
Phylogeographic Patterns and Genetic Diversity of Anopheles stephensi: Implications for Global Malaria Transmission

 , , ,
, , ,  , and
, and
Abstract
1. Introduction
2. Materials and Methods
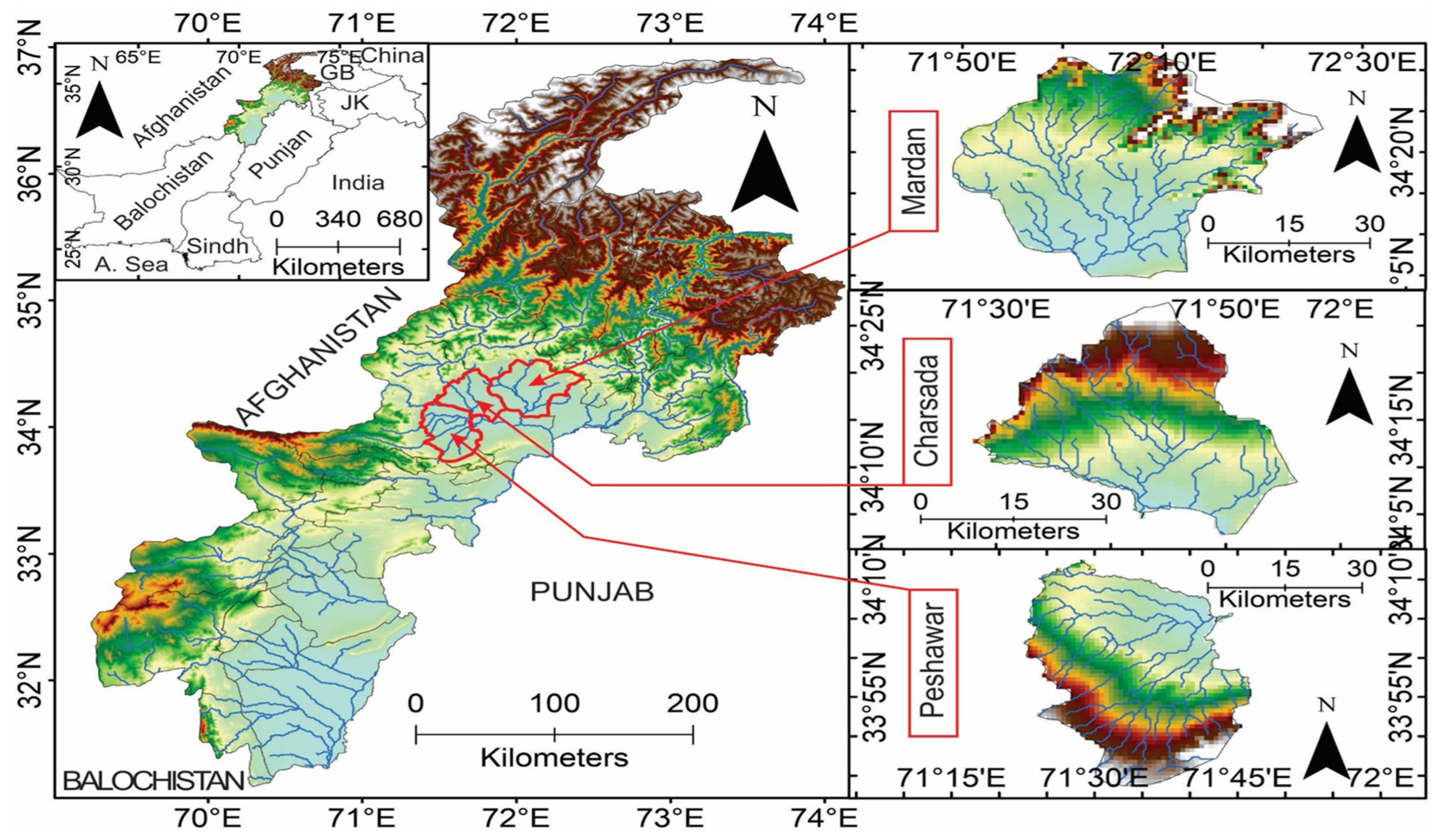
2.1. Study Area
2.2. Mosquito Sampling and Colony Maintenance
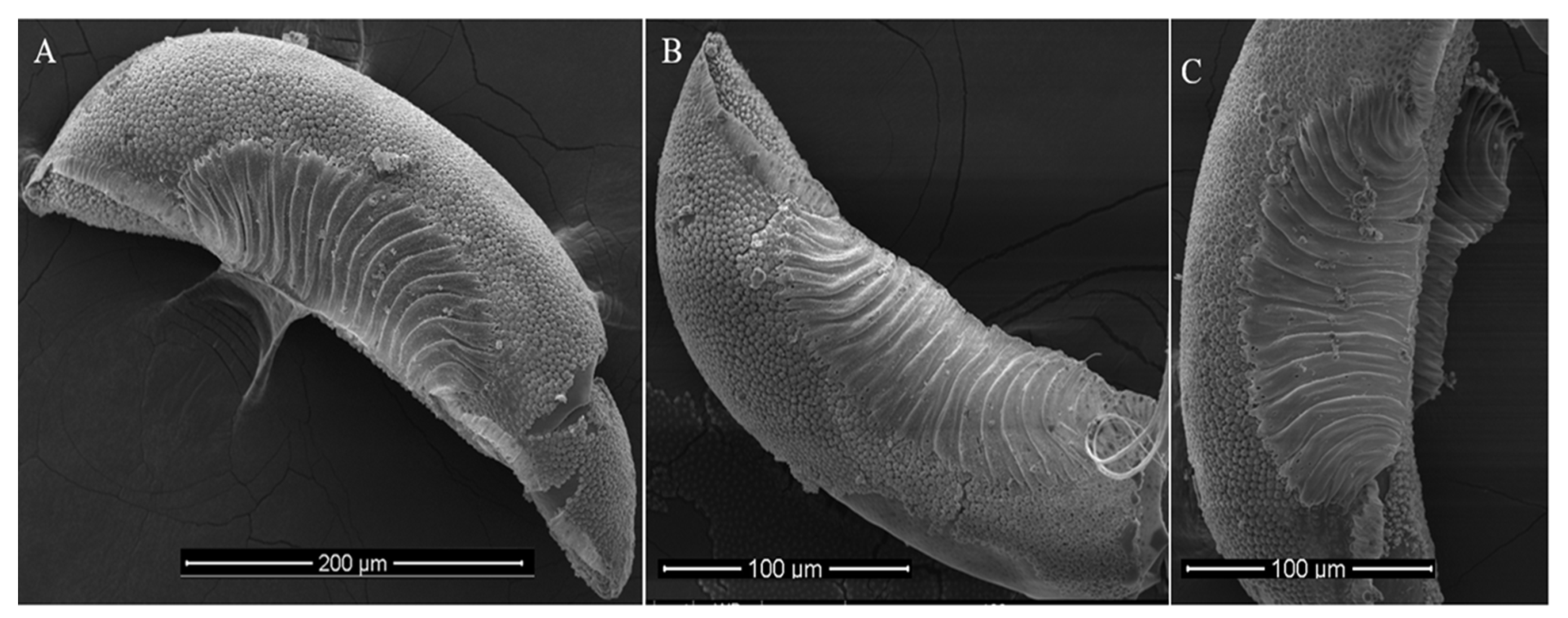
2.3. Morphological Study of Mosquito Egg
2.4. DNA Extraction
2.5. PCR Amplification and Sequencing
2.6. Sequences and Phylogenetic Analysis
2.7. Phylogeographic Clustering and Population Genetics Analysis
3. Results
3.1. Egg Morphometric Analysis
