It has also been described that NF-κB signaling can be induced by ROS through their ability to react with cysteines, mainly at the catalytic sites. In particular, ROS can activate the IKK complex (IκB Kinase), leading to the phosphorylation and degradation of IκB [108]. Reactive oxygen species are also known to inactivate specific phosphatase, resulting in the enhanced activity of kinases and other enzymes involved in the activation of the NF-κB pathway [108].
The interplay between ROS and inflammation has also been supported by experiments in which mdx mice have been treated with anti-oxidant drugs, such as vitamin E analogue or N-acetylcysteine (NAC), demonstrating a reduced activation of NF-κB and an amelioration of dystrophic symptoms [105,106,109].
Moreover, we demonstrated that the over-expression in mdx mice of a pro-inflammatory cytokine critical for the establishment of a chronic inflammation, namely IL-6, was sufficient to exacerbate the dystrophic phenotype [58], by enhancing the expression of NOX2, NF-κB, and other inflammatory mediators during the pre-necrotic stage of disease. These findings supported the hypothesis that NOX2-related ROS production might be a primary pathogenic mechanism, directly linked to the lack of dystrophin, whereas inflammation could be a critical amplifier of degenerative pathways, also fostering oxidative stress.
Indeed, during the progression of pathology, the increased plasma levels of IL-6 in mdx mice induced a progressive increase of several components of X-ROS signaling, including NOX2 and tubulins expression, amplifying muscle degeneration and thus reflecting the exacerbated dystrophic phenotype.
Moreover, the enhanced ROS production induced by increased levels of the pro-inflammatory cytokine IL-6 in mdx mice was also related to an altered redox status of dystrophic muscle due to the impinged Nrf2-mediated anti-oxidant response, closely approximating DMD progression in human patients [77,79].
5. Therapeutic Approaches Direct against Pathogenic Mechanisms Downstream in the Absence of Dystrophin Protein
Despite preclinical and clinical studies that have reported important advances, the development of an efficacious treatment is still lacking. However, several therapeutic approaches have been attempted and current intervention strategies targeting the primary defect, such as gene and cell therapy, have been shown to be promising in animal models, although have not been sufficiently effective in human patients. Probably, degenerative circuitries activated by secondary mechanisms of pathology (Figure 2) might interfere with and limit the efficacy of primary DMD approaches. In light of recent evidence suggesting a role for muscle environment in the regulation of stem cell survival and activity, the modulation of dystrophic muscle milieu could represent an important adjuvant supporting dystrophin-expression therapy. Treatments aimed to ameliorate secondary mechanisms associated with dystropathology are principally directed to reduce inflammation and oxidative stress, these being predominant mechanisms in dystrophic muscle milieu. To date, the standard of care for DMD utilizes anti-inflammatory glucocorticoids including prednisone and deflazacort, which have been recently approved [3,110]. With a general effect on inflammatory response, steroids have been proven to prolong the ambulatory ability of DMD patients with a modest improvement of muscle strength and cardiopulmonary function [3]. Although short-term clinical trials reported beneficial effects of corticosteroids, a chronic administration revealed a heterogeneous tolerance and response to treatment together with important side effects [111]. The aspecific mechanism of action of glucocorticoids could be a drawback of treatment, making unclear and controversial which molecular pathways provide the efficacy in DMD and which pathways are responsible for detrimental effects [111], hence the importance of identifying more specific drugs counteracting inflammation. Among these, the blockade of IL-6 activity in mdx mice, using a neutralizing antibody against the IL-6 receptor (moAb-IL6r), resulted in an enhanced robustness to dystrophic muscle, impeded the activation of a chronic inflammatory response, significantly reduced necrosis, prevented excessive ROS production, and activated the circuitry of muscle differentiation and maturation [28,79].
Moreover, antioxidant drugs can be an important therapeutic option to counteract the secondary signs of dystrophinopathy [68]. Clinical studies using the synthetic derivative of coenzyme Q10, namely idebenone, revealed the efficacy of antioxidant drugs for DMD treatment. Idebenone is a short-chain quinone stimulating mitochondrial electron reflux that has been reported to be cardioprotective in mdx mice and, most importantly, to reduce loss of respiratory function in DMD patients in a recent phase III clinical trial [112,113]. The safety of idebenone in human patients has been confirmed, however, an ongoing phase II extension trial is currently testing its safety and efficacy for use in long-term treatment. Unlike idebenone, which is lipophilic and thus quickly adsorbed, N-acetylcysteine (NAC) is a hydrophilic ROS scavenger that promotes the direct detoxification of free radicals; NAC is also currently being investigated for the anti-inflammatory effect that it showed in preclinical studies [114].
An interplay between oxidative stress and inflammatory response in DMD pathogenesis is further supported by pre-clinical studies on mdx mice, in which the treatment with anti-oxidant drugs have been shown to alleviate relevant DMD-related endpoints, including inflammation. In particular, sulforaphane (SFN), which is a Nrf2 activator, and the sulfur-containing amino acid taurine, induced an amelioration of mdx muscle phenotype modulating both redox markers and critical pro-inflammatory mediators, such as NF-κB [76,115].
Thus, a comprehensive understanding of pathogenic mechanisms interplaying in dystrophic milieu and contributing to muscle degeneration will provide the rationale to design a more appropriate and efficacious approach to treating DMD patients.
