Abstract
Duchenne muscular dystrophy (DMD) is a lethal X-linked pathology due to lack of dystrophin and characterized by progressive muscle degeneration, impaired locomotion and premature death. The chronic presence of inflammatory cells, fibrosis and fat deposition are hallmarks of DMD muscle tissue. Many different therapeutic approaches to DMD have been tested, including cell-based and gene-based approaches, exon skipping, induction of expression of the dystrophin paralogue, utrophin, and, most recently the application of the CASPR/Cas9 genome editing system. However, corticosteroid treatment remains the gold standard therapy, even if corticosteroids have shown multiple undesirable side effects. Sertoli cells (SeC) have long been known for their ability to produce immunomodulatory and trophic factors, and have been used in a plethora of experimental models of disease. Recently, microencapsulated porcine SeC (MC-SeC) injected intraperitoneally in dystrophic mice produced morphological and functional benefits in muscles thanks to their release into the circulation of anti-inflammatory factors and heregulin β1, a known inducer of utrophin expression, thus opening a new avenue in the treatment of DMD. In order to stress the potentiality of the use of MC-SeC in the treatment of DMD, here, we examine the principal therapeutic approaches to DMD, and the properties of SeC (either nude or encapsulated into alginate-based microcapsules) and their preclinical and clinical use. Finally, we discuss the potential and future development of this latter approach.
1. Duchenne Muscular Dystrophy (DMD)
Duchenne muscular dystrophy (DMD) is the most common muscular dystrophy. Muscular dystrophies are a group of inherited muscle diseases characterized by mutations in specific genes and resulting in muscle degeneration, impaired locomotion and premature death [1,2]. DMD is an X-linked recessive pathology caused by mutations in the dystrophin gene (DMD) usually resulting in the complete absence of this protein. Dystrophin is an essential component of the dystrophin-associated protein complex (DAPC) at the sarcolemma, a complex that ensures the structural and functional integrity of the myofibers during contraction representing a mechanical link between the intracellular cytoskeleton and the extracellular matrix. Absence of dystrophin or other components of the DAPC compromises the integrity of the DAPC itself leading to a susceptibility of myofibers to degeneration during contraction and consequent progressive loss of muscle efficiency [1,2]. The muscle damage subsequent to the absence of dystrophin determines massive infiltration of immune cells in muscle tissue, and the chronic activation of signaling pathways implicated in the inflammatory response [3]. Indeed, muscles of DMD patients are characterized by a condition of chronic inflammation and overexpression of inflammatory genes at the onset and during the progression of the pathology [3,4]. Moreover, as a consequence of continuous degeneration/regeneration cycles, during the progression of the disease an exhaustion of the regenerative potential occurs in dystrophic muscles. As a result of high demand for myogenesis and poor compensatory mechanisms, fibrous and fatty connective tissue progressively overtake the functional myofibers culminating in progressive muscle wasting and severely affecting skeletal muscle efficiency which ultimately results in premature death due to cardiac and respiratory failure [2]. Interestingly, DMD shares several features with other conditions characterized by loss of muscle mass and strength, specifically age-related sarcopenia [5] and cancer-associated cachexia, a multifactorial syndrome affecting large part of cancer patients [6]. Systemic and muscle inflammation, oxidative stress, ultrastructural abnormalities, alteration of the DAPC, and loss of muscle precursor cells are common hallmarks of DMD, sarcopenia and cachexia [7,8,9], suggesting that common approaches could be thought of to treat these conditions.
1.1. Therapeutic Approaches to DMD
1.1.1. Corticosteroids
Due to the deleterious role of inflammation in dystrophic muscles, anti-inflammatory steroids still represent the gold standard for the treatment of DMD, being able to improve the patients’ quality of life [10,11]. Prednisone and deflazacort delay the loss of muscle strength and functionality, the loss of ambulation, the onset of scoliosis, and respiratory and cardiac failure. However, besides their positive role in inhibiting inflammatory pathways that cause muscle necrosis and fibrosis, corticosteroids have shown limited activity and cause several adverse effects such as gain of weight, reduction of bone mineral density, increased risk of bones fractures gastrointestinal irritation, skin fragility, adrenal suppression, susceptibility to infections, metabolic disorders, hypertension, behavioural changes, and cushingoid appearance [12,13,14,15]. Moreover, definitive evidence is lacking about the role of corticosteroids on muscle regeneration [16], and glucocorticoids exert anti-myogenic effects on myoblasts by inducing the expression of glucocorticoid-induced leucine zipper (GILZ) and its newly identified isoform, long GILZ (L-GILZ) [17]. Recently, VBP15, an NF-κB inhibitor with high glucocorticoid receptor specificity, has been designed to reduce the side effects typical of steroids. In mdx mice, the oral administration of VBP15 resulted in improvement of the dystrophic phenotype, with less adverse effects compared with prednisone [18].
1.1.2. Cell Therapy
Two main cell types can be used to obtain new myofibers with functional dystrophin in DMD patients: satellite cells (myoblasts) isolated from healthy donors or genetically modified cells coming from the DMD patients themselves [19]. Once injected in mdx mice, an animal model of DMD, muscle precursor cells fuse with pre-existing or regenerating myofibers leading to dystrophin-positive fibers and partially restoring the muscle defect [20]. Consequently, a small number of DMD patients were injected intramuscularly with dystrophin-positive myoblasts, re-establishing the expression of dystrophin but without significant improvement in muscle morphology and functionality. This is because of the low survival and migration of injected myoblasts, and the immune rejection of transplanted cells [21]. Among the most innovative cell therapies is transplantation of mesoangioblasts, which are multipotent mesenchymal cells isolated from muscle vasculature. Mesoangioblasts can differentiate into myoblasts, and migrate to the surrounding damaged myofibers. Golden retriever muscular dystrophy (GRMD) dogs injected intra-arterially with mesoangioblasts isolated from healthy dogs showed expression of dystrophin in muscle tissue, and improved muscle morphology and functionality [19,22]. Unfortunately, injection of mesoangioblasts in DMD patients has demonstrated only minimal efficacy so far [23].
Another promising therapeutic tool is represented by the use of iPSC (induced pluripotent stem cells). Intramuscular transplantation of skeletal myogenic progenitors derived from human iPSC in dystrophic mice translated into appearance of dystrophin-positive myofibers and amelioration of muscle function [24], and miRNA cocktails promoting the myogenic potential of human mesodermal iPSC-derived progenitors have been recently defined [25].
1.1.3. Gene Therapy
Approaches to DMD extensively under study are represented by therapeutic intervention aimed at the delivery of a gene codifying for a functional dystrophin. Due to the very large dimension of the DMD gene (2.4 Mb and 79 exons, corresponding to about 0.1% of the human genome) [26], it is impossible to find a recombinant adeno-associated virus (rAAV) fitting with it. AAV vectors carried mini- or micro-dystrophins (i.e., different reduced parts of the DMD gene that still result in functional forms of the protein) have been used to treat animal models of DMD and DMD patients [19,27]. AAV vectors carrying minigenes have been injected in mdx mice and GRMD dogs demonstrating the efficient therapeutic effects of this treatment [28,29,30]. However, a phase I clinical trial using AAV-mini-dystrophin revealed only limited dystrophin expression and irrelevant muscle improvements due to the potential presence of AAV-neutralizing antibodies already present in humans, and the immune response against non-self-dystrophin [31], which requires this kind of approach be accompanied by immunosuppressive therapy.
1.1.4. Exon Skipping
More than 83% of the DMD patients, with deletions duplications or small mutations in the DMD gene could be treated with an exon skipping approach [32]. This therapy aims at restoring the dystrophin expression thanks to the possibility to skip the exon bearing the mutation, thus producing a shorter but functional protein. Antisense oligonucleotides (AONs) are the molecules used to obtain exon skipping. These AONs are composed of 20–30 nucleotides, specifically designed to match the pre-mRNA sequence to skip the DMD exon with the mutation, thus leading to truncated transcripts that are translated into functional proteins [11]. Phosphorothioate oligonucleotides (PS, Drisapersen) and morpholino phosphorodiamidate oligomers (PMO, Eteplirsen) are useful for patients carrying the mutation in exon 51; they can be injected locally or delivered systemically [33]. During phase II and III clinical trials, Drisapersen has not shown the expected results, and it is now under investigation in younger patients [19,34]. Instead, patients treated with Eteplirsen revealed an increased number of dystrophin-positive myofibers compared with placebo-treated patients, and improvement in functional tests [35] so that the US FDA accelerated the approval of this drug on September 2016. Eteplirsen has been used with good results also in patients carrying mutations in exon 53 in Japan [36]. However, the use of AONs have some aspects to be improved, such as the poor tissue uptake and the relative low rescue of dystrophin expression. Recently, tricyclo-DNA (tcDNA) has been tested in two different DMD mouse models inducing the restoration of dystrophin expression in several tissues, and improvement of respiratory and cardiac functions [37,38].
1.1.5. Induction of Utrophin Expression
Due to the high degree of sequence identity, utrophin is a paralogue of dystrophin, able to associate with members of the DAPC [26,39]. Thus, induction of utrophin expression is an investigated therapeutic approach to DMD. Dystrophin and utrophin show different expression patterns in healthy adult muscle fibers, with dystrophin being present along the entire sarcolemma and utrophin being confined to the myotendineous and the neuromuscular junctions (NMJs) [40,41]. However, high levels of utrophin are found at the sarcolemma during muscle development and muscle regeneration [42].
