Role of Peripheral Immune Cells in Multiple Sclerosis and Experimental Autoimmune Encephalomyelitis


 ,
, 
{kind=link}
{kind=link}
Abstract
:1. Introduction
Diagnosis of MS Disorder
2. Innate Immunity in MS
2.1. Role of TLRs in MS
2.2. NOD-Like Receptors in MS
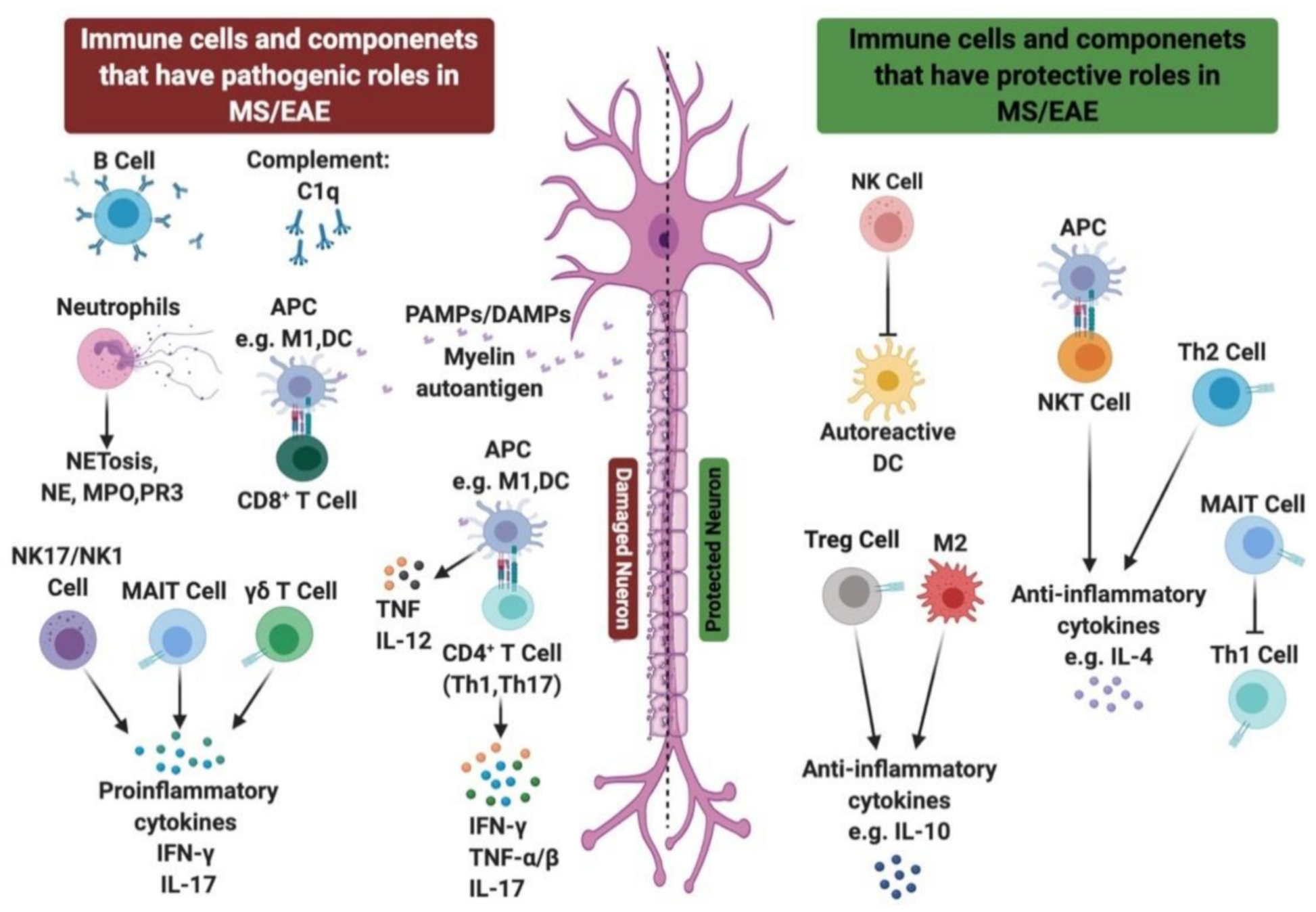
2.3. Role of Innate Immune Cells in MS Disease
2.3.1. Role of Neutrophils in MS/EAE
2.3.2. Role of NK Cells in MS/EAE
2.3.3. Role of NKT Cells in MS/EAE
2.3.4. Role of γδ T Cells in MS/EAE
2.3.5. Role of MAIT Cells in MS/EAE
2.4. Role of Complement System in MS
3. Role of Adaptive Immune Cells in MS
3.1. Role of CD4+ T Cells in MS and EAE
3.2. Role of CD8+ Cells in MS
3.3. Role of Regulatory T Cells (Tregs) in MS or EAE
3.4. Role of B Cells in MS/EAE
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
Abbreviations
| AIM2 | Absent in melanoma 2 |
| APCs | Antigen-presenting cells |
| ALLO | Allopregnanolone |
| ASC | Apoptosis-associated speck-like protein |
| BBB | Blood–brain barrier |
| CIS | Clinical isolated syndrome |
| CNS | Central nervous system |
| CSF | Cerebrospinal fluid |
| CTLA-4 | Cytotoxic T-lymphocyte-associated protein 4 |
| DAMPs | Danger-associated molecular patterns |
| DCs | Dendritic cells |
| DMF | Dimethyl fumarate |
| EAE | Experimental autoimmune encephalitis |
| G-CSF | Granulocyte-colony stimulating factor |
| GA | Glatiramer acetate |
| GABA-A | Gamma aminobutyric acid-A |
| GalCer | Galactosylceramide |
| GWAS | Genome-wide association studies |
| HLA | Human leukocyte antigen |
| IFN | Interferon |
| IL- | Interleukin |
| Ig | Immunoglobulin |
| LFA-1 | Lymphocyte function-associated antigen 1 |
| MAIT | Mucosal-associated invariant T |
| MBP | Myelin basic protein |
| MHC | Major histocompatibility complex |
| MMF | Monomethyl fumarate |
| MOG | Myelin oligodendrocyte glycoprotein |
| MPO | Myeloperoxidase |
| MR1 | MHC-related molecule 1 |
| MRI | Magnetic resonance imaging |
| MS | Multiple sclerosis |
| NETs | Neutrophils’ extracellular traps |
| NK | Natural killer |
| NKT | Natural killer T |
| NLRs | NOD-like receptors |
| NOD | Nucleotide binding oligomerization domain |
| PAMPs | Pathogen-associated molecular patterns |
| PPMS | Primary progressive multiple sclerosis |
| PRMS | Progressive relapsing multiple sclerosis |
| PRRs | Pattern recognition receptors |
| ROS | Reactive oxygen species |
| RRMS | Relapsing remitting multiple sclerosis |
| SPMS | Secondary progressive multiple sclerosis |
| TCR | T cell receptor |
| Th | T helper |
| TLR | Toll-like receptor |
| TNF | Tumor necrosis factor |
| Tregs | Regulatory T cells |
| VLA-4 | Very late antigen-4. |
References
- Kobelt, G.; Thompson, A.; Berg, J.; Gannedahl, M.; Eriksson, J. New insights into the burden and costs of multiple sclerosis in Europe. Mult. Scler. 2017, 23, 1123–1136. [Google Scholar] [CrossRef] [PubMed]
- Popescu, B.F.; Pirko, I.; Lucchinetti, C.F. Pathology of multiple sclerosis. Continuum 2013, 19, 901–921. [Google Scholar] [PubMed]
- Lublin, F.D.; Reingold, S.C.; Cohen, J.A.; Cutter, G.R.; Sorensen, P.S.; Thompson, A.J.; Wolinsky, J.S.; Balcer, L.J.; Banwell, B.; Barkhof, F.; et al. Defining the clinical course of multiple sclerosis: The 2013 revisions. Neurology 2014, 83, 278–286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, D.H.; Chard, D.T.; Ciccarelli, O. Clinically isolated syndromes. Lancet Neurol. 2012, 11, 157–169. [Google Scholar] [CrossRef]
- Gourraud, P.; Harbo, H.F.; Hauser, S.L.; Baranzini, S.E. The genetics of multiple sclerosis: An up-to-date review. Immunol. Rev. 2012, 248, 87–103. [Google Scholar] [CrossRef] [Green Version]
- Holmøy, T.; Hestvik, A.L. Multiple sclerosis: Immunopathogenesis and controversies in defining the cause. Curr. Opin. Infect. Dis. 2008, 21, 271–278. [Google Scholar] [CrossRef]
- Egg, R.; Reindl, M.; Deisenhammer, F.; Linington, C.; Berger, T. Anti-MOG and anti-MBP antibody subclasses in multiple sclerosis. Mult. Scler. 2001, 7, 285–289. [Google Scholar] [CrossRef]