Acknowledgments
We apologize to colleagues whose studies have been inadvertently omitted. The work in the authors’ laboratory has been supported by Fondazione Roma, Telethon, Agenzia Spaziale Italiana ASI, and progetti di Ateneo.
Author Contributions
Antonio Musarò conceptualized the study; Antonio Musarò and Laura Forcina wrote the original draft; Laura Pelosi and Carmen Miano wrote/reviewed and edited the text and figures.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Guiraud, S.; Chen, H.; Burns, D.T.; Davies, K.E. Advances in genetic therapeutic strategies for Duchenne muscular dystrophy. Exp. Physiol. 2015, 100, 1458–1467. [Google Scholar] [CrossRef] [PubMed]
- Guiraud, S.; Edwards, B.; Squire, S.E.; Babbs, A.; Shah, N.; Berg, A.; Chen, H.; Davies, K.E. Identification of serum protein biomarkers for utrophin based DMD therapy. Sci. Rep. 2017, 7, 43697. [Google Scholar] [CrossRef] [PubMed]
- Guiraud, S.; Davies, K.E. Pharmacological advances for treatment in Duchenne muscular dystrophy. Curr. Opin. Pharmacol. 2017, 34, 36–48. [Google Scholar] [CrossRef] [PubMed]
- Monaco, A.P.; Neve, R.L.; Colletti-Feener, C.; Bertelson, C.J.; Kurnit, D.M.; Kunkel, L.M. Isolation of candidate cDNAs for portions of the Duchenne muscular dystrophy gene. Nature 1986, 323, 646–650. [Google Scholar] [CrossRef] [PubMed]
- Consalvi, S.; Saccone, V.; Giordani, L.; Minetti, G.; Mozzetta, C.; Puri, P.L. Histone deacetylase inhibitors in the treatment of muscular dystrophies: Epigenetic drugs for genetic diseases. Mol. Med. 2011, 17, 457–465. [Google Scholar] [CrossRef] [PubMed]
- Glass, D.J. Two tales concerning skeletal muscle. J. Clin. Investig. 2007, 117, 2388–2391. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, K.F.; Kunkel, L.M. Dystrophin and muscular dystrophy: Past, present, and future. Mol. Genet. Metab. 2001, 74, 75–88. [Google Scholar] [CrossRef] [PubMed]
- Grounds, M.D.; Radley, H.G.; Lynch, G.S.; Nagaraju, K.; de Luca, A. Towards developing standard operating procedures for pre-clinical testing in the mdx mouse model of Duchenne muscular dystrophy. Neurobiol. Dis. 2008, 31, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Salam, E.; Abdel-Meguid, I.; Korraa, S.S. Markers of degeneration and regeneration in Duchenne muscular dystrophy. Acta Myol. 2009, 28, 94–100. [Google Scholar] [PubMed]
- Carnwath, J.W.; Shotton, D.M. Muscular dystrophy in the mdx mouse: Histopathology of the soleus and extensor digitorum longus muscles. J. Neurol. Sci. 1987, 80, 39–54. [Google Scholar] [CrossRef]
- Deconinck, N.; Dan, B. Pathophysiology of duchenne muscular dystrophy: Current hypotheses. Pediatr. Neurol. 2007, 36, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Evans, N.P.; Misyak, S.A.; Robertson, J.L.; Bassaganya-Riera, J.; Grange, R.W. Dysregulated intracellular signaling and inflammatory gene expression during initial disease onset in Duchenne muscular dystrophy. Am. J. Phys. Med. Rehabil. 2009, 88, 502–522. [Google Scholar] [CrossRef] [PubMed]
- Partridge, T.A. The mdx mouse model as a surrogate for Duchenne muscular dystrophy. FEBS J. 2013, 280, 4177–4186. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Long, C.; Yue, Y.; Duan, D. Sub-physiological sarcoglycan expression contributes to compensatory muscle protection in mdx mice. Hum. Mol. Genet. 2009, 18, 1209–1220. [Google Scholar] [CrossRef] [PubMed]
- Pescatori, M.; Broccolini, A.; Minetti, C.; Bertini, E.; Bruno, C.; D’amico, A.; Bernardini, C.; Mirabella, M.; Silvestri, G.; Giglio, V.; et al. Gene expression profiling in the early phases of DMD: A constant molecular signature characterizes DMD muscle from early postnatal life throughout disease progression. FASEB J. 2007, 21, 1210–1226. [Google Scholar] [CrossRef] [PubMed]
- Porter, J.D.; Khanna, S.; Kaminski, H.J.; Rao, J.S.; Merriam, A.P.; Richmonds, C.R.; Leahy, P.; Li, J.; Guo, W.; Andrade, F.H. A chronic inflammatory response dominates the skeletal muscle molecular signature in dystrophin-deficient mdx mice. Hum. Mol. Genet. 2002, 11, 263–272. [Google Scholar] [CrossRef] [PubMed]