SMT C1100 (2-arylbenzoxazole[5-(ethyl sulfonyl)-2-(naphthalen-2-yl)benzo(d)oxazole)] is a small molecule able to upregulate utrophin mRNA and protein after oral administration. The use of SMT C1100 translated into improvement of muscle morphology and functionality, with utrophin detected also in heart and diaphragm in mdx mice [43]. A phase I clinical trial showed that SMT C1100 is safe and well tolerated [44]. Neuregulin 1/heregulin beta 1 (NRG1/HRGβ1) is a factor that controls the expression of utrophin at the NMJ, through the interaction with erbB/HER receptor and the utrophin A promoter, after GABPα/β activation by the ERK pathway [45]. Repeated intraperitoneal (i.p.) injections of HRGβ1 translated into an increment of utrophin expression in myofibers, with concomitant improvement of muscle morphology in mdx mice [46].
1.1.6. Alternative Approaches
A number of molecules are under investigation, characterized by different mechanisms of action. Givinostat, an inhibitor of the histone deacetylase (HDAC) characterized by anti-inflammatory, anti-angiogenic and antineoplastic features, has been tested in mdx mice resulting in slowdown of pathology progression and improvement of muscle morphology in terms of increment of myofiber cross-sectional area and reduction of fibrosis and adipose tissue deposition [47,48]. Givinostat is currently in a phase II clinical trial to evaluate its safety and tolerability.
Tadalafil and Sildenafil are two phosphodiesterase-5 (PDE5) inhibitors able to induce relaxation of vascular smooth muscle cells, by increasing the levels of cyclic guanosine monophosphate (cGMP). The administration of Tadalafil or Sildenafil resulted in beneficial effects in mdx mice and DMD patients [19,49,50]. Unfortunately, disappointing results came out on February 2016 from the phase III trial of Tadalafil, due to failure to prove evidence for efficacy in slowing the decline in the walking ability of DMD boys.
Halofuginone (HT-100) is a molecule that reduces the fibrotic tissue deposition induced by TGF-β. Treatment with halofuginone translated into reduction of muscle fibrosis and amelioration of respiratory and cardiac functions in old mdx mice. Halofuginone also acts directly on muscle cells promoting fusion into myotubes in both normal and dystrophic conditions [51].
The benzoquinone, Idebenone (Catena/Raxone) stimulates both mitochondrial electron reflux and cellular energy production and inhibits lipid peroxidation [52]. Mdx mice treated with Idebenone were characterized by improvement of cardiac functions and exercise performance. In a phase II clinical trial, Idebenone showed positive effects on functional cardiac and respiratory parameters in DMD patients [52]. A phase III clinical trial showed reduction of loss of respiratory functions in DMD patients, even in the absence of glucocorticoid treatment [53,54].
Aminoglycosides (a group of drugs used in Gram-negative bacterial infections) might restore dystrophin expression, introducing a nucleotide sequence at the aminoacyl transfer RNA acceptor site and binding the decoding site of ribosomal RNA, which leads to the expression of a full-length functional dystrophin protein [55]. Because of the several disadvantages of aminoglycosides (i.e., high dosages and the renal and oto-toxicity), other molecules have been developed with the same activity but fewer side effects [11]. One of these molecules is Ataluren (Translarna, former PTC124), a nonaminoglycoside without antibacterial activity [11]. The expression of dystrophin is upregulated in skeletal muscles (including diaphragm) and hearth of mdx mice treated with Ataluren [56,57]. Two doses of Ataluren in patients with nonsense mutation were demonstrated efficacious and well tolerated during a phase IIb clinical trial [58]. Instead, the results obtained from a phase III clinical trial were not statistically significant, even if none of the Ataluren-treated group needed a wheelchair during the study, contrary to the placebo group in which four patients lost ambulation [59].
Myostatin is a negative regulator of skeletal muscle growth, and animals lacking myostatin are characterized by increased muscle mass [60]. Consequently, blockade of myostatin activity is under investigation as a potential therapy for muscle degenerative diseases. Dystrophic mice lacking myostatin or treated with an anti-myostatin blocking antibody showed increment of muscle mass and muscle strength, together with reduced fibrosis [61,62]. A phase II clinical trial is currently ongoing to analyze the pharmacokinetics and pharmacodynamics of Domagrozumab, a monoclonal antibody that binds myostatin, in pediatric DMD patients since a previous study revealed an increment of muscle mass in SCID (severe combined immunodeficiency) mice and healthy monkeys treated with this neutralizing antibody [63,64].
Recently, an AAV-based strategy has been developed to deliver (in mdx mice) the CRISPR/Cas9 genome editing system, in which the Cas9 nuclease cleaves DNA sequences targeted by a guide RNA [65] to remove the mutated exon 23 from the Dmd gene. This promising approach resulted in partial recovery of skeletal and cardiac muscle functionality, biochemistry and force, and generated a pool of endogenously corrected myogenic precursors in mdx muscles [66,67].
The different approaches used so far to counteract the DMD symptoms or correct the DMD mutations have revealed intrinsic limitations: rAAV are unable to accommodate the full-length DMD gene, and their use is accompanied with viral toxicity, immune response and limited persistence of the transgene expression; myoblast transplantation and stem cell therapy have shown low efficiency due to precocious death and scarce homing of the injected cells; and, AONs require repeated administrations. Moreover, most of the proposed approaches require immunosuppression in order to protect the foreign material used from the host immune system attack. For these reasons, the investigation in the DMD field is still active, and some authors encourage combinatorial approaches [68].
The mechanisms of action, the effects, and the trial status of these alternative approaches are listed in Table 1.

Table 1.
Mechanisms of action and effects of alternative approaches of Section 1.1.6.
2. Sertoli Cells
2.1. Sertoli Cells: Multiple Roles for a Single Cell Type
Sertoli cells (SeC) are the most abundant cells of the seminiferous tubules in the testis, where they protect the developing germ cells from the immune system attack by creating a physical barrier (the blood–testis barrier, BTB) made of adjacent SeC linked with tight junctions, which isolates the lumen of seminiferous tubules from the interstitial fluid. In addition, SeC secrete many trophic and maturative factors indispensable for the growth and differentiation of germ cells, as well as immunomodulatory factors that further contribute to create an immuneprivileged environment in the testis (Table 2) [74,75]. The latter is necessary to avoid auto-immune responses against the developing germ cells, which express novel molecules on their surface potentially recognized as foreign by the immune system. The immuneprivileged feature of the testis was first discovered in 1767, when John Hunter transplanted rooster testes into the abdominal cavity of a hen, and he found that they maintained their normal structure over time [76]. Several studies have demonstrated that SeC are the most relevant cell type of the testis that can protect testicular allogeneic and xenogeneic grafts from immune rejection, even if transplanted outside the BTB, thus confirming that humoral besides mechanic processes are involved in the immuneprivileged status of the testes [75,77,78]. Evidence that SeC are primarily responsible for the successful survival and function of transplanted tissues was provided when SeC were co-transplanted with allogeneic Langerhans islets, showing that SeC were able to protect islets from rejection in the absence of immunosuppression [79]. Later, several experiments confirmed the ability of SeC to protect from immune destruction allogeneic and xenogeneic pancreatic islets [80,81], and other tissues, including xenogeneic adrenal chromaffin cells [82], allogeneic dopaminergic neurons [83], allogeneic and xenogeneic skin grafts [81,84], and allogeneic heart grafts [85]. The ability of SeC to protect xenogeneic pancreatic islets from rejection was demonstrated also in human patients, in a controversial study with respect to ethical issues. Pancreatic islets were inserted together with neonatal porcine SeC in a porous chamber and placed subcutaneously in the anterior abdominal wall of young diabetic patients, without immunosuppressive treatment. The patients did not show any complications in a 7-years follow-up, and a half of the grafted patients significantly diminished their insulin doses [86,87].
Table 2.
List of the major factors secreted by SeC.
Finally, SeC have been employed in different experimental models of diseases to take advantage of their release of trophic and anti-inflammatory factors (Table 2). SeC have been implanted into the central nervous system of animal models of Parkinson’s disease, Huntington’s disease and amyotrophic lateral sclerosis, where they provided protection of the surrounding tissue through local release of trophic and antiinflammatory molecules [82,88,89].
2.2. The Immunomodulatory Properties of SeC
SeC inhibit the humoral and T-cell response thanks to their ability to secrete factors that block T lymphocyte proliferation and interleukin (IL)-2 production [90,91]. Moreover, FASL (CD95L) expressed on SeC might interact with the FAS receptor (CD95) on the T cell surface, inducing apoptosis of lymphocytes [92]. The immunomodulation exerted by SeC has been ascribed to the activity of tolerogenic factors, such as TGF-β and IDO (indoleamine 2,3-dioxygenase), a tryptophan-metabolizing enzyme. Suarez-Pinzon et al. showed that TGF-β secretion by SeC is necessary for islet graft survival when co-transplanted in NOD (non-obese diabetic) mice. TGF-β secreted by SeC might modulate T cell phenotype favoring their differentiation into a tolerogenic Th2 phenotype [93]. Injection (i.p.) of SeC in NOD mice partially reversed the pathology with the recovery of β cell function thanks to restoration of the systemic immune tolerance through a TGF-β/IDO-dependent mechanism that led to emergence of regulatory T cells (Tregs) [94]. Moreover, it has been demonstrated that TGF-β with IL-10 and activin-A, released by SeC, contribute to modulate the immune cell response in the testis, stimulating immune cells involved in the tolerogenic response, such as M2 (anti-inflammatory) macrophages, and Th2 and Treg cells [95]. Accordingly, most macrophages located in the testis show an M2 phenotype [96]. Recently, a soluble form of JAGGED1 (JAG1), which induces the generation of Treg cells, has been reported to be secreted by SeC [97]. Additional contribution to immune protection comes from the secretion by SeC of complement and inhibitors of granzyme, a cytolytic molecule released by cytotoxic T cells [98,99]. Interestingly, SeC are also able to respond to virus and bacteria, eliciting an inflammatory response. Indeed, they express toll-like receptors, whose activation in SeC induces the release of proinflammatory cytokines and chemokines [100].