3.2. Molecular Analysis
3.2.1. Cytochrome Oxidase Subunit I (COI)
3.2.2. Cytochrome Oxidase Subunit II (COII)
3.2.3. Internal Transcribed Spacer 2
3.3. Phylogeographic Dynamics of An. stephensi
3.3.1. Cytochrome Oxidase I
3.3.2. Cytochrome Oxidase II
3.3.3. Internal Transcribed Spacer 2
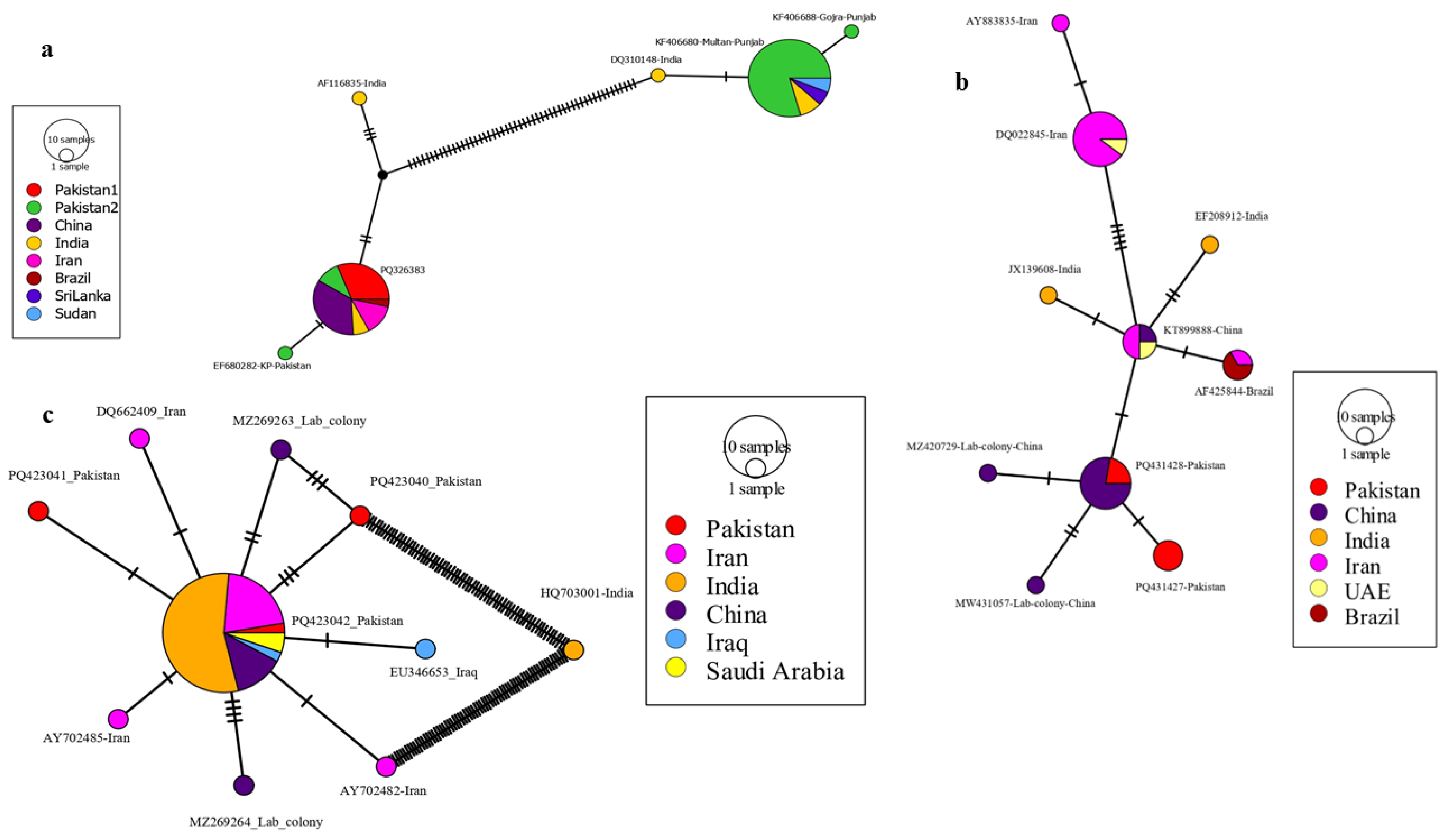
3.4. Haplotypes Distribution
3.4.1. COI-Based Network
3.4.2. COII-Based Network
3.4.3. ITS2-Based Network
3.5. An. stephensi Population Genetic Diversity
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Correction Statement
References
- WHO. World Malaria Report 2024. Available online: https://www.who.int/teams/global-malaria-programme/reports/world-malaria-report-2024 (accessed on 2 February 2025).
- WHO. Surveillance and Control of Anopheles stephensi: Country Experiences; World Health Organization: Geneva, Switzerland, 2024; Available online: https://www.who.int/publications/i/item/9789240094420 (accessed on 2 February 2025).
- Ministry of Health Pakistan. Epidemiology of Malaria in Pakistan. 2022. Available online: https://www.who.int/emergencies/disease-outbreak-news/item/2022-DON413 (accessed on 17 September 2024).