- Connolly, A.M.; Keeling, R.M.; Mehta, S.; Pestronk, A.; Sanes, J.R. Three mouse models of muscular dystrophy: The natural history of strength and fatigue in dystrophin-, dystrophin/utrophin-, and laminin α2-deficient mice. Neuromuscul. Disord. 2001, 11, 703–712. [Google Scholar] [CrossRef]
- Deconinck, N.; Tinsley, J.; De Backer, F.; Fisher, R.; Kahn, D.; Phelps, S.; Davies, K.; Gillis, J.M. Expression of truncated utrophin leads to major functional improvements in dystrophin-deficient muscles of mice. Nat. Med. 1997, 3, 1216–1221. [Google Scholar] [CrossRef] [PubMed]
- Grady, R.M.; Teng, H.; Nichol, M.C.; Cunningham, J.C.; Wilkinson, R.S.; Sanes, J.R. Skeletal and cardiac myopathies in mice lacking utrophin and dystrophin: A model for Duchenne muscular dystrophy. Cell 1997, 90, 729–738. [Google Scholar] [CrossRef]
- Megeney, L.A.; Kablar, B.; Garrett, K.; Anderson, J.E.; Rudnicki, M.A. MyoD is required for myogenic stem cell function in adult skeletal muscle. Genes Dev. 1996, 10, 1173–1183. [Google Scholar] [CrossRef] [PubMed]
- Raymackers, J.M.; Debaix, H.; Colson-Van Schoor, M.; De Backer, F.; Tajeddine, N.; Schwaller, B.; Gailly, P.; Gillis, J.M. Consequence of parvalbumin deficiency in the mdx mouse: Histological, biochemical and mechanical phenotype of a new double mutant. Neuromuscul. Disord. 2003, 13, 376–387. [Google Scholar] [CrossRef]
- Guo, C.; Willem, M.; Werner, A.; Raivich, G.; Emerson, M.; Neyses, L.; Mayer, U. Absence of α 7 integrin in dystrophin-deficient mice causes a myopathy similar to Duchenne muscular dystrophy. Hum. Mol. Genet. 2006, 15, 989–998. [Google Scholar] [CrossRef] [PubMed]
- Sacco, A.; Mourkioti, F.; Tran, R.; Choi, J.; Llewellyn, M.; Kraft, P.; Shkreli, M.; Delp, S.; Pomerantz, J.H.; Artandi, S.E.; et al. Short telomeres and stem cell exhaustion model Duchenne muscular dystrophy in mdx/mTR mice. Cell 2010, 143, 1059–1071. [Google Scholar] [CrossRef] [PubMed]
- Hathout, Y.; Marathi, R.L.; Rayavarapu, S.; Zhang, A.; Brown, K.J.; Seol, H.; Gordish-Dressman, H.; Cirak, S.; Bello, L.; Nagaraju, K.; et al. Discovery of serum protein biomarkers in the mdx mouse model and cross-species comparison to Duchenne muscular dystrophy patients. Hum. Mol. Genet. 2014, 23, 6458–6469. [Google Scholar] [CrossRef] [PubMed]
- Bentzinger, C.F.; von Maltzahn, J.; Dumont, N.A.; Stark, D.A.; Wang, Y.X.; Nhan, K.; Frenette, J.; Cornelison, D.D.; Rudnicki, M.A. Wnt7a stimulates myogenic stem cell motility and engraftment resulting in improved muscle strength. J. Cell Biol. 2014, 205, 97–111. [Google Scholar] [CrossRef] [PubMed]
- Consalvi, S.; Mozzetta, C.; Bettica, P.; Germani, M.; Fiorentini, F.; del Bene, F.; Rocchetti, M.; Leoni, F.; Monzani, V.; Mascagni, P.; et al. Preclinical studies in the mdx mouse model of duchenne muscular dystrophy with the histone deacetylase inhibitor givinostat. Mol. Med. 2013, 19, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Denti, M.A.; Rosa, A.; D’Antona, G.; Sthandier, O.; de Angelis, F.G.; Nicoletti, C.; Allocca, M.; Pansarasa, O.; Parente, V.; Musaro, A.; et al. Body-wide gene therapy of Duchenne muscular dystrophy in the mdx mouse model. Proc. Natl. Acad. Sci. USA 2006, 103, 3758–3763. [Google Scholar] [CrossRef] [PubMed]
- Pelosi, L.; Berardinelli, M.G.; De Pasquale, L.; Nicoletti, C.; D’amico, A.; Carvello, F.; Moneta, G.M.; Catizone, A.; Bertini, E.; de Benedetti, F.; et al. Functional and morphological improvement of dystrophic muscle by interleukin 6 receptor blockade. EBioMedicine 2015, 2, 285–293. [Google Scholar] [CrossRef] [PubMed]
- Quattrocelli, M.; Salamone, I.M.; Page, P.G.; Warner, J.L.; Demonbreun, A.R.; McNally, E.M. Intermittent glucocorticoid dosing improves muscle repair and function in mice with limb-girdle muscular dystrophy. Am. J. Pathol. 2017, 187, 2520–2535. [Google Scholar] [CrossRef] [PubMed]