2.3. The Encapsulation Chance
The preclinical use of SeC has been improved with the introduction of the encapsulation procedure. Microcapsules are spherical particles with size varying between 50 and 2 mm characterized by a cut-off permeability that allows the passage of oxygen, nutrients, therapeutic protein products, waste products and other molecules, including soluble factors released from the encapsulated cells themselves [101]. The biomaterial the microcapsules are made of has to be potentially invisible to the host immune system in order to prevent immune reactions, and should protect the capsule content as long as possible.
The research in the diabetes field has been particularly active at exploring the most efficacious biomaterials into which to encapsulate the insulin-producing pancreatic β cells, and in 1980s Lim and Sun showed efficacy of transplanted microencapsulated insulae for the treatment of diabetic mice [102]. Many biomaterials, including agarose, poly-cations (poly-l-lysine, poly-l-ornithine), poly-ethilen-glycole and alginate, have been tested so far to ensure the best immune-invisibility and long-lasting viability of the entrapped cells, together with a suitable capsule cut-off [103].
In particular, alginate has been studied enough to be defined safe for human applications [104,105,106]. Alginate is a linear anionic polysaccharide, distributed widely in the cell wall of brown algae and some bacteria [101,107]. It is composed of two types of uronic acids, β-d-mannuronic acid (M) and α-l-guluronic acid (G) (1,4)-linked to form blocks of consecutive G residues, consecutive M residues, and alternating M and G residues. The physical and chemical properties depend on G/M ratio, sequence, G-block length, and molecular weight (Figure 1).
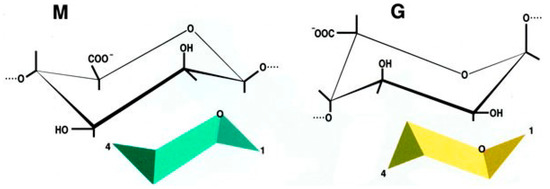
Figure 1.
Chemical structure of β-d-mannuronic acid (M) and α-l-guluronic acid (G).
Alginate is totally biocompatible and can exist in the form of a hydrogel. Different bivalent cations that bind alginate might be used to obtain the gel, with barium being the most used since it forms stronger gels with reduced permeability due to its high affinity to alginate [108,109]. The ability of alginate to chelate bivalent cations is linked to the G/M ratio. High G/M ratios lead to zig-zag structures that allow cations to insert inside perfect niches resulting in a more stable bond of alginate with cations, higher resistance, unvarying porosity and long-lasting durability. Ions belonging to the second group of the Mendeleev’s periodic table share the ability to ligate alginate. Barium alginate microcapsules are mechanically stable and easy to obtain, with a procedure that is substantially independent of the type of cells to be encapsulated [101,110].
Alginate-based microcapsules have shown long durability in terms of survival of the entrapped cells and have been used in rodents, dogs and monkeys [111,112,113,114]. Moreover, alginate-based microcapsules containing human pancreatic islets have been employed in clinical trials in which they were transplanted i.p. in non-immunosuppressed type-1 diabetic patients [104,105,115]. The procedure proved safe and painless, and no adverse effects were reported.
2.4. Pre-Clinical Studies Using Microencapsulated SeC
SeC entrapped into alginate-based microcapsules (MC-SeC) (Figure 2) have been tested in several experimental models of disease. Injection (i.p.) of microcapsules containing rat SeC together with rat pancreatic islets in induced diabetic mice resulted in prolongation of normoglycemia [116]. SeC co-encapsulated with human hepatocyte HepG2 cells were i.p.-injected in rats with acute hepatic failure in order to protect HepG2 cells from immune reaction, prolonging the survival of the treated animals [117]. MC-SeC have been injected i.p. in a mouse model of type-1 diabetes, resulting in successful diabetes prevention and reversion in the absence of additional β cells or insulin therapy, through a TGF-β/IDO-mediated restoration of the systemic tolerance and induction of neogenesis of β cells [94,118]. Injection (i.p.) of MC-SeC protected allogeneic skin graft from immune destruction in rats; the treated animals showed an increased presence of Tregs compared to animal injected with empty microcapsules (E-MC) [119]. A single i.p. injection of MC-SeC promoted body growth in a mouse model of the human Laron syndrome (dwarfism), characterized by mutations in GHR (growth hormone receptor) and reduced production of IGF-1, via the release into the circulation of IGF-1 by SeC. Interestingly, SeC-derived IGF-1 remained detectable in the serum of the treated mice up to one year after injection [113]. Similarly, a single i.p. injection of MC-SeC in an experimental model of Huntington’s disease resulted in longer survival and improved muscle functionality of the treated animals [120].
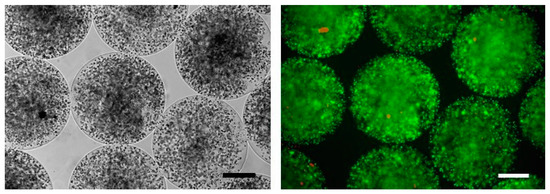
Figure 2.
MC-SeC as viewed by phase contrast microscopy (Left) or by epi-fluorescence after staining with ethidium bromide (orange) and fluorescein diacetate (green) to analyze cell viability (Right). Bars, 200 µm.
MC-SeC have also been tested in mammals. Obese rhesus macaques with spontaneous type-2 diabetes injected i.p. with a single dose of porcine MC-SeC showed reduction of plasma glucose and B lymphocytes together with absence of rejection and adverse effects at six months from injection [121].
2.5. Use of MC-SeC in DMD
Recently, we obtained encouraging results by treating dystrophic, mdx mice with a single i.p. injection of MC-SeC in the absence of any immunosuppressive regimen. SeC were purified from testis of SPF White Large piglet and entrapped into clinical grade alginate-based microcapsules with an M/G ratio of 1.093 and an average diameter of about 600 µm. We transplanted mdx mice in the acute phase of the pathology (four weeks of age) with an equivalent amount of 1.0 × 106 SeC/g body weight, and evaluated the effects in comparison with age-matched mdx mice injected with empty microcapsules (E-MC) [114]. Three weeks after the injection, the treated animals exhibited amelioration of muscle morphology in terms of reduction of infiltrating inflammatory cells (especially, macrophages), fibrosis and myofiber necrosis. This was accompanied by functional recovery and increased resistance to exercise-induced muscle damage. Interestingly, a single i.p. injection of porcine MC-SeC in acute mdx mice resulted in benefits detectable even in long-term (5 months) analysis. Diaphragm, which is the muscle that accumulates damage over time in the mdx model, showed reduced necrosis, inflammation and fibrosis, and reduced fat deposition in treated animals compared with control mice [114]. The i.p. injection of MC-SeC had similar positive effects even on muscles of chronic (12 months of age) mdx mice, and resulted protective against muscle necrosis and inflammation in presymptomatic (two weeks of age) animals [122].
Although an antiinflammatory effect exerted by SeC was to be expected based on the peculiarities of this cell type, an additional and unexpected induction of utrophin expression in muscle tissue after i.p. injection of MC-SeC was found. The expression of utrophin was induced by SeC-released porcine heregulin β1, which from the peritoneal cavity was able to reach each muscle through the circulation [114]. The mechanism of action and the effects of i.p. injection of MC-SeC in mdx mice are summarized in Table 3.
Table 3.
Effects of a single i.p. injection of MC-SeC in DMD animals.
3. Conclusions and Remarks
DMD is the most common muscular dystrophy, affecting one in 3600–5000 male live births worldwide [123]. In DMD the lack of dystrophin leads to progressive muscle degeneration, which creates a condition of chronic inflammation that in turn hampers compensatory mechanisms and favors the accumulation of fibrous and adipose tissues [3]. Despite the huge effort to find a cure, corticosteroids remain the gold standard therapy for DMD patients, since the numerous approaches investigated so far have shown several limitations, with most of them requiring a concomitant immunosuppressive treatment.
Immunosuppression achieved with specific drugs (such as, corticosteroids, calcineurin inhibitors and m-TOR inhibitors) represents a condition difficult to manage because of the severe side effects of the used drugs, including frequent infections, especially in life-long treatments. Consequently, over the past years many studies have aimed at finding alternative therapeutic approaches to treat or cure DMD pathology [11].
One of the most recent approaches is centered on the peculiarities of SeC, which have a physiological role in protecting the developing germ cells through the production of immunomodulatory factors, and in favoring germ cell maturation thanks to the secretion of trophic factors [75,78]. Purified SPF porcine SeC were encapsulated and injected into the peritoneal cavity of mdx mice. The encapsulation procedure has the advantage of confining the cells to a restricted three-dimensional space, avoiding their spreading throughout the host body, thus overcoming a big obstacle relative to the safety of allo- and xeno-engraftments in humans. When injected in a confined body region, such as the peritoneal cavity, the microcapsules can potentially be retrieved if unexpected adverse effects arise.
We chose alginate as a well-established biocompatible agent. However, it has been reported that alginate can attract immune cells, such as macrophages and neutrophils, over time leading to deposition of fibrotic scar tissue that hampers the functionality of the capsule in-out exchange and reduces the viability of the entrapped cells. This unfavorable event is not found when SeC are the encapsulated cell type because of their continuous release of immunomodulatory factors, making the injected capsules freely floating, morphologically intact, and without fibrotic tissue overgrowth even after months from injection [114].