- Taylor, R.; Messenger, L.A.; Abeku, T.A.; Clarke, S.E.; Yadav, R.S.; Lines, J. Invasive Anopheles stephensi in Africa: Insights from Asia. Trends Parasitol. 2024, 40, 731–743. [Google Scholar] [CrossRef] [PubMed]
- Thomas, S.; Ravishankaran, S.; Justin, N.A.; Asokan, A.; Mathai, M.T.; Valecha, N.; Montgomery, J.; Thomas, M.B.; Eapen, A. Resting and feeding preferences of Anopheles stephensi in an urban setting, perennial for malaria. Malar. J. 2017, 16, 111. [Google Scholar] [CrossRef]
- Djadid, N.D.; Gholizadeh, S.; Aghajari, M.; Zehi, A.H.; Raeisi, A.; Zakeri, S. Genetic analysis of rDNA-ITS2 and RAPD loci in field populations of the malaria vector, Anopheles stephensi (Diptera: Culicidae): Implications for the control program in Iran. Acta Trop. 2006, 97, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Oshaghi, M.; Yaghoobi, F.; Vatandoost, H.; Abai, M.; Akbarzadeh, K. Anopheles stephensi biological forms, geographical distribution, and malaria transmission in malarious regions in Iran. Pak. J. Biol. Sci. 2006, 9, 294–298. [Google Scholar] [CrossRef]
- Firooziyan, S.; Djadid, N.D.; Gholizadeh, S. Speculation on the possibility for introducing Anopheles stephensi as a species complex: Preliminary evidence based on odorant-binding protein 1 intron I sequence. Malar. J. 2018, 17, 366. [Google Scholar] [CrossRef]
- Gholizadeh, S.; Firooziyan, S.; Ladonni, H.; Hajipirloo, H.M.; Djadid, N.D.; Hosseini, A.; Raz, A. The Anopheles stephensi odorant binding protein 1 (AsteObp1) gene: A new molecular marker for biological forms diagnosis. Acta Trop. 2015, 146, 101–113. [Google Scholar] [CrossRef]
- Gholizadeh, S.; Zakeri, S.; Djadid, N.D. Genotyping Plasmodium vivax isolates infecting Anopheles stephensi, an Asian main malaria vector. Exp. Parasitol. 2013, 134, 48–51. [Google Scholar] [CrossRef]
- Gholizadeh, S.; Djadid, N.D.; Nouroozi, B.; Bekmohammadi, M. Molecular phylogenetic analysis of Anopheles and Cellia subgenus anophelines (Diptera: Culicidae) in temperate and tropical regions of Iran. Acta Trop. 2013, 126, 63–74. [Google Scholar] [CrossRef]
- Subbarao, S.K.; Vasantha, K.; Adak, T.; Sharma, V.P.; Curtis, C.F. Egg float ridge number in Anopheles stephensi: Ecological variation and genetic analysis. Med. Vet. Entomol. 1987, 1, 265–271. [Google Scholar] [CrossRef]
- Sweet, W.C.; Rao, B.A. Races of Anopheles stephensi Liston. Indian Med. Gaz. 1937, 72, 665–674. [Google Scholar]
- Rao, B.A.; Sweet, W.C.; Subbarao, A.M. Ova measurements of Anopheles stephensi type and Anopheles stephensi var. mysorensis. J. Malar. Inst. India 1938, 1, 261–266. [Google Scholar]
- Khan, J.; Gholizadeh, S.; Zhang, D.; Wang, G.; Guo, Y.; Zheng, X.; Wu, Z.; Wu, Y. Identification of a biological form in the Anopheles stephensi laboratory colony using the odorant-binding protein 1 intron I sequence. PLoS ONE 2022, 17, e0263836. [Google Scholar] [CrossRef] [PubMed]
- Sinka, M.E.; Bangs, M.J.; Manguin, S.; Chareonviriyaphap, T.; Patil, A.P.; Temperley, W.H.; Gething, P.W.; Elyazar, I.R.; Kabaria, C.W.; Harbach, R.E.; et al. The dominant Anopheles vectors of human malaria in the Asia-Pacific region: Occurrence data, distribution maps and bionomic precis. Parasit. Vectors 2011, 4, 89. [Google Scholar] [CrossRef]