- Sarathy, A.; Wuebbles, R.D.; Fontelonga, T.M.; Tarchione, A.R.; Mathews Griner, L.A.; Heredia, D.J.; Nunes, A.M.; Duan, S.; Brewer, P.D.; van Ry, T.; et al. SU9516 Increases α7β1 Integrin and ameliorates disease progression in the mdx mouse model of duchenne muscular dystrophy. Mol. Ther. 2017, 25, 1395–1407. [Google Scholar] [CrossRef] [PubMed]
- Tedesco, F.S.; Hoshiya, H.; D’Antona, G.; Gerli, M.F.; Messina, G.; Antonini, S.; Tonlorenzi, R.; Benedetti, S.; Berghella, L.; Torrente, Y.; et al. Stem cell-mediated transfer of a human artificial chromosome ameliorates muscular dystrophy. Sci. Transl. Med. 2011, 3, 96ra78. [Google Scholar] [CrossRef] [PubMed]
- Vojnits, K.; Pan, H.; Dai, X.; Sun, H.; Tong, Q.; Darabi, R.; Huard, J.; Li, Y. Functional neuronal differentiation of injury-induced muscle-derived stem cell-like cells with therapeutic implications. Sci. Rep. 2017, 7, 1177. [Google Scholar] [CrossRef] [PubMed]
- Blau, H.M.; Webster, C.; Chiu, C.P.; Guttman, S.; Chandler, F. Differentiation properties of pure populations of human dystrophic muscle cells. Exp. Cell Res. 1983, 144, 495–503. [Google Scholar] [CrossRef]
- Dadgar, S.; Wang, Z.; Johnston, H.; Kesari, A.; Nagaraju, K.; Chen, Y.W.; Hill, D.A.; Partridge, T.A.; Giri, M.; Freishtat, R.J.; et al. Asynchronous remodeling is a driver of failed regeneration in Duchenne muscular dystrophy. J. Cell Biol. 2014, 207, 139–158. [Google Scholar] [CrossRef] [PubMed]
- Chang, N.C.; Chevalier, F.P.; Rudnicki, M.A. Satellite cells in muscular dystrophy—Lost in polarity. Trends Mol. Med. 2016, 22, 479–496. [Google Scholar] [CrossRef] [PubMed]
- Mauro, A. Satellite cell of skeletal muscle fibers. J. Biophys. Biochem. Cytol. 1961, 9, 493–495. [Google Scholar] [CrossRef] [PubMed]
- Musaro, A. The basis of muscle regeneration. Adv. Biol. 2014, 1–16. [Google Scholar] [CrossRef]
- Almada, A.E.; Wagers, A.J. Molecular circuitry of stem cell fate in skeletal muscle regeneration, ageing and disease. Nat. Rev. Mol. Cell Biol. 2016, 17, 267–279. [Google Scholar] [CrossRef] [PubMed]
- Scicchitano, B.M.; Sica, G.; Musaro, A. Stem cells and tissue niche: Two faces of the same coin of muscle regeneration. Eur. J. Transl. Myol. 2016, 26, 6125. [Google Scholar] [CrossRef] [PubMed]
- Wada, E.; Tanihata, J.; Iwamura, A.; Takeda, S.; Hayashi, Y.K.; Matsuda, R. Treatment with the anti-IL-6 receptor antibody attenuates muscular dystrophy via promoting skeletal muscle regeneration in dystrophin-/utrophin-deficient mice. Skelet. Muscle 2017, 7, 23. [Google Scholar] [CrossRef] [PubMed]
- Dumont, N.A.; Wang, Y.X.; von Maltzahn, J.; Pasut, A.; Bentzinger, C.F.; Brun, C.E.; Rudnicki, M.A. Dystrophin expression in muscle stem cells regulates their polarity and asymmetric division. Nat. Med. 2015, 21, 1455–1463. [Google Scholar] [CrossRef] [PubMed]
- Kottlors, M.; Kirschner, J. Elevated satellite cell number in Duchenne muscular dystrophy. Cell Tissue Res. 2010, 340, 541–548. [Google Scholar] [CrossRef] [PubMed]
- Seale, P.; Sabourin, L.A.; Girgis-Gabardo, A.; Mansouri, A.; Gruss, P.; Rudnicki, M.A. Pax7 is required for the specification of myogenic satellite cells. Cell 2000, 102, 777–786. [Google Scholar] [CrossRef]
- Brenman, J.E.; Chao, D.S.; Xia, H.; Aldape, K.; Bredt, D.S. Nitric oxide synthase complexed with dystrophin and absent from skeletal muscle sarcolemma in Duchenne muscular dystrophy. Cell 1995, 82, 743–752. [Google Scholar] [CrossRef]
- Cacchiarelli, D.; Martone, J.; Girardi, E.; Cesana, M.; Incitti, T.; Morlando, M.; Nicoletti, C.; Santini, T.; Sthandier, O.; Barberi, L.; et al. MicroRNAs involved in molecular circuitries relevant for the Duchenne muscular dystrophy pathogenesis are controlled by the dystrophin/nNOS pathway. Cell Metab. 2010, 12, 341–351. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.W.; Nagaraju, K.; Bakay, M.; McIntyre, O.; Rawat, R.; Shi, R.; Hoffman, E.P. Early onset of inflammation and later involvement of TGF β in Duchenne muscular dystrophy. Neurology 2005, 65, 826–834. [Google Scholar] [CrossRef] [PubMed]