Data obtained from the engraftment of MC-SeC in mdx mice revealed that MC-SeC act as a micro-biofactory that secrete anti-inflammatory/immunomodulatory factors and trophic factors, especially heregulin β1, thus contributing to the amelioration of muscle morphology and performance by at least a dual mechanism: the activation of a positive anti-inflammatory loop leading to reduction of necrosis, fibrosis and adipose tissue deposition; and the induction of utrophin expression functionally mimicking the lacking dystrophin (Figure 3). Additional trophic factors known to be secreted by SeC may also concur to the amelioration of dystrophic muscle morphology. As an example, it has been demonstrated that SeC secrete IGF-1 [113], which has an essential role in muscle growth during development and regeneration [124] suggesting a possible involvement of IGF-1 in improving muscle regeneration in MC-SeC-treated dystrophic mice (Figure 3).
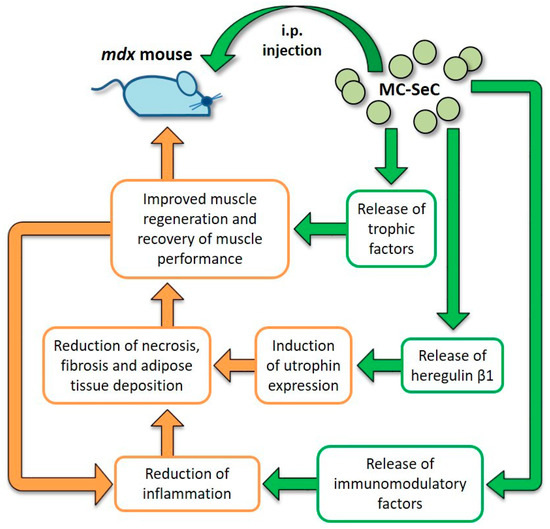
Figure 3.
Diagram of the effects exerted by MC-SeC once injected i.p. into mdx mice. From inside the microcapsules, SeC release immunomodulatory factors that reduce muscle inflammation thus reducing necrosis of the myofibers, and fibrous and adipose tissue deposition. This is also favored by secretion by SeC of heregulin β1 and subsequent heregulin β1-dependent induction of utrophin expression. Moreover, additional SeC-derived trophic factors (such as IGF-1) might concur to improve muscle regeneration and the recovery of muscle performance, which in turn reduces local inflammation. Straight green arrows and boxes indicate secretion by MC-SeC and point to the effects of the secreted factors; orange boxes and arrows highlight the beneficial effects ignited by secreted factors.
Thus, at the basis of the beneficial effects of the treatment with MC-SeC there is the production by SeC of a cocktail of factors rather than a single factor, that makes this treatment the equivalent of a combinatorial approach in which anti-inflammatory and trophic factors are mixed together and act towards the same target by affecting different molecular and cellular hallmarks of the pathology.
Another important aspect of the use of MC-SeC is that, unlike several approaches to DMD that are mutation-specific (e.g., exon skipping approaches) it represents a universal approach, potentially applicable to the entire cohort of DMD patients.
These intriguing results have opened a new avenue in the treatment of DMD. Future investigation should evaluate the correct dosage of MC-SeC (i.e., by establishing the minimum efficacious dose), the specific mechanism through which SeC restrain inflammation (e.g., by using utrophin-deficient mdx/Utrn−/− mice), and the immunomodulatory rather than immunosuppressive role of SeC treatment (e.g., by investigating the effects of i.p. injection of MC-SeC in experimental models of infection and cancer). In the case of positive results, and after validation in mammals, the i.p. injection of MC-SeC might become a concrete approach to counteract muscle inflammation and induce utrophin expression in muscles of DMD patients and, potentially, patients suffering from other myopathies characterized by muscle inflammation or immune disorders, such as autoimmune myositis.
Acknowledgments
Laura Salvadori is recipient of a fellowship by Parent Project Onlus, Italy. Giovanni Luca and Riccardo Calafiore were supported by a grant from Gary Harlem (Altucell, NY, USA). Rosario Donato was supported by the AFM (Project 15679). Guglielmo Sorci is supported by Parent Project Onlus, Italy.
Conflicts of Interest
The authors declare no conflict of interest. The founding sponsors had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, and in the decision to publish the results.
Abbreviations
| AAV | Adeno-associated virus |
| AON | Antisense oligonucleotide |
| BTB | Blood-testis barrier |
| DAPC | Dystrophin-associated protein complex |
| DMD | Duchenne muscular dystrophy |
| E-MC | Empty microcapsules |
| GRMD | Golden retriever muscular dystrophy |
| HDAC | Histone deacetylase |
| HRGβ1 | Heregulin β 1 |
| IDO | Indoleamine 2,3-dioxygenase |
| IGF-1 | Insulin-like growth factor 1 |
| iPSC | Induced pluripotent stem cells |
| MC-SeC | Microencapsulated Sertoli cells |
| NMJ | Neuromuscular junction |
| NOD | Non-obese diabetic |
| NRG1 | Neuregulin 1 |
| PDE5 | Phosphodiesterase 5 |
| SeC | Sertoli cells |
| SPF | Specific-pathogens free |
| TGF-β | Transforming growth factor β |
References
- Dalkilic, I.; Kunkel, L.M. Muscular dystrophies: Genes to pathogenesis. Curr. Opin. Genet. Dev. 2003, 13, 231–238. [Google Scholar] [CrossRef]
- Davies, K.E.; Nowak, K.J. Molecular mechanisms of muscular dystrophies: Old and new players. Nat. Rev. Mol. Cell Biol. 2006, 10, 762–773. [Google Scholar] [CrossRef] [PubMed]
- Evans, N.P.; Misyak, S.A.; Robertson, J.L.; Bassaganya−Riera, J.; Grange, R.W. Immune mediated mechanisms potentially regulate the disease time course of Duchenne muscular dystrophy and provide targets for therapeutic intervention. Am. J. Phys. Med. Rehabil. 2009, 1, 755–768. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.W.; Zhao, P.; Borup, R.; Hoffman, E.P. Expression profiling in the muscular dystrophies: Identification of novel aspects of molecular pathophysiology. J. Cell Biol. 2000, 151, 1321–1336. [Google Scholar] [CrossRef] [PubMed]
- Narici, M.V.; Maffulli, N. Sarcopenia: Characteristics, mechanisms and functional significance. Br. Med. Bull. 2010, 95, 139–159. [Google Scholar] [CrossRef] [PubMed]
- Fearon, K.; Strasser, F.; Anker, S.D.; Bosaeus, I.; Bruera, E.; Fainsinger, R.L.; Jatoi, A.; Loprinzi, C.; MacDonald, N.; Mantovani, G.; et al. Definition and classification of cancer cachexia: An international consensus. Lancet Oncol. 2011, 12, 489–495. [Google Scholar] [CrossRef]
- Meng, S.-J.; Yu, L.-J. Oxidative stress, molecular inflammation and sarcopenia. Int. J. Mol. Sci. 2010, 11, 1509–1526. [Google Scholar] [CrossRef] [PubMed]
- Musumeci, G. Sarcopenia and exercise “The state of the art”. J. Funct. Morphol. Kinesiol. 2017, 2, 40. [Google Scholar] [CrossRef]
- Berardi, E. Muscular dystrophies and cancer cachexia: Similarities in chronic skeletal muscle degeneration. J. Funct. Morphol. Kinesiol. 2017, 2, 39. [Google Scholar] [CrossRef]
- Muntoni, F.; Fisher, I.; Morgan, J.E.; Abraham, D. Steroids in Duchenne muscular dystrophy: From clinical trials to genomic research. Neuromuscul. Disord. 2002, 12, S162–S165. [Google Scholar] [CrossRef]
- Falzarano, M.S.; Scotton, C.; Passarelli, C.; Ferlini, A. Duchenne Muscular Dystrophy: From Diagnosis to Therapy. Molecules 2015, 20, 18168–18184. [Google Scholar] [CrossRef] [PubMed]
- Fenichel, G.M.; Florence, J.M.; Pestronk, A.; Mendell, J.R.; Moxley, R.T., 3rd; Griggs, R.C.; Brooke, M.H.; Miller, J.P.; Robison, J.; King, W.; et al. Long-term benefit from prednisone therapy in Duchenne muscular dystrophy. Neurology 1991, 41, 1874–1877. [Google Scholar] [CrossRef] [PubMed]
- Mozzetta, C.; Minetti, G.; Puri, P.L. Regenerative pharmacology in the treatment of genetic diseases: The paradigm of muscular dystrophy. Int. J. Biochem. Cell Biol. 2008, 41, 701–710. [Google Scholar] [CrossRef] [PubMed]
- Ichim, T.E.; Alexandrescu, D.T.; Solano, F.; Lara, F.; De Necochea Campion, R.; Paris, E.; Woods, E.J.; Murphy, M.P.; Dasanu, C.A.; Patel, A.N.; et al. Mesenchymal stem cells as anti−inflammatories: Implications for treatment of Duchenne muscular dystrophy. Cell. Immunol. 2010, 260, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Angelini, C.; Peterle, E. Old and new therapeutic developments in steroid treatment in Duchenne muscular dystrophy. Acta Myol. 2012, 31, 9–15. [Google Scholar] [PubMed]
- Manzur, A.Y.; Kuntzer, T.; Pike, M.; Swan, A. Glucocorticoid corticosteroids for Duchenne muscular dystrophy. Cochrane Database Syst. Rev. 2008, 1, CD003725. [Google Scholar] [CrossRef]