- Robinson, A.P.; Harp, C.T.; Noronha, A.; Miller, S.D. The experimental autoimmune encephalomyelitis (EAE) model of MS: Utility for understanding disease pathophysiology and treatment. Handb. Clin. Neurol. 2014, 122, 173–189. [Google Scholar]
- Giralt, M.; Molinero, A.; Hidalgo, J. Active induction of experimental autoimmune encephalomyelitis (EAE) with MOG35-55 in the mouse. Methods Mol. Biol. 2018, 1791, 227–232. [Google Scholar]
- Farooqi, N.; Gran, B.; Constantinescu, C.S. Are current disease-modifying therapeutics in multiple sclerosis justified on the basis of studies in experimental autoimmune encephalomyelitis? J. Neurochem. 2010, 115, 829–844. [Google Scholar] [CrossRef]
- Polman, C.H.; O’Connor, P.W.; Havrdova, E.; Hutchinson, M.; Kappos, L.; Miller, D.H.; Phillips, J.T.; Lublin, F.D.; Giovannoni, G.; Sandrock, A.W.; et al. A randomized, placebo-controlled trial of natalizumab for relapsing multiple sclerosis. N. Engl. J. Med. 2006, 354, 899–910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yednock, T.A.; Cannon, C.; Fritz, L.C.; Sanchez-Madrid, F.; Steinman, L.; Karin, N. Prevention of experimental autoimmune encephalomyelitis by antibodies against alpha 4 beta 1 integrin. Nature 1992, 356, 63–66. [Google Scholar] [CrossRef] [PubMed]
- Ridge, S.C.; Sloboda, A.E.; McReynolds, R.A.; Levine, S.; Oronsky, A.L.; Kerwar, S.S. Suppression of experimental allergic encephalomyelitis by mitoxantrone. J. Clin. Immunol. Immunopathol. Res. 1985, 35, 35–42. [Google Scholar] [CrossRef]
- Huang, W.; Chen, W.; Zhang, X. Multiple sclerosis: Pathology, diagnosis and treatments. Exp. Ther. Med. 2017, 13, 3163–3166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghasemi, N.; Razavi, S.; Nikzad, E. Multiple sclerosis: Pathogenesis, symptoms, diagnoses and cell-based therapy. Cell J. 2016, 19, 1–10. [Google Scholar]
- Calabresi, P.A. Diagnosis and management of multiple sclerosis. Am. Fam. Physician 2004, 70, 1935–1944. [Google Scholar]
- Olson, J.K.; Miller, S.D. The innate immune response affects the development of the autoimmune response in Theiler’s virus-induced demyelinating disease. J. Immunol. 2009, 182, 5712–5722. [Google Scholar] [CrossRef]
- Tosi, M.F. Innate immune responses to infection. J. Allergy Clin. Immunol. 2005, 116, 241–250. [Google Scholar] [CrossRef]
- Deerhake, M.E.; Biswas, D.D.; Barclay, W.E.; Shinohara, M.L. Pattern recognition receptors in multiple sclerosis and its animal models. Front. Immunol. 2019, 10, 2644. [Google Scholar] [CrossRef] [Green Version]
- Pone, E.J.; Zan, H.; Zhang, J.; Al-Qahtani, A.; Xu, Z.; Casali, P. Toll-like receptors and B-cell receptors synergize to induce immunoglobulin class-switch DNA recombination: Relevance to microbial antibody responses. Crit. Rev. Immunol. 2010, 30, 1–29. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Meng, Z.; Jiang, M.; Zhang, E.; Trippler, M.; Broering, R.; Bucchi, A.; Krux, F.; Dittmer, U.; Yang, D.; et al. Toll-like receptor-induced innate immune responses in non-parenchymal liver cells are cell type-specific. Immunology 2010, 129, 363–374. [Google Scholar] [CrossRef] [PubMed]
- Suresh, R.; Mosser, D.M. Pattern recognition receptors in innate immunity, host defense, and immunopathology. Adv. Physiol. Educ. 2013, 37, 284–291. [Google Scholar] [CrossRef] [PubMed]
- Kigerl, K.A.; Lai, W.; Rivest, S.; Hart, R.P.; Satoskar, A.R.; Popovich, P.G. Toll-like receptor (TLR)-2 and TLR-4 regulate inflammation, gliosis, and myelin sparing after spinal cord injury. J. Neurochem. 2007, 102, 37–50. [Google Scholar] [CrossRef] [PubMed]
- Sloane, J.A.; Batt, C.; Ma, Y.; Harris, Z.; Trapp, B.; Vartanian, T. Hyaluronan blocks oligodendrocyte progenitor maturation and remyelination through TLR2. Proc. Natl. Acad. Sci. USA 2010, 107, 11555–11560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanafy, K.H.; Sloane, J.A. Regulation of remyelination in multiple sclerosis. FEBS Lett. 2011, 585, 3821–3828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Back, S.A.; Tuohy, T.M.; Chen, H.; Wallingford, N.; Craig, A.; Struve, J.; Luo, N.; Banine, F.; Liu, Y.; Chang, A.; et al. Hyaluronan accumulates in demyelinated lesions and inhibits oligodendrocyte progenitor maturation. Nat. Med. 2005, 11, 966–972. [Google Scholar] [CrossRef]