- Neafsey, D.E.; Waterhouse, R.M.; Abai, M.R.; Aganezov, S.S.; Alekseyev, M.A.; Allen, J.E.; Amon, J.; Arcà, B.; Arensburger, P.; Artemov, G.; et al. Highly evolvable malaria vectors: The genomes of 16 Anopheles mosquitoes. Science 2015, 347, 1258522. [Google Scholar] [CrossRef] [PubMed]
- Yared, S.; Gebressielasie, A.; Damodaran, L.; Bonnell, V.; Lopez, K.; Janies, D.; Carter, T.E. Insecticide resistance in Anopheles stephensi in Somali Region, eastern Ethiopia. Malar. J. 2020, 19, 180. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.A.; Aaqil, M.; Hafeez, M. Genetic and morphological variations in a national population of the malaria mosquito, Anopheles stephensi from Karachi, Pakistan. Biologia 1972, 18, 29–41. [Google Scholar]
- Afridi, M.K.; Talibi, S.A.; Rashid, S.A.; Hussain, M.Z.Y. Identification of races of Anopheles stephensi prevalent in the federal Karachi area by measurement of their ova. Pak. J. Health 1958, 8, 71–76. [Google Scholar]
- Klinkenberg, E.; Konradsen, F.; Herrel, N.; Mukhtar, M.; van der Hoek, W.; Amerasinghe, F.P. Malaria vectors in the changing environment of the southern Punjab, Pakistan. Trans. R. Soc. Trop. Med. Hyg. 2004, 98, 442–449. [Google Scholar] [CrossRef]
- Khan, J.; Adil, M.; Wang, G.; Tsheten, T.; Zhang, D.; Pan, W.; Khan, M.A.; Rehman, I.U.; Zheng, X.; Wu, Z.; et al. A cross-sectional study to assess the epidemiological situation and associated risk factors of dengue fever; knowledge, attitudes, and practices about dengue prevention in Khyber Pakhtunkhwa Province, Pakistan. Front. Public Health 2022, 10, 923277. [Google Scholar] [CrossRef]
- Malik, S.M.; Awan, H.; Khan, N. Mapping vulnerability to climate change and its repercussions on human health in Pakistan. Glob. Health 2012, 8, 1–10. [Google Scholar] [CrossRef]
- Pakistan Meteorological Department. Available online: https://rmckpk.pmd.gov.pk/ (accessed on 1 November 2024).
- Ali, N.; Noreen, S.; Khan, K.; Wahid, S. Population dynamics of mosquitoes and malaria vector incrimination in district Charsadda, Khyber Pakhtunkhwa (KP), Pakistan. Acta Trop. 2015, 141 Pt A, 25–31. [Google Scholar] [CrossRef]
- Rowland, M.; Hewitt, S.; Durrani, N.; Bano, N.; Wirtz, R. Transmission and control of vivax malaria in Afghan refugee settlements in Pakistan. Trans. R. Soc. Trop. Med. Hyg. 1997, 91, 252–255. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Zhang, D.; Li, Y.; Wu, Y.; Liang, K.; Liang, X.; Liang, Y.; Pan, X.; Hu, L.; Sun, Q.; et al. Incompatible and sterile insect techniques combined eliminate mosquitoes. Nature 2019, 572, 56. [Google Scholar] [CrossRef] [PubMed]
- Amerasinghe, F.P.; Mukhtar, M.; Herrel, N. Keys to the Anopheline mosquitoes (Diptera: Culicidae) of Pakistan. J. Med. Entomol. 2002, 39, 28–35. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Larkin, M.A.; Blackshields, G.; Brown, N.P.; Chenna, R.; McGettigan, P.A.; McWilliam, H.; Valentin, F.; Wallace, I.M.; Wilm, A.; Lopez, R.; et al. ClustalW and ClustalX version 2. Bioinformatics 2007, 23, 2947–2948. [Google Scholar] [CrossRef]
- Tamura, K.; Peterson, D.; Peterson, N.; Stecher, G.; Nei, M.; Kumar, S. MEGA5: Molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol. 2011, 28, 2731–2739. [Google Scholar] [CrossRef]
- Nguyen, L.T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Hoang, D.T.; Chernomor, O.; von Haeseler, A.; Minh, B.Q.; Vinh, L.S. UFBoot2: Improving the ultrafast bootstrap approximation. Mol. Biol. Evol. 2018, 35, 518–522. [Google Scholar] [CrossRef]