- Bruscoli, S.; Donato, V.; Velardi, E.; Di Sante, M.; Migliorati, G.; Donato, R.; Riccardi, C. Glucocorticoid-induced leucine zipper (GILZ) and long GILZ inhibit myogenic differentiation and mediate anti-myogenic effects of glucocorticoids. J. Biol. Chem. 2010, 285, 10385–10396. [Google Scholar] [CrossRef] [PubMed]
- Heier, C.R.; Damsker, J.M.; Yu, Q.; Dillingham, B.C.; Huynh, T.; van der Meulen, J.H.; Sali, A.; Miller, B.K.; Phadke, A.; Scheffer, L.; et al. VBP15, a novel anti-inflammatory and membrane-stabilizer, improves muscular dystrophy without side effects. EMBO Mol. Med. 2013, 5, 1569–1585. [Google Scholar] [CrossRef] [PubMed]
- Shimizu-Motohashi, Y.; Miyatake, S.; Komaki, H.; Takeda, S.; Aoki, Y. Recent advances in innovative therapeutic approaches for Duchenne muscular dystrophy: From discovery to clinical trials. Am. J. Transl. Res. 2016, 8, 2471–2489. [Google Scholar] [PubMed]
- Partridge, T.A.; Morgan, J.E.; Coulton, G.R.; Hoffman, E.P.; Kunkel, L.M. Conversion of mdx myofibres from dystrophin-negative to positive by injection of normal myoblasts. Nature 1989, 337, 176–179. [Google Scholar] [CrossRef] [PubMed]
- Skuk, D.; Goulet, M.; Roy, B.; Chapdelaine, P.; Bouchard, J.P.; Roy, R.; Dugre, F.J.; Sylvain, M.; Lachance, J.G.; Deschenes, L.; et al. Dystrophin expression in muscles of duchenne muscular dystrophy patients after high-density injections of normal myogenic cells. J. Neuropathol. Exp. Neurol. 2006, 65, 371–386. [Google Scholar] [CrossRef] [PubMed]
- Sampaolesi, M.; Blot, S.; D’Antona, G.; Granger, N.; Tonlorenzi, R.; Innocenzi, A.; Mognol, P.; Thibaud, J.L.; Galvez, B.G.; Barthelemy, I.; et al. Mesoangioblast stem cells ameliorate muscle function in dystrophic dogs. Nature 2006, 444, 574–579. [Google Scholar] [CrossRef] [PubMed]
- Cossu, G.; Previtali, S.C.; Napolitano, S.; Cicalese, M.P.; Tedesco, F.S.; Nicastro, F.; Noviello, M.; Roostalu, U.; Natali Sora, M.G.; Scarlato, M.; et al. Intra-arterial transplantation of HLA-matched donor mesoangioblasts in Duchenne muscular dystrophy. EMBO Mol. Med. 2015, 7, 1513–1528. [Google Scholar] [CrossRef] [PubMed]
- Darabi, R.; Arpke, R.W.; Irion, S.; Dimos, J.T.; Grskovic, M.; Kyba, M.; Perlingeiro, R.C. Human ES- and iPS-derived myogenic progenitors restore DYSTROPHIN and improve contractility upon transplantation in dystrophic mice. Cell Stem Cell 2012, 10, 610–619. [Google Scholar] [CrossRef] [PubMed]
- Giacomazzi, G.; Holvoet, B.; Trenson, S.; Caluwé, E.; Kravic, B.; Grosemans, H.; Cortés-Calabuig, Á.; Deroose, C.M.; Huylebroeck, D.; Hashemolhosseini, S.; et al. MicroRNAs promote skeletal muscle differentiation of mesodermal iPSC-derived progenitors. Nat. Commun. 2017, 8, 1249. [Google Scholar] [CrossRef] [PubMed]
- Blake, D.J.; Weir, A.; Newey, S.E.; Davies, K.E. Function and genetics of dystrophin and dystrophin related proteins in muscle. Physiol. Rev. 2002, 82, 291–329. [Google Scholar] [CrossRef] [PubMed]
- Harper, S.Q.; Hauser, M.A.; DelloRusso, C.; Duan, D.; Crawford, R.W.; Phelps, S.F.; Harper, H.A.; Robinson, A.S.; Engelhardt, J.F.; Brooks, S.V.; et al. Modular flexibility of dystrophin: Implications for gene therapy of Duchenne muscular dystrophy. Nat. Med. 2002, 8, 253–261. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Li, J.; Xiao, X. Adeno-associated virus vector carrying human minidystrophin genes effectively ameliorates muscular dystrophy in mdx mouse model. Proc. Natl. Acad. Sci. USA 2000, 97, 13714–13719. [Google Scholar] [CrossRef] [PubMed]
- Gregorevic, P.; Allen, J.M.; Minami, E.; Blankinship, M.J.; Haraguchi, M.; Meuse, L.; Finn, E.; Adams, M.E.; Froehner, S.C.; Murry, C.E.; et al. rAAV6-microdystrophin preserves muscle function and extends lifespan in severely dystrophic mice. Nat. Med. 2006, 12, 787–789. [Google Scholar] [CrossRef] [PubMed]
- Yue, Y.; Pan, X.; Hakim, C.H.; Kodippili, K.; Zhang, K.; Shin, J.H.; Yang, H.T.; McDonald, T.; Duan, D. Safe and bodywide muscle transduction in young adult Duchenne muscular dystrophy dogs with adeno-associated virus. Hum. Mol. Genet. 2015, 24, 5880–5890. [Google Scholar] [CrossRef] [PubMed]
- Bowles, D.E.; McPhee, S.W.; Li, C.; Gray, S.J.; Samulski, J.J.; Camp, A.S.; Li, J.; Wang, B.; Monahan, P.E.; Rabinowitz, J.E.; et al. Phase 1 gene therapy for Duchenne muscular dystrophy using a translational optimized AAV vector. Mol. Ther. 2012, 20, 443–455. [Google Scholar] [CrossRef] [PubMed]
- Aartsma-Rus, A.; Fokkema, I.; Verschuuren, J.; Ginjaar, I.; van Deutekom, J.; van Ommen, G.J.; den Dunnen, J.T. Theoretic applicability of antisense-mediated exon skipping for Duchenne muscular dystrophy mutations. Hum. Mutat. 2009, 30, 293–299. [Google Scholar] [CrossRef] [PubMed]
- Lu, Q.L.; Cirak, S.; Partridge, T. What Can We Learn From Clinical Trials of Exon Skipping for DMD? Mol. Ther. Nucleic Acids 2014, 3, e152. [Google Scholar] [CrossRef] [PubMed]
- Voit, T.; Topaloglu, H.; Straub, V.; Muntoni, F.; Deconinck, N.; Campion, G.; De Kimpe, S.J.; Eagle, M.; Guglieri, M.; Hood, S.; et al. Safety and efficacy of drisapersen for the treatment of Duchenne muscular dystrophy (DEMAND II): An exploratory, randomised, placebo-controlled phase 2 study. Lancet Neurol. 2014, 13, 987–996. [Google Scholar] [CrossRef]
- Mendell, J.R.; Rodino-Klapac, L.R.; Sahenk, Z.; Roush, K.; Bird, L.; Lowes, L.P.; Alfano, L.; Gomez, A.M.; Lewis, S.; Kota, J.; et al. Eteplirsen for the treatment of Duchenne muscular dystrophy. Ann. Neurol. 2013, 74, 637–647. [Google Scholar] [CrossRef] [PubMed]
- Lim, K.R.; Maruyama, R.; Yokota, T. Eteplirsen in the treatment of Duchenne muscular dystrophy. Drug Des. Dev. Ther. 2017, 11, 533–545. [Google Scholar] [CrossRef] [PubMed]
- Goyenvalle, A.; Griffith, G.; Babbs, A.; El Andaloussi, S.; Ezzat, K.; Avril, A.; Dugovic, B.; Chaussenot, R.; Ferry, A.; Voit, T.; et al. Functional correction in mouse models of muscular dystrophy using exon-skipping tricyclo-DNA oligomers. Nat. Med. 2015, 21, 270–275. [Google Scholar] [CrossRef] [PubMed]
- Relizani, K.; Griffith, G.; Echevarría, L.; Zarrouki, F.; Facchinetti, P.; Vaillend, C.; Leumann, C.; Garcia, L.; Goyenvalle, A. Efficacy and Safety Profile of Tricyclo-DNA Antisense Oligonucleotides in Duchenne Muscular Dystrophy Mouse Model. Mol. Ther. Nucleic Acids 2017, 8, 144–157. [Google Scholar] [CrossRef] [PubMed]
- Cohn, R.D.; Campbell, K.P. Molecular basis of muscular dystrophies. Muscle Nerve 2000, 23, 1456–1471. [Google Scholar] [CrossRef]
- Khurana, T.S.; Watkins, S.C.; Chafey, P.; Chelly, J.; Tomé, F.M.; Fardeau, M.; Kaplan, J.C.; Kunkel, L.M. Immunolocalization and developmental expression of dystrophin related protein in skeletal muscle. Neuromuscul. Disord. 1991, 1, 185–194. [Google Scholar] [CrossRef]
- Nguyen, T.M.; Ellis, J.M.; Love, D.R.; Davies, K.E.; Gatter, K.C.; Dickson, G.; Morris, G.E. Localization of the DMDL gene-encoded dystrophin-related protein using a panel of nineteen monoclonal antibodies: Presence at neuromuscular junctions, in the sarcolemma of dystrophic skeletal muscle, in vascular and other smooth muscles, and in proliferating brain cell lines. J. Cell Biol. 1991, 115, 1695–1700. [Google Scholar] [PubMed]
- Clerk, A.; Morris, G.E.; Dubowitz, V.; Davies, K.E.; Sewry, C.A. Dystrophin-related protein, utrophin, in normal and dystrophic human fetal skeletal muscle. Histochem. J. 1993, 25, 554–561. [Google Scholar] [CrossRef] [PubMed]
- Tinsley, J.M.; Fairclough, R.J.; Storer, R.; Wilkes, F.J.; Potter, A.C.; Squire, S.E.; Powell, D.S.; Cozzoli, A.; Capogrosso, R.F.; Lambert, A.; et al. Daily treatment with SMTC1100, a novel small molecule utrophin upregulator, dramatically reduces the dystrophic symptoms in the mdx mouse. PLoS ONE 2011, 6, e19189. [Google Scholar] [CrossRef] [PubMed]