- Zheng, C.; Chen, J.; Chu, F.; Zhu, J.; Jin, T. Inflammatory role of TLR-Myd88 signaling in multiple sclerosis. Front. Mol. Neurosci. 2020, 12, 314. [Google Scholar] [CrossRef]
- Marta, M. Toll-like receptors in multiple sclerosis mouse experimental models. Ann. N. Y. Acad. Sci. 2009, 1173, 458–462. [Google Scholar] [CrossRef]
- Touil, T.; Fitzgerald, D.; Zhang, G.; Rostami, A.; Gran, B. Cutting Edge: TLR3 stimulation suppresses experimental autoimmune encephalomyelitis by inducing endogenous IFN-β. J. Immunol. 2006, 177, 7505–7509. [Google Scholar] [CrossRef] [Green Version]
- Giacomini, E.; Severa, M.; Rizzo, F.; Mechelli, R.; Annibali, V.; Ristori, G.; Riccieri, V.; Salvetti, M.; Coccia, E.M. IFN-β therapy modulates B-cell and monocyte crosstalk via TLR7 in multiple sclerosis patients. Eur. J. Immunol. 2013, 43, 1963–1972. [Google Scholar] [CrossRef]
- Zhang, X.; Jin, J.; Tang, Y.; Speer, D.; Sujkowska, D.; Markovic-Plese, S. IFN-β1a inhibits the secretion of Th17-polarizing cytokines in human dendritic cells via TLR7 up-regulation. J. Immunol. 2009, 182, 3928–3936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, Y.; Shin, J.; Nahm, M.H. NOD-like receptors in infection, immunity, and diseases. Yonsei Med. J. 2016, 57, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hachim, M.Y.; Khalil, B.A.; Elemam, N.M.; Maghazachi, A.A. Pyroptosis: The missing puzzle among innate and adaptive immunity crosstalk. J. Leukoc. Biol. 2020, 108, 323–338. [Google Scholar] [CrossRef] [PubMed]
- Franchi, L.; Eigenbrod, T.; Muñoz-Planillo, R.; Nuñez, G. The inflammasome: A caspase-1-activation platform that regulates immune responses and disease pathogenesis. Nat. Immunol. 2009, 10, 241–247. [Google Scholar] [CrossRef]
- Hernández-Pedro, N.Y.; Espinosa-Ramirez, G.; de la Cruz, V.; Pineda, B.; Sotelo, J. Initial immunopathogenesis of multiple sclerosis: Innate immune response. Clin. Dev. Immunol. 2013, 2013, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Muhammad, J.S.; Jayakumar, M.N.; Elemam, N.M.; Venkatachalam, T.; Raju, T.K.; Hamoudi, R.A.; Maghazachi, A.A. Gasdermin D hypermethylation inhibits pyroptosis and LPS-induced IL-1β release from NK92 cells. ImmunoTargets Ther. 2019, 8, 29–41. [Google Scholar] [CrossRef] [Green Version]
- Yap, J.K.; Pickard, B.S.; Chan, E.W.; Gan, S. The role of neuronal NLRP1 inflammasome in Alzheimer’s disease: Bringing neurons into the neuroinflammation game. Mol. Neurobiol. 2019, 56, 7741–7753. [Google Scholar] [CrossRef]
- Tan, M.; Yu, J.; Jiang, T.; Zhu, X.; Tan, L. The NLRP3 inflammasome in alzheimer’s disease. Mol. Neurobiol. 2013, 48, 875–882. [Google Scholar] [CrossRef]
- Liu, F.; Li, Z.; He, X.; Yu, H.; Feng, J. Ghrelin attenuates neuroinflammation and demyelination in experimental autoimmune encephalomyelitis involving NLRP3 inflammasome signaling pathway and pyroptosis. Front. Pharmacol. 2019, 10, 1320. [Google Scholar] [CrossRef] [Green Version]
- Gaudino, S.J.; Kumar, P. Cross-talk between antigen presenting cells and T cells impacts intestinal homeostasis, bacterial infections, and tumorigenesis. Front. Immunol. 2019, 10, 360. [Google Scholar] [CrossRef] [Green Version]
- Raphael, I.; Nalawade, S.; Eagar, T.N.; Forsthuber, T.G. T cell subsets and their signature cytokines in autoimmune and inflammatory diseases. Cytokine 2015, 74, 5–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mayo, L.; Quintana, F.J.; Weiner, H.L. The innate immune system in demyelinating disease. Immunol. Rev. 2012, 248, 170–187. [Google Scholar] [CrossRef] [PubMed]
- Vogel, D.Y.; Vereyken, E.J.; Glim, J.E.; Heijnen, P.D.; Moeton, M.; van der Valk, P.; Amor, S.; Teunissen, C.E.; van Horssen, J.; Dijkstra, C.D. Macrophages in inflammatory multiple sclerosis lesions have an intermediate activation status. J. Neuroinflamm. 2013, 10, 809. [Google Scholar] [CrossRef] [Green Version]