- Villalta, S.A.; Nguyen, H.X.; Deng, B.; Gotoh, T.; Tidball, J.G. Shifts in macrophage phenotypes and macrophage competition for arginine metabolism affect the severity of muscle pathology in muscular dystrophy. Hum. Mol. Genet. 2009, 18, 482–496. [Google Scholar] [CrossRef] [PubMed]
- Grounds, M.D.; Radley, H.G.; Gebski, B.L.; Bogoyevitch, M.A.; Shavlakadze, T. Implications of cross-talk between tumour necrosis factor and insulin-like growth factor-1 signalling in skeletal muscle. Clin. Exp. Pharmacol. Physiol. 2008, 35, 846–851. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, M.; Echigoya, Y.; Fukada, S.I.; Yokota, T. Current Translational research and murine models for Duchenne muscular dystrophy. J. Neuromuscul. Dis. 2016, 3, 29–48. [Google Scholar] [CrossRef] [PubMed]
- Douglas, M.R.; Morrison, K.E.; Salmon, M.; Buckley, C.D. Why does inflammation persist: A dominant role for the stromal microenvironment? Expert Rev. Mol. Med. 2002, 4, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Scheller, J.; Chalaris, A.; Schmidt-Arras, D.; Rose-John, S. The pro- and anti-inflammatory properties of the cytokine interleukin-6. Biochim. Biophys. Acta 2011, 1813, 878–888. [Google Scholar] [CrossRef] [PubMed]
- Kurek, J.B.; Nouri, S.; Kannourakis, G.; Murphy, M.; Austin, L. Leukemia inhibitory factor and interleukin-6 are produced by diseased and regenerating skeletal muscle. Muscle Nerve 1996, 19, 1291–1309. [Google Scholar] [CrossRef]
- Serrano, A.L.; Baeza-Raja, B.; Perdiguero, E.; Jardi, M.; Munoz-Canoves, P. Interleukin-6 is an essential regulator of satellite cell-mediated skeletal muscle hypertrophy. Cell Metab. 2008, 7, 33–44. [Google Scholar] [CrossRef] [PubMed]
- Goodman, M.N. Interleukin-6 induces skeletal muscle protein breakdown in rats. Proc. Soc. Exp. Biol. Med. 1994, 205, 182–185. [Google Scholar] [CrossRef] [PubMed]
- Haddad, F.; Zaldivar, F.; Cooper, D.M.; Adams, G.R. IL-6-induced skeletal muscle atrophy. J. Appl. Physiol. 2005, 98, 911–917. [Google Scholar] [CrossRef] [PubMed]
- Tsujinaka, T.; Ebisui, C.; Fujita, J.; Kishibuchi, M.; Morimoto, T.; Ogawa, A.; Katsume, A.; Ohsugi, Y.; Kominami, E.; Monden, M. Muscle undergoes atrophy in association with increase of lysosomal cathepsin activity in interleukin-6 transgenic mouse. Biochem. Biophys. Res. Commun. 1995, 207, 168–174. [Google Scholar] [CrossRef] [PubMed]
- Messina, S.; Vita, G.L.; Aguennouz, M.; Sframeli, M.; Romeo, S.; Rodolico, C.; Vita, G. Activation of NF-kappaB pathway in Duchenne muscular dystrophy: Relation to age. Acta Myol. 2011, 30, 16–23. [Google Scholar] [PubMed]
- Pelosi, L.; Berardinelli, M.G.; Forcina, L.; Spelta, E.; Rizzuto, E.; Nicoletti, C.; Camilli, C.; Testa, E.; Catizone, A.; De Benedetti, F.; et al. Increased levels of interleukin-6 exacerbate the dystrophic phenotype in mdx mice. Hum. Mol. Genet. 2015, 24, 6041–6053. [Google Scholar] [CrossRef] [PubMed]
- Kostek, M.C.; Nagaraju, K.; Pistilli, E.; Sali, A.; Lai, S.H.; Gordon, B.; Chen, Y.W. IL-6 signaling blockade increases inflammation but does not affect muscle function in the mdx mouse. BMC Musculoskelet. Disord. 2012, 13, 106. [Google Scholar] [CrossRef] [PubMed]
- Price, F.D.; von Maltzahn, J.; Bentzinger, C.F.; Dumont, N.A.; Yin, H.; Chang, N.C.; Wilson, D.H.; Frenette, J.; Rudnicki, M.A. Inhibition of JAK-STAT signaling stimulates adult satellite cell function. Nat. Med. 2014, 20, 1174–1181. [Google Scholar] [CrossRef] [PubMed]
- Tierney, M.T.; Aydogdu, T.; Sala, D.; Malecova, B.; Gatto, S.; Puri, P.L.; Latella, L.; Sacco, A. STAT3 signaling controls satellite cell expansion and skeletal muscle repair. Nat. Med. 2014, 20, 1182–1186. [Google Scholar] [CrossRef] [PubMed]
- Scott, H.R.; McMillan, D.C.; Crilly, A.; McArdle, C.S.; Milroy, R. The relationship between weight loss and interleukin 6 in non-small-cell lung cancer. Br. J. Cancer 1996, 73, 1560–1562. [Google Scholar] [CrossRef] [PubMed]
- Spencer, M.J.; Marino, M.W.; Winckler, W.M. Altered pathological progression of diaphragm and quadriceps muscle in TNF-deficient, dystrophin-deficient mice. Neuromuscul. Disord. 2000, 10, 612–619. [Google Scholar] [CrossRef]