- Ricotti, V.; Spinty, S.; Roper, H.; Hughes, I.; Tejura, B.; Robinson, N.; Layton, G.; Davies, K.; Muntoni, F.; Tinsley, J. Safety, tolerability, and pharmacokinetics of SMT C1100, a 2-arylbenzoxazole utrophin modulator, following single- and multiple-dose administration to pediatric patients with duchenne muscular dystrophy. PLoS ONE 2016, 11, e0152840. [Google Scholar] [CrossRef] [PubMed]
- Basu, U.; Gyrd-Hansen, M.; Baby, S.M.; Lozynska, O.; Krag, T.O.; Jensen, C.J.; Frödin, M.; Khurana, T.S. Heregulin-induced epigenetic regulation of the utrophin-A promoter. FEBS Lett. 2007, 581, 4153–4158. [Google Scholar] [CrossRef] [PubMed]
- Krag, T.O.; Bogdanovich, S.; Jensen, C.J.; Fischer, M.D.; Hansen-Schwartz, J.; Javazon, E.H.; Flake, A.W.; Edvinsson, L.; Khurana, T.S. Heregulin ameliorates the dystrophic phenotype in mdx mice. Proc. Natl. Acad. Sci. USA 2004, 101, 13856–13860. [Google Scholar] [CrossRef] [PubMed]
- Colussi, C.; Mozzetta, C.; Gurtner, A.; Illi, B.; Rosati, J.; Straino, S.; Ragone, G.; Pescatori, M.; Zaccagnini, G.; Antonini, A.; et al. HDAC2 blockade by nitric oxide and histone deacetylase inhibitors reveals a common target in Duchenne muscular dystrophy treatment. Proc. Natl. Acad. Sci. USA 2009, 105, 19183–19187. [Google Scholar] [CrossRef] [PubMed]
- Consalvi, S.; Mozzetta, C.; Bettica, P.; Germani, M.; Fiorentini, F.; del Bene, F.; Rocchetti, M.; Leoni, F.; Monzani, V.; Mascagni, P.; et al. Preclinical studies in the mdx mouse model of duchenne muscular dystrophy with the histone deacetylase inhibitor givinostat. Mol. Med. 2013, 19, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Percival, J.M.; Whitehead, N.P.; Adams, M.E.; Adamo, C.M.; Beavo, J.A.; Froehner, S.C. Sildenafil reduces respiratory muscle weakness and fibrosis in the mdx mouse model of Duchenne muscular dystrophy. J. Pathol. 2012, 228, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Nelson, M.D.; Rader, F.; Tang, X.; Tavyev, J.; Nelson, S.F.; Miceli, M.C.; Elashoff, R.M.; Sweeney, H.L.; Victor, R.G. PDE5 inhibition alleviates functional muscle ischemia in boys with Duchenne muscular dystrophy. Neurology 2014, 82, 2085–2091. [Google Scholar] [CrossRef] [PubMed]
- Bodanovsky, A.; Guttman, N.; Barzilai-Tutsch, H.; Genin, O.; Levy, O.; Pines, M.; Halevy, O. Halofuginone improves muscle-cell survival in muscular dystrophies. Biochim. Biophys. Acta 2014, 1843, 1339–1347. [Google Scholar] [CrossRef] [PubMed]
- Buyse, G.M.; Goemans, N.; van den Hauwe, M.; Meier, T. Effects of glucocorticoids and idebenone on respiratory function in patients with duchenne muscular dystrophy. Pediatr. Pulmonol. 2013, 48, 912–920. [Google Scholar] [CrossRef] [PubMed]
- Buyse, G.M.; Voit, T.; Schara, U.; Straathof, C.S.; D’Angelo, M.G.; Bernert, G.; Cuisset, J.M.; Finkel, R.S.; Goemans, N.; McDonald, C.M.; et al. Efficacy of idebenone on respiratory function in patients with Duchenne muscular dystrophy not using glucocorticoids (DELOS): A double-blind randomised placebo-controlled phase 3 trial. Lancet 2015, 385, 1748–1757. [Google Scholar] [CrossRef]
- McDonald, C.M.; Meier, T.; Voit, T.; Schara, U.; Straathof, C.S.; D’Angelo, M.G.; Bernert, G.; Cuisset, J.M.; Finkel, R.S.; Goemans, N.; et al. DELOS Study Group. Idebenone reduces respiratory complications in patients with Duchenne muscular dystrophy. Neuromuscul. Disord. 2016, 26, 473–480. [Google Scholar] [CrossRef] [PubMed]
- Kimura, S.; Ito, K.; Miyagi, T.; Hiranuma, T.; Yoshioka, K.; Ozasa, S.; Matsukura, M.; Ikezawa, M.; Matsuo, M.; Takeshima, Y.; et al. A novel approach to identify Duchenne muscular dystrophy patients for aminoglycoside antibiotics therapy. Brain Dev. 2005, 27, 400–405. [Google Scholar] [CrossRef] [PubMed]
- Kayali, R.; Ku, J.M.; Khitrov, G.; Jung, M.E.; Prikhodko, O.; Bertoni, C. Read-through compound 13 restores dystrophin expression and improves muscle function in the mdx mouse model for Duchenne muscular dystrophy. Hum. Mol. Genet. 2012, 21, 4007–4020. [Google Scholar] [CrossRef] [PubMed]
- Finkel, R.S.; Flanigan, K.M.; Wong, B.; Bonnemann, C.; Sampson, J.; Sweeney, H.L.; Reha, A.; Northcutt, V.J.; Elfring, G.; Barth, J.; et al. Phase 2a study of ataluren-mediated dystrophin production in patients with nonsense mutation Duchenne muscular dystrophy. PLoS ONE 2013, 8, e81302. [Google Scholar] [CrossRef] [PubMed]
- Bushby, K.; Finkel, R.; Wong, B.; Barohn, R.; Campbell, C.; Comi, G.P.; Connolly, A.M.; Day, J.W.; Flanigan, K.M.; Goemans, N.; et al. Ataluren treatment of patients with nonsense mutation dystrophinopathy. Muscle Nerve 2014, 50, 477–487. [Google Scholar] [CrossRef] [PubMed]
- McDonald, C.M.; Campbell, C.; Torricelli, R.E.; Finkel, R.S.; Flanigan, K.M.; Goemans, N.; Heydemann, P.; Kaminska, A.; Kirschner, J.; Muntoni, F.; et al. Clinical Evaluator Training Group; ACT DMD Study Group. Ataluren in patients with nonsense mutation Duchenne muscular dystrophy (ACTDMD): A multicentre, randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2017, 390, 1489–1498. [Google Scholar] [CrossRef]
- McPherron, A.C.; Lawler, A.M.; Lee, S.J. Regulation of skeletal muscle mass in mice by a new TGF-β superfamily member. Nature 1997, 387, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Wagner, K.R.; McPherron, A.C.; Winik, N.; Lee, S.J. Loss of myostatin attenuates severity of muscular dystrophy in mdx mice. Ann. Neurol. 2002, 52, 832–836. [Google Scholar] [CrossRef] [PubMed]
- Bogdanovich, S.; Krag, T.O.; Barton, E.R.; Morris, L.D.; Whittemore, L.A.; Ahima, R.S.; Khurana, T.S. Functional improvement of dystrophic muscle by myostatin blockade. Nature 2002, 420, 418–421. [Google Scholar] [CrossRef] [PubMed]
- Singh, P.; Rong, H.; Gordi, T.; Bosley, J.; Bhattacharya, I. Translational Pharmacokinetic/Pharmacodynamic Analysis of MYO-029 Antibody for Muscular Dystrophy. Clin. Transl. Sci. 2016, 9, 302–310. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, I.; Manukyan, Z.; Chan, P.; Heatherington, A.; Harnisch, L. Application of Quantitative Pharmacology Approaches in Bridging Pharmacokinetics and Pharmacodynamics of Domagrozumab From Adult Healthy Subjects to Pediatric Patients With Duchenne Muscular Disease. J. Clin. Pharmacol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Ran, F.A.; Cong, L.; Yan, W.X.; Scott, D.A.; Gootenberg, J.S.; Kriz, A.J.; Zetsche, B.; Shalem, O.; Wu, X.; Makarova, K.S.; et al. In vivo genome editing using Staphylococcus aureus Cas9. Nature 2015, 520, 186–191. [Google Scholar] [CrossRef] [PubMed]
- Nelson, C.E.; Hakim, C.H.; Ousterout, D.G.; Thakore, P.I.; Moreb, E.A.; Castellanos Rivera, R.M.; Madhavan, S.; Pan, X.; Ran, F.A.; Yan, W.X.; et al. In vivo genome editing improves muscle function in a mouse model of Duchenne muscular dystrophy. Science 2016, 351, 403–407. [Google Scholar] [CrossRef] [PubMed]
- Tabebordbar, M.; Zhu, K.; Cheng, J.K.; Chew, W.L.; Widrick, J.J.; Yan, W.X.; Maesner, C.; Wu, E.Y.; Xiao, R.; Ran, F.A.; et al. In vivo gene editing in dystrophic mouse muscle and muscle stem cells. Science 2016, 351, 407–411. [Google Scholar] [CrossRef] [PubMed]
- Van Deutekom, J.C.; van Ommen, G.J. Advances in Duchenne muscular dystrophy gene therapy. Nat. Rev. Genet. 2003, 4, 774–783. [Google Scholar] [CrossRef] [PubMed]
- Bettica, P.; Petrini, S.; D’Oria, V.; D’Amico, A.; Catteruccia, M.; Pane, M.; Sivo, S.; Magri, F.; Brajkovic, S.; Messina, S.; et al. Histological effects of givinostat in boys with Duchenne muscular dystrophy. Neuromuscul. Disord. 2016, 26, 643–649. [Google Scholar] [CrossRef] [PubMed]
- Victor, R.G.; Sweeney, H.L.; Finkel, R.; McDonald, C.M.; Byrne, B.; Eagle, M.; Goemans, N.; Vandenborne, K.; Dubrovsky, A.L.; Topaloglu, H.; et al. Tadalafil DMD Study Group. A phase 3 randomized placebo-controlled trial of tadalafil for Duchenne muscular dystrophy. Neurology 2017, 89, 1811–1820. [Google Scholar] [CrossRef] [PubMed]
- Akashi Administration. Dosing and Enrollment in HT-100 Trial Suspended. Available online: http://akashirx.com/news/dosing-and-enrollment-in-ht-100-trial-suspended (accessed on 14 December 2017).