- Vainchtein, I.D.; Vinet, J.; Brouwer, N.; Brendecke, S.; Biagini, G.; Biber, K.; Boddeke, H.W.; Eggen, B.J. In acute experimental autoimmune encephalomyelitis, infiltrating macrophages are immune activated, whereas microglia remain immune suppressed. Glia 2014, 62, 1724–1735. [Google Scholar] [CrossRef] [PubMed]
- Chu, F.; Shi, M.; Zheng, C.; Shen, D.; Zhu, J.; Zheng, X.; Cui, L. The roles of macrophages and microglia in multiple sclerosis and experimental autoimmune encephalomyelitis. J. Neuroimmunol. 2018, 318, 1–7. [Google Scholar] [CrossRef]
- Karni, A.; Abraham, M.; Monsonego, A.; Cai, G.; Freeman, G.J.; Hafler, D.; Khoury, S.J.; Weiner, H.L. Innate immunity in multiple sclerosis: Myeloid dendritic cells in secondary progressive multiple sclerosis are activated and drive a proinflammatory immune response. J. Immunol. 2006, 177, 4196–4202. [Google Scholar] [CrossRef] [Green Version]
- Noorbakhsh, F.; Baker, G.B.; Power, C. Allopregnanolone and neuroinflammation: A focus on multiple sclerosis. Front. Cell Neurosci. 2014, 8, 134. [Google Scholar] [CrossRef] [Green Version]
- Reyes-García, M.G.; Hernández-Hernández, F.; Hernández-Téllez, B.; García-Tamayo, F. GABA receptor subunits RNA expression in mice peritoneal macrophages modulate their IL-6/IL-12 production. J. Neuroimmunol. 2007, 188, 64–68. [Google Scholar] [CrossRef]
- Bhat, R.; Axtell, R.; Mitra, A.; Miranda, M.; Lock, C.; Tsien, R.W.; Steinman, L. Inhibitory role for GABA in autoimmune inflammation. Proc. Natl. Acad. Sci. USA 2010, 107, 2580–2585. [Google Scholar] [CrossRef] [Green Version]
- Legroux, L.; Arbour, N. Multiple sclerosis and T lymphocytes: An entangled story. J. Neuroimmune Pharmacol. 2015, 10, 528–546. [Google Scholar] [CrossRef] [Green Version]
- Rumble, J.M.; Huber, A.K.; Krishnamoorthy, G.; Srinivasan, A.; Giles, D.A.; Zhang, X.; Wang, L.; Segal, B.M. Neutrophil-related factors as biomarkers in EAE and MS. J. Exp. Med. 2015, 212, 23–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, Y.; Yang, F.; Sun, E. Neutrophil extracellular traps in autoimmune diseases. Chin. Med. J. 2018, 131, 1513–1519. [Google Scholar] [CrossRef] [PubMed]
- Strzepa, A.; Dittel, B.N. Inflammatory disease severity is ameliorated by inhibition of neutrophil-derived MPO that supports endothelial/epithelial integrity. J. Immunol. 2017, 198, 127. [Google Scholar]
- Yu, G.; Zheng, S.; Zhang, H. Inhibition of myeloperoxidase by N-acetyl lysyltyrosylcysteine amide reduces experimental autoimmune encephalomyelitis-induced injury and promotes oligodendrocyte regeneration and neurogenesis in a murine model of progressive multiple sclerosis. NeuroReport 2018, 29, 208–213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Ray, A.; Miller, N.M.; Hartwig, D.; Pritchard, K.A.; Dittel, B.N. Inhibition of myeloperoxidase at the peak of experimental autoimmune encephalomyelitis restores blood-brain barrier integrity and ameliorates disease severity. J. Neurochem. 2015, 136, 826–836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herges, K.; de Jong, B.A.; Kolkowitz, I.; Dunn, C.; Mandelbaum, G.; Ko, R.M.; Maini, A.; Han, M.H.; Killestein, J.; Polman, C.; et al. Protective effect of an elastase inhibitor in a neuromyelitis optica-like disease driven by a peptide of myelin oligodendroglial glycoprotein. Mult. Scler. 2012, 18, 398–408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minohara, M.; Matsuoka, T.; Li, W.; Osoegawa, M.; Ishizu, T.; Ohyagi, Y.; Kira, J. Upregulation of myeloperoxidase in patients with opticospinal multiple sclerosis: Positive correlation with disease severity. J. Neuroimmunol. 2006, 178, 156–160. [Google Scholar] [CrossRef]
- Maghazachi, A.A. Compartmentalization of human natural killer cells. Mol. Immunol. 2005, 42, 523–529. [Google Scholar] [CrossRef]
- Glimcher, L.; Shen, F.W.; Cantor, H. Identification of a cell-surface antigen selectively expressed on the natural killer cell. J. Exp. Med. 1977, 145, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Maghazachi, A.A. Role of natural killer cells in multiple sclerosis. ISRN Immunol. 2012, 2012, 1–14. [Google Scholar] [CrossRef]