- Arthur, P.G.; Grounds, M.D.; Shavlakadze, T. Oxidative stress as a therapeutic target during muscle wasting: Considering the complex interactions. Curr. Opin. Clin. Nutr. Metab. Care 2008, 11, 408–416. [Google Scholar] [CrossRef] [PubMed]
- Moylan, J.S.; Reid, M.B. Oxidative stress, chronic disease, and muscle wasting. Muscle Nerve 2007, 35, 411–429. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Kwak, H.B.; Thompson, L.V.; Lawler, J.M. Contribution of oxidative stress to pathology in diaphragm and limb muscles with Duchenne muscular dystrophy. J. Muscle Res. Cell Motil. 2013, 34, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Rando, T.A. Oxidative stress and the pathogenesis of muscular dystrophies. Am. J. Phys. Med. Rehabil. 2002, 81, S175–S186. [Google Scholar] [CrossRef] [PubMed]
- Malik, V.; Rodino-Klapac, L.R.; Mendell, J.R. Emerging drugs for Duchenne muscular dystrophy. Expert Opin. Emerg. Drugs 2012, 17, 261–277. [Google Scholar] [CrossRef] [PubMed]
- Motohashi, H.; Yamamoto, M. Nrf2-Keap1 defines a physiologically important stress response mechanism. Trends Mol. Med. 2004, 10, 549–557. [Google Scholar] [CrossRef] [PubMed]
- Musaro, A.; Fulle, S.; Fano, G. Oxidative stress and muscle homeostasis. Curr. Opin. Clin. Nutr. Metab. Care 2010, 13, 236–242. [Google Scholar] [CrossRef] [PubMed]
- Scicchitano, B.M.; Pelosi, L.; Sica, G.; Musaro, A. The physiopathologic role of oxidative stress in skeletal muscle. Mech. Ageing Dev. 2017. [Google Scholar] [CrossRef] [PubMed]
- Binder, H.J.; Herting, D.C.; Hurst, V.; Finch, S.C.; Spiro, H.M. Tocopherol deficiency in man. N. Engl. J. Med. 1965, 273, 1289–1297. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Yamamoto, M. Molecular basis of the Keap1-Nrf2 system. Free Radic. Biol. Med. 2015, 88, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Yang, C.; Xue, R.; Li, S.; Zhang, T.; Pan, L.; Ma, X.; Wang, L.; Li, D. Sulforaphane alleviates muscular dystrophy in mdx mice by activation of Nrf2. J. Appl. Physiol. 2015, 118, 224–237. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Li, S.; Li, D. Sulforaphane mitigates muscle fibrosis in mdx mice via Nrf2-mediated inhibition of TGF-β/Smad signaling. J. Appl. Physiol. 2016, 120, 377–390. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.C.; Li, S.J.; Yang, C.L.; Xue, R.L.; Xi, Y.Y.; Wang, L.; Zhao, Q.L.; Li, D.J. Sulforaphane Attenuates Muscle Inflammation in Dystrophin-deficient mdx Mice via NF-E2-related Factor 2 (Nrf2)-mediated Inhibition of NF-κB Signaling Pathway. J. Biol. Chem. 2015, 290, 17784–17795. [Google Scholar] [CrossRef] [PubMed]
- Petrillo, S.; Pelosi, L.; Piemonte, F.; Travaglini, L.; Forcina, L.; Catteruccia, M.; Petrini, S.; Verardo, M.; D’amico, A.; Musaro, A.; et al. Oxidative stress in Duchenne muscular dystrophy: Focus on the NRF2 redox pathway. Hum. Mol. Genet. 2017, 26, 2781–2790. [Google Scholar] [CrossRef] [PubMed]
- Khairallah, R.J.; Shi, G.; Sbrana, F.; Prosser, B.L.; Borroto, C.; Mazaitis, M.J.; Hoffman, E.P.; Mahurkar, A.; Sachs, F.; Sun, Y.; et al. Microtubules underlie dysfunction in duchenne muscular dystrophy. Sci. Signal. 2012, 5, ra56. [Google Scholar] [CrossRef] [PubMed]
- Pelosi, L.; Forcina, L.; Nicoletti, C.; Scicchitano, B.M.; Musaro, A. Increased circulating levels of Interleukin-6 Induce Perturbation in redox-regulated signaling cascades in muscle of dystrophic mice. Oxid. Med. Cell Longev. 2017. [Google Scholar] [CrossRef] [PubMed]
- Canton, M.; Menazza, S.; Di Lisa, F. Oxidative stress in muscular dystrophy: From generic evidence to specific sources and targets. J. Muscle Res. Cell Motil. 2014, 35, 23–36. [Google Scholar] [CrossRef] [PubMed]
- Prosser, B.L.; Ward, C.W.; Lederer, W.J. X-ROS signaling: Rapid mechano-chemo transduction in heart. Science 2011, 333, 1440–1445. [Google Scholar] [CrossRef] [PubMed]
- Prosser, B.L.; Khairallah, R.J.; Ziman, A.P.; Ward, C.W.; Lederer, W.J. X-ROS signaling in the heart and skeletal muscle: Stretch-dependent local ROS regulates [Ca2+]i. J. Mol. Cell Cardiol. 2013, 58, 172–181. [Google Scholar] [CrossRef] [PubMed]