- Bhattacharya, I.; Pawlak, S.; Marraffino, S.; Christensen, J.; Sherlock, S.P.; Alvey, C.; Morris, C.; Arkin, S.; Binks, M. Safety, tolerability, pharmacokinetics, and pharmacodynamics of domagrozumab (PF-06252616), an antimyostatin monoclonal antibody, in healthy subjects. Clin. Pharmacol. Drug Dev. 2017. [Google Scholar] [CrossRef] [PubMed]
- Bengtsson, N.E.; Hall, J.K.; Odom, G.L.; Phelps, M.P.; Andrus, C.R.; Hawkins, R.D.; Hauschka, S.D.; Chamberlain, J.R.; Chamberlain, J.S. Muscle-specific CRISPR/Cas9 dystrophin gene editing ameliorates pathophysiology in a mouse model for Duchenne muscular dystrophy. Nat. Commun. 2017, 8, 14454. [Google Scholar] [CrossRef] [PubMed]
- Russell, L.D.; Griswold, M.D. The Sertoli Cell; Cache River Press: Clearwater, FL, USA, 1993. [Google Scholar]
- Mital, P.; Kaur, G.; Dufour, J.M. Immunoprotective Sertoli cells: Making allogeneic and xenogeneic transplantation feasible. Reproduction 2010, 139, 495–504. [Google Scholar] [CrossRef] [PubMed]
- Qvist, G. John Hunter 1728–1793; William Heinemann Medical Books Ltd.: London, UK, 1981; ISBN 13:9780433270959. [Google Scholar]
- Meinhardt, A.; Hedger, M.P. Immunological, paracrine and endocrine aspects of testicular immune privilege. Mol. Cell. Endocrinol. 2011, 335, 60–68. [Google Scholar] [CrossRef] [PubMed]
- Kaur, G.; Thompson, L.A.; Dufour, J.M. Sertoli cells—Immunological sentinels of spermatogenesis. Semin. Cell Dev. Biol. 2014, 30, 36–44. [Google Scholar] [CrossRef] [PubMed]
- Selawry, H.P.; Cameron, D.F. Sertoli cell-enriched fractions in successful islet cell transplantation. Cell Transplant. 1993, 2, 123–129. [Google Scholar] [CrossRef] [PubMed]
- Dufour, J.M.; Rajotte, R.V.; Kin, T.; Korbutt, G.S. Immunoprotection of rat islet xenografts by cotransplantation with Sertoli cells and a single injection of antilymphocyte serum. Transplantation 2003, 75, 1594–1596. [Google Scholar] [CrossRef] [PubMed]
- Shamekh, R.; El-Badri, N.S.; Saporta, S.; Pascual, C.; Sanberg, P.R.; Cameron, D.F. Sertoli cells induce systemic donor-specific tolerance in xenogenic transplantation model. Cell Transplant. 2006, 15, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Sanberg, P.R.; Borlongan, C.V.; Saporta, S.; Cameron, D.F. Testis-derived Sertoli cells survive and provide localized immunoprotection for xenografts in rat brain. Nat. Biotechnol. 1996, 14, 1692–1695. [Google Scholar] [CrossRef] [PubMed]
- Willing, A.E.; Sudberry, J.J.; Othberg, A.I.; Saporta, S.; Poulos, S.G.; Poulos, S.G.; Cameron, D.F.; Freeman, T.B.; Sanberg, P.R. Sertoli cells decrease microglial response and increase engraftment of human hNT neurons in the hemiparkinsonian rat striatum. Brain Res. Bull. 1999, 48, 441–444. [Google Scholar] [CrossRef]
- Lee, H.M.; Lim, H.G.; Oh, B.C.; Park, C.S.; Lee, D.S.; Lee, J.R. Systemic immune modulation using chemokine receptor 7 expressing porcine Sertoli cells. Xenotransplantation 2007, 14, 619–626. [Google Scholar] [CrossRef] [PubMed]
- Lim, H.G.; Lee, H.M.; Oh, B.C.; Lee, J.R. Cell-mediated immunomodulation of chemokine receptor 7-expressing porcine sertoli cells in murine heterotopic heart transplantation. J. Heart Lung Transplant. 2009, 28, 72–78. [Google Scholar] [CrossRef] [PubMed]
- Valdés-González, R.A.; White, D.J.; Dorantes, L.M.; Terán, L.; Garibay-Nieto, G.N.; Bracho-Blanchet, E.; Dávila-Pérez, R.; Evia-Viscarra, L.; Ormsby, C.E.; Ayala-Sumuano, J.T.; et al. Three-yr follow-up of a type 1 diabetes mellitus patient with an islet xenotransplant. Clin. Transplant. 2007, 21, 352–357. [Google Scholar] [CrossRef] [PubMed]
- Esquivel-Pérez, R.; Rodriguez-Ventura, A.L.; Dorantes, L.M.; Ramírez-González, B.; López-Santos, M.G.; Valdés-Gonzalez, R. Correlation between insulin requirements and anti-galactose antibodies in patients with type 1 diabetes transplanted with neonatal pig islets. Clin. Exp. Immunol. 2011, 165, 104–109. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, A.I.; Willing, A.E.; Saporta, S.; Cameron, D.F.; Sanberg, P.R. Effects of Sertoli cells transplants in a 3 nitroproprionic acid model of early Huntington’s disease: A preliminary study. Neurotox. Res. 2003, 5, 443–450. [Google Scholar] [CrossRef] [PubMed]
- Hemendinger, R.; Wang, J.; Malik, S.; Persinski, R.; Copeland, J.; Emerich, D.; Gores, P.; Halberstadt, C.; Rosenfeld, J. Sertoli cells improve survival of motor neurons in SOD1 transgenic mice, a model of amyotrophic lateral sclerosis. Exp. Neurol. 2005, 196, 235–243. [Google Scholar] [CrossRef] [PubMed]
- Selawry, H.P.; Kotb, M.; Herrod, H.G.; Lu, Z.N. Production of a factor, or factors, suppressing IL-2 production and T cell proliferation by Sertoli cell-enriched preparations. A potential role for islet transplantation in an immunologically privileged site. Transplantation 1991, 52, 846–850. [Google Scholar] [CrossRef] [PubMed]
- De Cesaris, P.; Filippini, A.; Cervelli, C.; Riccioli, A.; Muci, S.; Starace, G.; Stefanini, M.; Ziparo, E. Immunosuppressive molecules produced by Sertoli cells cultured in vitro: Biological effects on lymphocytes. Biochem. Biophys. Res. Commun. 1992, 186, 1639–1646. [Google Scholar] [CrossRef]
- Takeda, Y.; Gotoh, M.; Dono, K.; Nishihara, M.; Grochowiecki, T.; Kimura, F.; Yoshida, T.; Ohta, Y.; Ota, H.; Ohzato, H.; et al. Protection of islet allografts transplanted together with Fas ligand expressing testicular allografts. Diabetologia 1998, 41, 315–321. [Google Scholar] [CrossRef] [PubMed]
- Suarez-Pinzon, W.; Korbutt, G.S.; Power, R.; Hooton, J.; Rajotte, R.V.; Rabinovitch, A. Testicular Sertoli cells protect islet β-cells from autoimmune destruction in NOD mice by a transforming growth factor beta1- dependent mechanism. Diabetes 2000, 49, 1810–1818. [Google Scholar] [CrossRef] [PubMed]
- Fallarino, F.; Luca, G.; Calvitti, M.; Mancuso, F.; Nastruzzi, C.; Fioretti, M.C.; Grohmann, U.; Becchetti, E.; Burgevin, A.; Kratzer, R.; et al. Therapy of experimental type 1 diabetes by isolated Sertoli cell xenografts alone. J. Exp. Med. 2009, 206, 2511–2526. [Google Scholar] [CrossRef] [PubMed]
- Winnall, W.R.; Muir, J.A.; Hedger, M.P. Rat resident testicular macrophages have an alternatively activated phenotype and constitutively produce interleukin-10 in vitro. J. Leukoc. Biol. 2011, 90, 133–143. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Feng, X.; Han, D. Mechanisms of testicular immune privilege. Front. Biol. 2011, 6, 19–30. [Google Scholar] [CrossRef]
- Campese, A.F.; Grazioli, P.; de Cesaris, P.; Riccioli, A.; Bellavia, D.; Pelullo, M.; Padula, F.; Noce, C.; Verkhovskaia, S.; Filippini, A.; et al. Mouse Sertoli cells sustain de novo generation of regulatory T cells by triggering the notch pathway through soluble JAGGED1. Biol. Reprod. 2014, 90, 53. [Google Scholar] [CrossRef] [PubMed]
- Dufour, J.M.; Hamilton, M.; Rajotte, R.V.; Korbutt, G.S. Neonatal porcine Sertoli cells inhibit human natural antibody-mediated lysis. Biol. Reprod. 2005, 72, 1224–1231. [Google Scholar] [CrossRef] [PubMed]