- Høglund, R.A.; Maghazachi, A.A. Multiple sclerosis and the role of immune cells. World J. Exp. Med. 2014, 4, 27. [Google Scholar] [CrossRef] [PubMed]
- Pandya, A.D.; Al-Jaderi, Z.; Høglund, R.A.; Holmøy, T.; Harbo, H.F.; Norgauer, J.; Maghazachi, A.A. Identification of human NK17/NK1 cells. PLoS ONE 2011, 6, e26780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sand, K.L.; Knudsen, E.; Rolin, J.; Al-Falahi, Y.; Maghazachi, A.A. Modulation of natural killer cell cytotoxicity and cytokine release by the drug glatiramer acetate. Cell. Mol. Life Sci. 2009, 66, 1446–1456. [Google Scholar] [CrossRef] [PubMed]
- Al-Jaderi, Z.; Maghazachi, A.A. Effects of vitamin D3, calcipotriol and FTY720 on the expression of surface molecules and cytolytic activities of human natural killer cells and dendritic cells. Toxins 2013, 5, 1932–1947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maghazachi, A.A.; Sand, K.L.; Al-Jaderi, Z. Glatiramer acetate, dimethyl fumarate, and monomethyl fumarate upregulate the expression of CCR10 on the surface of natural killer cells and enhance their chemotaxis and cytotoxicity. Front. Immunol. 2016, 7, 437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vego, H.; Sand, K.L.; Høglund, R.A.; Fallang, L.; Gundersen, G.; Holmøy, T.; Maghazachi, A.A. Monomethyl fumarate augments NK cell lysis of tumor cells through degranulation and the upregulation of Nkp46 and CD107a. Cell. Mol. Immunol. 2016, 13, 57–64. [Google Scholar] [CrossRef]
- Al-Jaderi, Z.; Maghazachi, A.A. Vitamin D3 and monomethyl fumarate enhance natural killer cell lysis of dendritic cells and ameliorate the clinical score in mice suffering from experimental autoimmune encephalomyelitis. Toxins 2015, 7, 4730–4744. [Google Scholar] [CrossRef]
- Munger, K.L.; Zhang, S.M.; O’Reilly, E.; Hernán, M.A.; Olek, M.J.; Willett, W.C.; Ascherio, A. Vitamin D intake and incidence of multiple sclerosis. Neurology 2004, 62, 60–65. [Google Scholar] [CrossRef]
- Munger, K.L.; Levin, L.I.; Hollis, B.W.; Howard, N.S.; Ascherio, A. Serum 25-hydroxyvitamin D levels and risk of multiple sclerosis. JAMA 2006, 296, 2832–2838. [Google Scholar] [CrossRef] [Green Version]
- Høglund, R.A.; Holmøy, T.; Harbo, H.F.; Maghazachi, A.A. A one year follow-up study of natural killer and dendritic cells activities in multiple sclerosis patients receiving glatiramer acetate (GA). PLoS ONE 2013, 8, e62237. [Google Scholar]
- Balato, A.; Unutmaz, D.; Gaspari, A.A. Natural killer T cells: An unconventional T-cell subset with diverse effector and regulatory functions. J. Investig. Dermatol. 2009, 129, 1628–1642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, Y.; Wan, Q. NKT cells in neurological diseases. Front. Cell. Neurosci. 2019, 13, 245. [Google Scholar] [CrossRef] [PubMed]
- Latha, T.S.; Reddy, M.C.; Durbaka, P.V.; Rachamallu, A.; Pallu, R.; Lomada, D. γδ T cell-mediated immune responses in disease and therapy. Front. Immunol. 2014, 5, 571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajan, A.J.; Gao, Y.L.; Raine, C.S.; Brosnan, C.F. A pathogenic role for gamma delta T cells in relapsing-remitting experimental allergic encephalomyelitis in the SJL mouse. J. Immunol. 1996, 157, 941–949. [Google Scholar] [PubMed]
- Spahn, T.W.; Issazadah, S.; Salvin, A.J.; Weiner, H.L. Decreased severity of myelin oligodendrocyte glycoprotein peptide 33–35-induced experimental autoimmune encephalomyelitis in mice with a disrupted TCR δ chain gene. Eur. J. Immunol. 1999, 29, 4060–4071. [Google Scholar] [CrossRef]
- Treiner, E.; Liblau, R.S. Mucosal-associated invariant T cells in multiple sclerosis: The jury is still out. Front. Immunol. 2015, 6, 503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Bourhis, L.; Guerri, L.; Dusseaux, M.; Martin, E.; Soudais, C.; Lantz, O. Mucosal-associated invariant T cells: Unconventional development and function. Trends Immunol. 2011, 32, 212–218. [Google Scholar] [CrossRef] [PubMed]