- Hidalgo, C.; Sanchez, G.; Barrientos, G.; Aracena-Parks, P. A transverse tubule NADPH oxidase activity stimulates calcium release from isolated triads via ryanodine receptor type 1 S-glutathionylation. J. Biol. Chem. 2006, 281, 26473–26482. [Google Scholar] [CrossRef] [PubMed]
- Fong, P.Y.; Turner, P.R.; Denetclaw, W.F.; Steinhardt, R.A. Increased activity of calcium leak channels in myotubes of Duchenne human and mdx mouse origin. Science 1990, 250, 673–676. [Google Scholar] [CrossRef] [PubMed]
- Franco, A., Jr.; Lansman, J.B. Calcium entry through stretch-inactivated ion channels in mdx myotubes. Nature 1990, 344, 670–673. [Google Scholar] [CrossRef] [PubMed]
- Gumerson, J.D.; Michele, D.E. The dystrophin-glycoprotein complex in the prevention of muscle damage. J. Biomed. Biotechnol. 2011, 2011, 210797. [Google Scholar] [CrossRef] [PubMed]
- Turner, P.R.; Westwood, T.; Regen, C.M.; Steinhardt, R.A. Increased protein degradation results from elevated free calcium levels found in muscle from mdx mice. Nature 1988, 335, 735–738. [Google Scholar] [CrossRef] [PubMed]
- Yeung, E.W.; Whitehead, N.P.; Suchyna, T.M.; Gottlieb, P.A.; Sachs, F.; Allen, D.G. Effects of stretch-activated channel blockers on [Ca2+]i and muscle damage in the mdx mouse. J. Physiol. 2005, 562, 367–380. [Google Scholar] [CrossRef] [PubMed]
- Isaeva, E.V.; Shirokova, N. Metabolic regulation of Ca2+ release in permeabilized mammalian skeletal muscle fibres. J. Physiol. 2003, 547, 453–462. [Google Scholar] [CrossRef] [PubMed]
- Isaeva, E.V.; Shkryl, V.M.; Shirokova, N. Mitochondrial redox state and Ca2+ sparks in permeabilized mammalian skeletal muscle. J. Physiol. 2005, 565, 855–872. [Google Scholar] [CrossRef] [PubMed]
- Millay, D.P.; Sargent, M.A.; Osinska, H.; Baines, C.P.; Barton, E.R.; Vuagniaux, G.; Sweeney, H.L.; Robbins, J.; Molkentin, J.D. Genetic and pharmacologic inhibition of mitochondrial-dependent necrosis attenuates muscular dystrophy. Nat. Med. 2008, 14, 442–447. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, T.; Shimizu, S.; Watanabe, T.; Yamaguchi, O.; Otsu, K.; Yamagata, H.; Inohara, H.; Kubo, T.; Tsujimoto, Y. Cyclophilin D-dependent mitochondrial permeability transition regulates some necrotic but not apoptotic cell death. Nature 2005, 434, 652–658. [Google Scholar] [CrossRef] [PubMed]
- Orrenius, S.; Zhivotovsky, B.; Nicotera, P. Regulation of cell death: The calcium-apoptosis link. Nat. Rev. Mol. Cell Biol. 2003, 4, 552–565. [Google Scholar] [CrossRef] [PubMed]
- Shkryl, V.M.; Martins, A.S.; Ullrich, N.D.; Nowycky, M.C.; Niggli, E.; Shirokova, N. Reciprocal amplification of ROS and Ca2+ signals in stressed mdx dystrophic skeletal muscle fibers. Pflugers Arch. 2009, 458, 915–928. [Google Scholar] [CrossRef] [PubMed]
- Kalogeris, T.; Bao, Y.; Korthuis, R.J. Mitochondrial reactive oxygen species: A double edged sword in ischemia/reperfusion vs. preconditioning. Redox Biol. 2014, 2, 702–714. [Google Scholar] [CrossRef] [PubMed]
- Prins, K.W.; Humston, J.L.; Mehta, A.; Tate, V.; Ralston, E.; Ervasti, J.M. Dystrophin is a microtubule-associated protein. J. Cell Biol. 2009, 186, 363–369. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, A.; Yoshida, M.; Yamamoto, H.; Ozawa, E. Glycoprotein-binding site of dystrophin is confined to the cysteine-rich domain and the first half of the carboxy-terminal domain. FEBS Lett. 1992, 308, 154–160. [Google Scholar] [CrossRef]
- Tidball, J.G.; Wehling-Henricks, M. The role of free radicals in the pathophysiology of muscular dystrophy. J. Appl. Physiol. 2007, 102, 1677–1686. [Google Scholar] [CrossRef] [PubMed]
- Becker, C.; Bray-French, K.; Drewe, J. Pharmacokinetic evaluation of idebenone. Expert Opin. Drug Metab. Toxicol. 2010, 6, 1437–1444. [Google Scholar] [CrossRef] [PubMed]
- Bodmer, M.; Vankan, P.; Dreier, M.; Kutz, K.W.; Drewe, J. Pharmacokinetics and metabolism of idebenone in healthy male subjects. Eur. J. Clin. Pharmacol. 2009, 65, 493–501. [Google Scholar] [CrossRef] [PubMed]