- Sipione, S.; Simmen, K.C.; Lord, S.J.; Motyka, B.; Ewen, C.; Shostak, I.; Rayat, G.R.; Dufour, J.M.; Korbutt, G.S.; Rajotte, R.V.; et al. Identification of a novel human granzyme B inhibitor secreted by cultured sertoli cells. J. Immunol. 2006, 177, 5051–5058. [Google Scholar] [CrossRef] [PubMed]
- Riccioli, A.; Starace, D.; Galli, R.; Fuso, A.; Scarpa, S.; Palombi, F.; de Cesaris, P.; Ziparo, E.; Filippini, A. Sertoli cells initiate testicular innate immune responses through TLR activation. J. Immunol. 2006, 177, 7122–7130. [Google Scholar] [CrossRef] [PubMed]
- Paredes Juárez, G.A.; Spasojevic, M.; Faas, M.M.; de Vos, P. Immunological and technical considerations in application of alginate-based microencapsulation systems. Front. Bioeng. Biotechnol. 2014, 2, 26. [Google Scholar] [CrossRef] [PubMed]
- Lim, F.; Sun, A.M. Microencapsulated islets as bioartificial endocrine pancreas. Science 1980, 210, 908–910. [Google Scholar] [CrossRef] [PubMed]
- Orive, G.; Hernández, R.M.; Rodríguez Gascón, A.; Calafiore, R.; Chang, T.M.; de Vos, P.; Hortelano, G.; Hunkeler, D.; Lacík, I.; Pedraz, J.L. History, challenges and perspectives of cell microencapsulation. Trends Biotechnol. 2004, 22, 87–92. [Google Scholar] [CrossRef] [PubMed]
- Calafiore, R.; Basta, G.; Luca, G.; Lemmi, A.; Montanucci, M.P.; Calabrese, G.; Racanicchi, L.; Mancuso, F.; Brunetti, P. Microencapsulated pancreatic islet allografts into nonimmunosuppressed patients with type 1 diabetes: First two cases. Diabetes Care 2006, 29, 137–138. [Google Scholar] [CrossRef] [PubMed]
- Calafiore, R.; Basta, G.; Luca, G.; Lemmi, A.; Racanicchi, L.; Mancuso, F.; Montanucci, M.P.; Brunetti, P. Standard technical procedures for microencapsulation of human islets for graft into nonimmunosuppressed patients with type 1 diabetes mellitus. Transplant. Proc. 2006, 38, 1156–1157. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.Y.; Mooney, D.J. Alginate: Properties and biomedical applications. Prog. Polym. Sci. 2012, 37, 106–126. [Google Scholar] [CrossRef] [PubMed]
- Remminghorst, U.; Rehm, B.H. Bacterial alginates: From biosynthesis to applications. Biotechnol. Lett. 2006, 28, 1701–1712. [Google Scholar] [CrossRef] [PubMed]
- Orive, G.; Ponce, S.; Hernández, R.M.; Gascón, A.R.; Igartua, M.; Pedraz, J.L. Biocompatibility of microcapsules for cell immobilization elaborated with different type of alginates. Biomaterials 2002, 23, 3825–3831. [Google Scholar] [CrossRef]
- Zimmermann, H.; Shirley, S.G.; Zimmermann, U. Alginate-based encapsulation of cells: Past, present, and future. Curr. Diabetes Rep. 2007, 7, 314–320. [Google Scholar] [CrossRef]
- Luca, G.; Calvitti, M.; Nastruzzi, C.; Bilancetti, L.; Becchetti, E.; Angeletti, G.; Mancuso, F.; Calafiore, R. Encapsulation, in vitro characterization, and in vivo biocompatibility of Sertoli cells in alginate-based microcapsules. Tissue Eng. 2007, 13, 641–648. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, T.; Aomatsu, Y.; Iwata, H.; Kin, T.; Kanehiro, H.; Hisanga, M.; Ko, S.; Nagao, M.; Harb, G.; Nakajima, Y. Survival of microencapsulated islets at 400 days posttransplantation in the omental pouch of NOD mice. Cell Transplant. 2006, 15, 359–365. [Google Scholar] [CrossRef] [PubMed]
- Basta, G.; Montanucci, P.; Luca, G.; Boselli, C.; Noya, G.; Barbaro, B.; Qi, M.; Kinzer, K.P.; Oberholzer, J.; Calafiore, R. Long-term metabolic and immunological follow-up of nonimmunosuppressed patients with type 1 diabetes treated with microencapsulated islet allografts: Four cases. Diabetes Care 2011, 34, 2406–2409. [Google Scholar] [CrossRef] [PubMed]
- Luca, G.; Calvitti, M.; Mancuso, F.; Falabella, G.; Arato, I.; Bellucci, C.; List, E.O.; Bellezza, E.; Angeli, G.; Lilli, C.; et al. Reversal of experimental Laron Syndrome by xenotransplantation of microencapsulated porcine Sertoli cells. J. Controll. Release 2013, 165, 75–81. [Google Scholar] [CrossRef] [PubMed]
- Chiappalupi, S.; Luca, G.; Mancuso, F.; Madaro, L.; Fallarino, F.; Nicoletti, C.; Calvitti, M.; Arato, I.; Falabella, G.; Salvadori, L.; et al. Intraperitoneal injection of microencapsulated Sertoli cells restores muscle morphology and performance in dystrophic mice. Biomaterials 2016, 75, 313–326. [Google Scholar] [CrossRef] [PubMed]
- Soon-Shiong, P.; Heintz, R.E.; Merideth, N.; Yao, Q.X.; Yao, Z.; Zheng, T.; Murphy, M.; Moloney, M.K.; Schmehl, M.; Harris, M.; et al. Insulin independence in a type 1 diabetic patient after encapsulated islet transplantation. Lancet 1994, 343, 950–951. [Google Scholar] [CrossRef]
- Luca, G.; Calafiore, R.; Basta, G.; Ricci, M.; Calvitti, M.; Neri, L.; Nastruzzi, C.; Becchetti, E.; Capitani, S.; Brunetti, P.; et al. Improved function of rat islets upon co-microencapsulation with Sertoli’s cells in alginate/poly-Lornithine. AAPS PharmSciTech 2001, 2, E15. [Google Scholar] [CrossRef] [PubMed]
- Rahman, T.M.; Diakanov, I.; Selden, C.; Hodgson, H. Cotransplantation of encapsulated HepG2 and rat Sertoli cells improves outcome in a thioacetamide induced rat model of acute hepatic failure. Transplant. Int. 2005, 18, 1001–1009. [Google Scholar] [CrossRef] [PubMed]
- Luca, G.; Fallarino, F.; Calvitti, M.; Mancuso, F.; Nastruzzi, C.; Arato, I.; Falabella, G.; Grohmann, U.; Becchetti, E.; Puccetti, P.; et al. Xenograft of microencapsulated sertoli cells reverses T1DM in NOD mice by inducing neogenesis of β-cells. Transplantation 2010, 90, 1352–1357. [Google Scholar] [CrossRef] [PubMed]
- Bistoni, G.; Calvitti, M.; Mancuso, F.; Arato, I.; Falabella, G.; Cucchia, R.; Fallarino, F.; Becchetti, A.; Baroni, T.; Mazzitelli, S.; et al. Prolongation of skin allograft survival in rats by the transplantation of microencapsulated xenogeneic neonatal porcine Sertoli cells. Biomaterials 2012, 33, 5333–5340. [Google Scholar] [CrossRef] [PubMed]
- Luca, G.; Bellezza, I.; Arato, I.; di Pardo, A.; Mancuso, F.; Calvitti, M.; Falabella, G.; Bartoli, S.; Maglione, V.; Amico, E.; et al. Terapeutic Potential of Microencapsulated Sertoli Cells in Huntington Disease. CNS Neurosci. Ther. 2016, 22, 686–690. [Google Scholar] [CrossRef] [PubMed]
- Luca, G.; Cameron, D.F.; Arato, I.; Mancuso, F.; Linden, E.H.; Calvitti, M.; Falabella, G.; Szekeres, K.; Bodo, M.; Ricci, G.; et al. Xenograft of microencapsulated Sertoli cells for the cell therapy of type 2 diabetes mellitus in spontaneously diabetic nonhuman primates: Preliminary data. Transplant. Proc. 2014, 46, 1999–2001. [Google Scholar] [CrossRef] [PubMed]
- Chiappalupi, S.; Luca, G.; Mancuso, F.; Madaro, L.; Fallarino, F.; Nicoletti, C.; Calvitti, M.; Arato, I.; Falabella, G.; Salvadori, L.; et al. Effects of intraperitoneal injection of microencapsulated Sertoli cells on chronic and presymptomatic dystrophic mice. Data Brief 2015, 15, 1015–1021. [Google Scholar] [CrossRef] [PubMed]
- Emery, A.E.H. Duchenne Muscular Dystrophy, 2nd ed.; Oxford University Press: Oxford, UK, 1993. [Google Scholar]
- Musarò, A.; McCullagh, K.; Paul, A.; Houghton, L.; Dobrowolny, G.; Molinaro, M.; Barton, E.R.; Sweeney, H.L.; Rosenthal, N. Localized IGF-1 transgene expression sustains hypertrophy and regeneration in senescent skeletal muscle. Nat. Genet. 2001, 27, 195–200. [Google Scholar] [CrossRef] [PubMed]



© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).