- Leigh, J.W.; Bryant, D. PopArt: Full-feature software for haplotype network construction. Methods Ecol. Evol. 2015, 6, 1110–1116. [Google Scholar] [CrossRef]
- Rozas, J.; Ferrer-Mata, A.; Sánchez-DelBarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sánchez-Gracia, A. DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef] [PubMed]
- Nagpal, B.N.; Srivastava, A.; Kalra, N.L.; Subbarao, S.K. Spiracular indices in Anopheles stephensi: A taxonomic tool to identify ecological variants. J. Med. Entomol. 2003, 40, 747–749. [Google Scholar] [CrossRef] [PubMed]
- Abubakr, M.; Sami, H.; Mahdi, I.; Altahir, O.; Abdelbagi, H.; Mohamed, N.S.; Ahmed, A. The phylodynamic and spread of the invasive Asian malaria vectors, Anopheles stephensi, in Sudan. Biology 2022, 11, 409. [Google Scholar] [CrossRef]
- Gakhar, S.K.; Sharma, R.; Sharma, A. Population genetic structure of malaria vector Anopheles stephensi Liston (Diptera: Culicidae). Indian J. Exp. Biol. 2013, 51, 273–279. [Google Scholar]
- Alam, M.Z.; Niaz Arifin, S.M.; Al-Amin, H.M.; Alam, M.S.; Rahman, M.S. A spatial agent-based model of Anopheles vagus for malaria epidemiology: Examining the impact of vector control interventions. Malar. J. 2017, 16, 432. [Google Scholar] [CrossRef] [PubMed]
- Sharma, R.; Sharma, A.; Kumar, A.; Dube, M.; Gakhar, S.K. Population genetic structure of urban malaria vector Anopheles stephensi in India. Infect. Genet. Evol. 2016, 39, 35–44. [Google Scholar] [CrossRef]
- Sinka, M.E.; Pironon, S.; Massey, N.C.; Longbottom, J.; Hemingway, J.; Moyes, C.L.; Willis, K.J. A new malaria vector in Africa: Predicting the expansion range of and identifying the urban populations at risk. Proc. Natl. Acad. Sci. USA 2020, 117, 24900–24908. [Google Scholar] [CrossRef]
- Carter, T.E.; Yared, S.; Getachew, D.; Spear, J.; Choi, S.H.; Samake, J.N.; Mumba, P.; Dengela, D.; Yohannes, G.; Chibsa, S.; et al. Genetic diversity of Anopheles stephensi in Ethiopia provides insight into patterns of spread. Parasit. Vectors 2021, 14, 602. [Google Scholar] [CrossRef]
- Beebe, N.W. DNA barcoding mosquitoes: Advice for potential prospectors. Parasitology 2018, 145, 622–633. [Google Scholar] [CrossRef]
- Hillis, D.M.; Dixon, M.T. Ribosomal DNA: Molecular evolution and phylogenetic inference. Q. Rev. Biol. 1991, 66, 411–453. [Google Scholar] [CrossRef]
- Thakare, A.; Ghosh, C.; Alalamath, T.; Kumar, N.; Narang, H.; Whadgar, S.; Paul, K.; Shrotri, S.; Kumar, S.; Soumya, M.; et al. The genome trilogy of Anopheles stephensi, an urban malaria vector, reveals structure of a locus associated with adaptation to environmental heterogeneity. Sci. Rep. 2022, 12, 3610. [Google Scholar] [CrossRef]
- Subbarao, S.K.; Nanda, N.; Rahi, M.; Raghavendra, K. Biology and bionomics of malaria vectors in India: Existing information and what more needs to be known for strategizing elimination of malaria. Malar. J. 2019, 18, 396. [Google Scholar] [CrossRef]
- Dharmasiri, G.A.; Perera, A.Y.; Harishchandra, J.; Herath, H.; Aravindan, K.; Jayasooriya, H.T.R.; Ranawaka, G.R.; Hewavitharane, M. First record of Anopheles stephensi in Sri Lanka: A potential challenge for prevention of malaria reintroduction. Malar. J. 2017, 16, 326. [Google Scholar] [CrossRef] [PubMed]