- Miyazaki, Y.; Miyake, S.; Chiba, A.; Lantz, O.; Yamamura, T. Mucosal-associated invariant T cells regulate Th1 response in multiple sclerosis. Int. Immunol. 2011, 23, 529–535. [Google Scholar] [CrossRef] [PubMed]
- Barnum, S.R. Complement biosynthesis in the central nervous system. Crit. Rev. Oral Biol. Med. 1995, 6, 132–146. [Google Scholar] [CrossRef]
- Wren, D.R.; Noble, M. Oligodendrocytes and oligodendrocyte/type-2 astrocyte progenitor cells of adult rats are specifically susceptible to the lytic effects of complement in absence of antibody. Proc. Natl. Acad. Sci. USA 1989, 86, 9025–9029. [Google Scholar] [CrossRef] [Green Version]
- Johns, T.G.; Bernard, C.C. Binding of complement component Clq to myelin oligodendrocyte glycoprotein: A novel mechanism for regulating CNS inflammation. Mol. Immunol. 1997, 34, 33–38. [Google Scholar] [CrossRef]
- Bhat, R.; Steinman, L. Innate and adaptive autoimmunity directed to the central nervous system. Neuron 2009, 64, 123–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goverman, J. Autoimmune T cell responses in the central nervous system. Nat. Rev. Immunol. 2009, 9, 393–407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Constantinescu, C.S.; Farooqi, N.; O’Brien, K.; Gran, B. Experimental autoimmune encephalomyelitis (EAE) as a model for multiple sclerosis (MS). Br. J. Pharmacol. 2011, 164, 1079–1106. [Google Scholar] [CrossRef] [PubMed]
- Fujinami, R.S.; Oldstone, M.B. Amino acid homology between the encephalitogenic site of myelin basic protein and virus: Mechanism for autoimmunity. Science 1985, 230, 1043–1045. [Google Scholar] [CrossRef] [PubMed]
- Getts, D.R.; Chastain, E.M.; Terry, R.L.; Miller, S.D. Virus infection, antiviral immunity, and autoimmunity. Immunol. Rev. 2013, 255, 197–209. [Google Scholar] [CrossRef] [Green Version]
- Keller, C.W.; Sina, C.; Kotur, M.B.; Ramelli, G.; Mundt, S.; Quast, I.; Ligeon, L.; Weber, P.; Becher, B.; Münz, C.; et al. ATG-dependent phagocytosis in dendritic cells drives myelin-specific CD4+ T cell pathogenicity during CNS inflammation. Proc. Natl. Acad. Sci. USA 2017, 114, e11228–e11237. [Google Scholar] [CrossRef] [Green Version]
- Lovett-Racke, A.E.; Yang, Y.; Racke, M.K. Th1 versus Th17: Are T cell cytokines relevant in multiple sclerosis? Biochim. Biophys. Acta 2011, 1812, 246–251. [Google Scholar] [CrossRef] [Green Version]
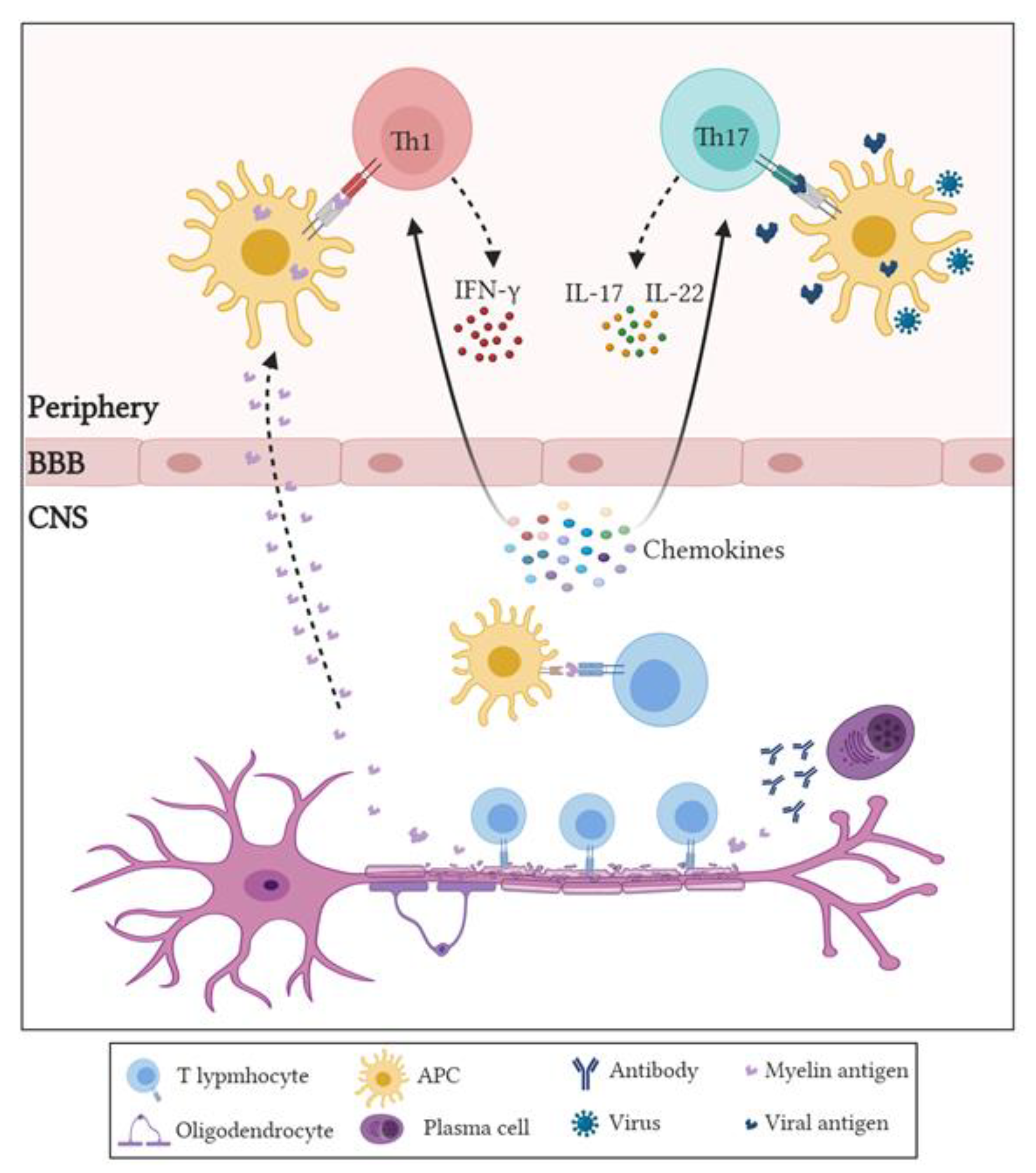
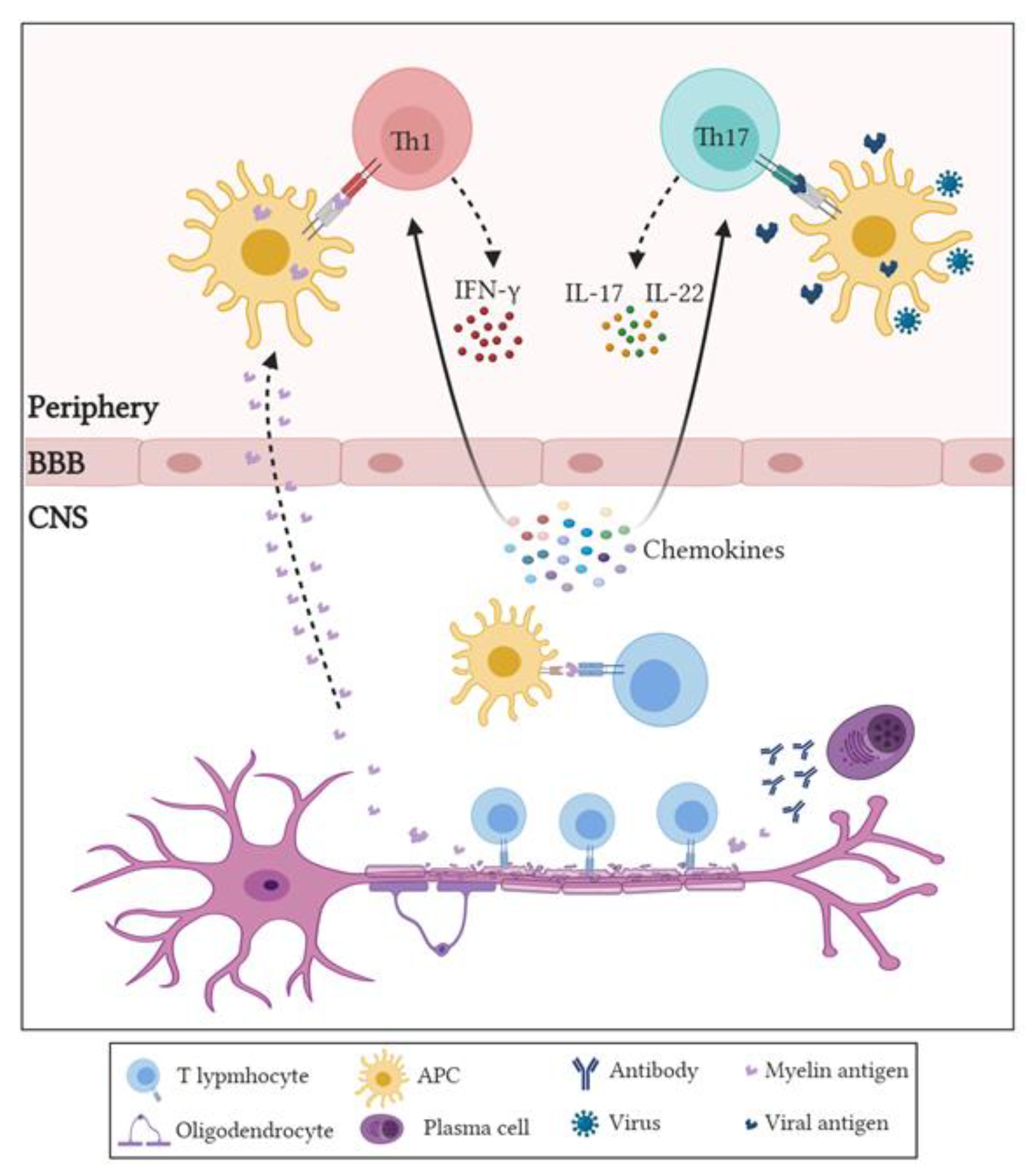
- Takeshita, Y.; Ransohoff, R.M. Inflammatory cell trafficking across the blood-brain barrier: Chemokine regulation and in vitro models. Immunol. Rev. 2012, 248, 228–239. [Google Scholar] [CrossRef] [Green Version]
- Gutcher, I.; Becher, B. APC-derived cytokines and T cell polarization in autoimmune inflammation. J. Clin. Investig. 2007, 117, 1119–1127. [Google Scholar] [CrossRef] [Green Version]
- Panitch, H.S.; Hirsch, R.L.; Haley, A.S.; Johnson, K.P. Exacerbations of multiple sclerosis in patients treated with gamma interferon. Lancet 1987, 1, 893–895. [Google Scholar] [CrossRef]
- Korn, T.; Bettelli, E.; Oukka, M.; Kuchroo, V.K. IL-17 and Th17 cells. Annu. Rev. Immunol. 2009, 27, 485–517. [Google Scholar] [CrossRef] [PubMed]
- Lock, C.; Hermans, G.; Pedotti, R.; Brendolan, A.; Schadt, E.; Garren, H.; Langer-Gould, A.; Strober, S.; Cannella, B.; Allard, J.; et al. Gene-microarray analysis of multiple sclerosis lesions yields new targets validated in autoimmune encephalomyelitis. Nat. Med. 2002, 8, 500–508. [Google Scholar] [CrossRef]
- Langrish, C.L.; Chen, Y.; Blumenschein, W.M.; Mattson, J.; Basham, B.; Sedgwick, J.D.; McClanahan, T.; Kastelein, R.A.; Cua, D.J. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J. Exp. Med. 2005, 201, 233–240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fletcher, J.M.; Lalor, S.J.; Sweeney, C.M.; Tubridy, N.; Mills, K.H. T cells in multiple sclerosis and experimental autoimmune encephalomyelitis. Clin. Exp. Immunol. 2010, 162, 1–11. [Google Scholar] [CrossRef]
- Segal, B.M. The diversity of encephalitogenic CD4+ T cells in multiple sclerosis and its animal models. J. Clin. Med. 2019, 8, 120. [Google Scholar] [CrossRef] [Green Version]