- Buyse, G.M.; Van der, M.G.; Erb, M.; D’hooge, J.; Herijgers, P.; Verbeken, E.; Jara, A.; Van Den, B.A.; Mertens, L.; Courdier-Fruh, I.; et al. Long-term blinded placebo-controlled study of SNT-MC17/idebenone in the dystrophin deficient mdx mouse: Cardiac protection and improved exercise performance. Eur. Heart J. 2009, 30, 116–124. [Google Scholar] [CrossRef] [PubMed]
- Gillis, J.C.; Benefield, P.; McTavish, D. Idebenone, A review of its pharmacodynamic and pharmacokinetic properties, and therapeutic use in age-related cognitive disorders. Drugs Aging 1994, 5, 133–152. [Google Scholar] [CrossRef] [PubMed]
- Karin, M. How NF-kappaB is activated: The role of the I κB kinase (IKK) complex. Oncogene 1999, 18, 6867–6874. [Google Scholar] [CrossRef] [PubMed]
- Karin, M.; Delhase, M. The I kappa B kinase (IKK) and NF-kappa B: Key elements of proinflammatory signalling. Semin. Immunol. 2000, 12, 85–98. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Boriek, A.M. Mechanical stress activates the nuclear factor-kappaB pathway in skeletal muscle fibers: A possible role in Duchenne muscular dystrophy. FASEB J. 2003, 17, 386–396. [Google Scholar] [CrossRef] [PubMed]
- Messina, S.; Altavilla, D.; Aguennouz, M.; Seminara, P.; Minutoli, L.; Monici, M.C.; Bitto, A.; Mazzeo, A.; Marini, H.; Squadrito, F.; et al. Lipid peroxidation inhibition blunts nuclear factor-kappaB activation, reduces skeletal muscle degeneration, and enhances muscle function in mdx mice. Am. J. Pathol. 2006, 168, 918–926. [Google Scholar] [CrossRef] [PubMed]
- Senftleben, U.; Karin, M. The IKK/NF-kappa B pathway. Crit. Care Med. 2002, 30, S18–S26. [Google Scholar] [CrossRef] [PubMed]
- Morgan, M.J.; Liu, Z.G. Crosstalk of reactive oxygen species and NF-κB signaling. Cell Res. 2011, 21, 103–115. [Google Scholar] [CrossRef] [PubMed]
- Messina, S.; Bitto, A.; Aguennouz, M.; Minutoli, L.; Monici, M.C.; Altavilla, D.; Squadrito, F.; Vita, G. Nuclear factor kappa-B blockade reduces skeletal muscle degeneration and enhances muscle function in Mdx mice. Exp. Neurol. 2006, 198, 234–241. [Google Scholar] [CrossRef] [PubMed]
- Traynor, K. Deflazacort approved for Duchenne muscular dystrophy. Am. J. Health Syst. Pharm. 2017, 74, 368. [Google Scholar] [CrossRef] [PubMed]
- Miyatake, S.; Shimizu-Motohashi, Y.; Takeda, S.; Aoki, Y. Anti-inflammatory drugs for Duchenne muscular dystrophy: Focus on skeletal muscle-releasing factors. Drug Des. Dev. Ther. 2016, 10, 2745–2758. [Google Scholar] [CrossRef]
- Buyse, G.M.; Voit, T.; Schara, U.; Straathof, C.S.; D’Angelo, M.G.; Bernert, G.; Cuisset, J.M.; Finkel, R.S.; Goemans, N.; McDonald, C.M.; et al. Efficacy of idebenone on respiratory function in patients with Duchenne muscular dystrophy not using glucocorticoids (DELOS): A double-blind randomised placebo-controlled phase 3 trial. Lancet 2015, 385, 1748–1757. [Google Scholar] [CrossRef]
- Buyse, G.M.; Voit, T.; Schara, U.; Straathof, C.S.; D’Angelo, M.G.; Bernert, G.; Cuisset, J.M.; Finkel, R.S.; Goemans, N.; Rummey, C.; et al. Treatment effect of idebenone on inspiratory function in patients with Duchenne muscular dystrophy. Pediatr. Pulmonol. 2017, 52, 508–515. [Google Scholar] [CrossRef] [PubMed]
- Sadowska, A.M.; Manuel, Y.K.; De Backer, W.A. Antioxidant and anti-inflammatory efficacy of NAC in the treatment of COPD: Discordant in vitro and in vivo dose-effects: A review. Pulm. Pharmacol. Ther. 2007, 20, 9–22. [Google Scholar] [CrossRef] [PubMed]
- Capogrosso, R.F.; Cozzoli, A.; Mantuano, P.; Camerino, G.M.; Massari, A.M.; Sblendorio, V.T.; De Bellis, M.; Tamma, R.; Giustino, A.; Nico, B.; et al. Assessment of resveratrol, apocynin and taurine on mechanical-metabolic uncoupling and oxidative stress in a mouse model of duchenne muscular dystrophy: A comparison with the gold standard, α-methyl prednisolone. Pharmacol. Res. 2016, 106, 101–113. [Google Scholar] [CrossRef] [PubMed]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).