- Salou, M.; Nicol, B.; Garcia, A.; Laplaud, D.A. Involvement of CD8+ T cells in multiple sclerosis. Front. Immunol. 2015, 6, 604. [Google Scholar] [CrossRef] [Green Version]
- Babbe, H.; Roers, A.; Waisman, A.; Lassmann, H.; Goebels, N.; Hohlfeld, R.; Friese, M.; Schröder, R.; Deckert, M.; Schmidt, S.; et al. Clonal expansions of Cd8+ T cells dominate the T cell infiltrate in active multiple sclerosis lesions as shown by micromanipulation and single cell polymerase chain reaction. J. Exp. Med. 2000, 192, 393–404. [Google Scholar] [CrossRef] [Green Version]
- Chevalier, G.; Suberbielle, E.; Monnet, C.; Duplan, V.; Martin-Blondel, G.; Farrugia, F.; Le Masson, G.; Liblau, R.; Gonzalez-Dunia, D. Neurons are MHC class I-dependent targets for CD8 T cells upon neurotropic viral infection. PLoS Pathog 2011, 7, e1002393. [Google Scholar] [CrossRef]
- Booss, J.; Esiri, M.M.; Tourtellotte, W.W.; Mason, D.Y. Immunohistological analysis of T lymphocyte subsets in the central nervous system in chronic progressive multiple sclerosis. J. Neurol Sci. 1983, 62, 219–232. [Google Scholar] [CrossRef]
- Lucchinetti, C.F.; Popescu, B.F.; Bunyan, R.F.; Moll, N.M.; Roemer, S.F.; Lassmann, H.; Brück, W.; Parisi, J.E.; Scheithauer, B.W.; Weigand, S.D.; et al. Inflammatory cortical demyelination in early multiple sclerosis. N. Engl. J. Med. 2011, 365, 2188–2197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Battistini, L.; Piccio, L.; Rossi, B.; Bach, S.; Galgani, S.; Gasperini, C.; Ottoboni, L.; Ciabini, D.; Caramia, M.D.; Bernardi, G.; et al. CD8+ T cells from patients with acute multiple sclerosis display selective increase of adhesiveness in brain venules: A critical role for P-selectin glycoprotein ligand-1. Blood 2003, 101, 4775–4782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, D.; Whitaker, J.N.; Huang, Z.; Liu, D.; Coleclough, C.; Wekerle, H.; Raine, C.S. Myelin antigen-specific CD8+ T cells are encephalitogenic and produce severe disease in C57BL/6 mice. J. Immunol. 2001, 166, 7579–7587. [Google Scholar]
- Najafian, N.; Chitnis, T.; Salama, A.D.; Zhu, B.; Benou, C.; Yuan, X.; Clarkson, M.R.; Sayegh, M.H.; Khoury, S.J. Regulatory functions of CD8+CD28-T cells in an autoimmune disease model. J. Clin. Investig. 2003, 112, 1037–1048. [Google Scholar] [CrossRef] [PubMed]
- York, N.R.; Mendoza, J.P.; Ortega, S.B.; Benagh, A.; Tyler, A.F.; Firan, M.; Karandikar, N.J. Immune regulatory CNS-reactive CD8+T cells in experimental autoimmune encephalomyelitis. J. Autoimmun. 2010, 35, 33–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sinha, S.; Boyden, A.W.; Itani, F.R.; Crawford, M.P.; Karandikar, N.J. CD8(+) T-cells as immune regulators of multiple sclerosis. Front. Immunol. 2015, 6, 619. [Google Scholar] [CrossRef] [Green Version]
- He, F.; Balling, R. The role of regulatory T cells in neurodegenerative diseases. Wiley Interdiscip. Rev. Syst. Biol. Med. 2013, 5, 153–180. [Google Scholar] [CrossRef]
- Danikowski, K.M.; Jayaraman, S.; Prabhakar, B.S. Regulatory T cells in multiple sclerosis and myasthenia gravis. J. Neuroinflamm. 2017, 14, 117. [Google Scholar] [CrossRef] [Green Version]
- Kipnis, J.; Avidan, H.; Caspi, R.R.; Schwartz, M. Dual effect of CD4+CD25+ regulatory T cells in neurodegeneration: A dialogue with microglia. Proc. Natl. Acad. Sci. USA 2004, 101, 14663–14669. [Google Scholar] [CrossRef] [Green Version]
- Walsh, J.T.; Kipnis, J. Regulatory T cells in CNS injury: The simple, the complex and the confused. Trends Mol. Med. 2011, 17, 541–547. [Google Scholar] [CrossRef] [Green Version]
- Calvo-Barreiro, L.; Eixarch, H.; Montalban, X.; Espejo, C. Combined therapies to treat complex diseases: The role of the gut microbiota in multiple sclerosis. Autoimmun. Rev. 2018, 17, 165–174. [Google Scholar] [CrossRef] [PubMed]
- Cekanaviciute, E.; Yoo, B.B.; Runia, T.F.; Debelius, J.W.; Singh, S.; Nelson, C.A.; Kanner, R.; Bencosme, Y.; Lee, Y.K.; Hauser, S.L.; et al. Gut bacteria from multiple sclerosis patients modulate human T cells and exacerbate symptoms in mouse models. Proc. Natl. Acad. Sci. USA 2017, 114, 10713–10718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wurtman, R. Multiple sclerosis, melatonin, and neurobehavioral diseases. Front. Endocrinol. 2017, 8, 280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hardy, R.R.; Carmack, C.E.; Shinton, S.A.; Kemp, J.D.; Hayakawa, K. Resolution and characterization of pro-B and pre–pro-B cell stages in normal mouse bone marrow. J. Exp. Med. 1991, 173, 1213–1225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michel, L.; Touil, H.; Pikor, N.B.; Gommerman, J.L.; Prat, A.; Bar-Or, A. B cells in the multiple sclerosis central nervous system: Trafficking and contribution to CNS-compartmentalized inflammation. Front. Immunol. 2015, 6, 636. [Google Scholar] [CrossRef] [Green Version]
- Al-ani, M.R.; Raju, T.K.; Hachim, M.Y.; Hachim, I.Y.; Elemam, N.M.; Guimei, M.; Bendardaf, R.; Maghazachi, A.A. Rituximab prevents the development of experimental autoimmune encephalomyelitis (EAE): Comparison with prophylactic, therapeutic or combinational regimens. J. Inflamm. Res. 2020, 13, 151–164. [Google Scholar] [CrossRef] [Green Version]
- Lin, M.; Wang, Z.; Han, X. B cells with regulatory function in animal models of autoimmune and non-autoimmune diseases. Open J. Immunol. 2015, 5, 9–17. [Google Scholar] [CrossRef] [Green Version]
- Kowarik, M.C.; Cepok, S.; Sellner, J.; Grummel, V.; Weber, M.S.; Korn, T.; Berthele, A.; Hemmer, B. CXCL13 is the major determinant for B cell recruitment to the CSF during neuroinflammation. J. Neuroinflamm. 2012, 9, 93. [Google Scholar] [CrossRef] [Green Version]
- Häusser-Kinzel, S.; Weber, M.S. The role of B Cells and antibodies in multiple sclerosis, neuromyelitis optica, and related disorders. Front. Immunol. 2019, 10, 201. [Google Scholar] [CrossRef]
- Duddy, M.; Niino, M.; Adatia, F.; Hebert, S.; Freedman, M.; Atkins, H.; Kim, H.J.; Bar-Or, A. Distinct effector cytokine profiles of memory and naive human B cell subsets and implication in multiple sclerosis. J. Immunol. 2007, 178, 6092–6099. [Google Scholar] [CrossRef] [Green Version]
- Pikor, N.B.; Prat, A.; Bar-Or, A.; Gommerman, J.L. Meningeal tertiary lymphoid tissues and multiple sclerosis: A gathering place for diverse types of immune cells during CNS autoimmunity. Front. Immunol. 2016, 6, 657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Negron, A.; Robinson, R.R.; Stüve, O.; Forsthuber, T.G. The role of B cells in multiple sclerosis: Current and future therapies. Cell Immunol. 2019, 339, 10–23. [Google Scholar] [CrossRef] [PubMed]
- Wolf, S.D.; Dittel, B.N.; Hardardottir, F.; Janeway, C.A., Jr. Experimental autoimmune encephalomyelitis induction in genetically B cell-deficient mice. J. Exp. Med. 1996, 184, 2271–2278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fillatreau, S.; Sweenie, C.H.; McGeachy, M.J.; Gray, D.; Anderton, S.M. B cells regulate autoimmunity by provision of IL-10. Nat. Immunol. 2002, 3, 944–950. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dhaiban, S.; Al-Ani, M.; Elemam, N.M.; Al-Aawad, M.H.; Al-Rawi, Z.; Maghazachi, A.A. Role of Peripheral Immune Cells in Multiple Sclerosis and Experimental Autoimmune Encephalomyelitis. Sci 2021, 3, 12. https://doi.org/10.3390/sci3010012
Dhaiban S, Al-Ani M, Elemam NM, Al-Aawad MH, Al-Rawi Z, Maghazachi AA. Role of Peripheral Immune Cells in Multiple Sclerosis and Experimental Autoimmune Encephalomyelitis. Sci. 2021; 3(1):12. https://doi.org/10.3390/sci3010012
Chicago/Turabian StyleDhaiban, Sarah, Mena Al-Ani, Noha Mousaad Elemam, Mahmood H. Al-Aawad, Zeinab Al-Rawi, and Azzam A. Maghazachi. 2021. "Role of Peripheral Immune Cells in Multiple Sclerosis and Experimental Autoimmune Encephalomyelitis" Sci 3, no. 1: 12. https://doi.org/10.3390/sci3010012
APA StyleDhaiban, S., Al-Ani, M., Elemam, N. M., Al-Aawad, M. H., Al-Rawi, Z., & Maghazachi, A. A. (2021). Role of Peripheral Immune Cells in Multiple Sclerosis and Experimental Autoimmune Encephalomyelitis. Sci, 3(1), 12. https://doi.org/10.3390/sci3010012