- Balkew, M.; Mumba, P.; Yohannes, G.; Abiy, E.; Getachew, D.; Yared, S.; Worku, A.; Gebresilassie, A.; Tadesse, F.G.; Gadisa, E.; et al. An update on the distribution, bionomics, and insecticide susceptibility of Anopheles stephensi in Ethiopia, 2018–2020. Malar. J. 2021, 20, 263. [Google Scholar] [CrossRef] [PubMed]
- Surendran, S.N.; Sivabalakrishnan, K.; Gajapathy, K.; Arthiyan, S.; Jayadas, T.T.; Karvannan, K.; Raveendran, S.; Parakrama Karunaratne, S.H.P.; Ramasamy, R. Genotype and biotype of invasive Anopheles stephensi in Mannar Island of Sri Lanka. Parasit. Vectors 2018, 11, 3. [Google Scholar] [CrossRef] [PubMed]
- Teshome, A.; Erko, B.; Golassa, L.; Yohannes, G.; Irish, S.R.; Zohdy, S.; Yoshimizu, M.; Dugassa, S. Resistance of Anopheles stephensi to selected insecticides used for indoor residual spraying and long-lasting insecticidal nets in Ethiopia. Malar. J. 2023, 22, 218. [Google Scholar] [CrossRef]
- Mnzava, A.; Monroe, A.C.; Okumu, F. Anopheles stephensi in Africa requires a more integrated response. Malar. J. 2022, 21, 156. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sampling Site (Isolate) | District | Geographic Coordinates | Trap Location | Climate Zone | Annual Mean Temperature ( °C) | Annual Precipitation (mm) |
|---|---|---|---|---|---|---|
| P1 | Peshawar | 33.9437° N, 71.6199° E | Location 1 | |||
| P2 | 33.9982° N, 71.4862° E | Location 2 | ||||
| P3 | 33.9744° N, 71.4359° E | Location 3 | ||||
| P4 | 34.0156° N, 71.7127° E | Location 4 | semi-arid | 22.7 | 1110 | |
| P5 | 34.0259° N, 71.5601° E | Location 5 | ||||
| P6 | 34.0054° N, 71.7237° E | Location 6 | ||||
| M7 | Mardan | 34.3100° N, 72.0468° E | Location 1 | |||
| M8 | 34.1500° N, 72.0379° E | Location 2 | ||||
| M9 | 34.2876° N, 71.9342° E | Location 3 | hot semi-arid | 22.2 | 1000 | |
| M10 | 34.3521° N, 72.0764° E | Location 4 | ||||
| M11 | 34.3410° N, 72.2897° E | Location 5 | ||||
| C12 | Charsadda | 34.1986° N, 71.7385° E | Location 1 | |||
| C13 | 34.3040° N, 71.6555° E | Location 2 | ||||
| C14 | 34.2186° N, 71.5546° E | Location 3 | semi-arid | 19.74 | 1000 | |
| C15 | 34°16′47 N, 71°33′59° E | Location 4 | ||||
| C16 | 34.1435° N, 71.7370° E | Location 5 | ||||
| C17 | 34°19′7° N, 71°35′35° E | Location 6 |
| COI-based Haplotypes Distribution | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Haplotypes | Pakistan | Iran | India | China | Brazil | Sudan | Sri Lanka | Total | % Prevalence |
| Hap_1 | 12 | 4 | 2 | 10 | 1 | 0 | 0 | 29 | 43.3 |
| Hap_2 | 27 | 0 | 3 | 0 | 0 | 2 | 2 | 34 | 50.7 |
| Hap_3 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 1.5 |
| Hap_4 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 1.5 |
| Hap_5 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 1 | 1.5 |
| Hap_6 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 1 | 1.5 |
| COII-based Haplotypes Distribution | |||||||||
| Haplotypes | Pakistan | Iran | India | China | Brazil | UAE | - | Total | % Prevalence |
| Hap_1 | 3 | 0 | 0 | 0 | 0 | 0 | - | 3 | 8.8 |
| Hap_2 | 2 | 0 | 0 | 7 | 0 | 0 | - | 9 | 26.5 |
| Hap_3 | 0 | 0 | 0 | 1 | 0 | 0 | - | 1 | 2.9 |
| Hap_4 | 0 | 0 | 0 | 1 | 0 | 0 | - | 1 | 2.9 |
| Hap_5 | 0 | 2 | 0 | 1 | 0 | 1 | - | 4 | 11.8 |
| Hap_6 | 0 | 1 | 0 | 0 | 2 | 0 | - | 3 | 8.8 |
| Hap_7 | 0 | 9 | 0 | 0 | 0 | 1 | - | 10 | 29.4 |
| Hap_8 | 0 | 1 | 0 | 0 | 0 | 0 | - | 1 | 2.9 |
| Hap_9 | 0 | 0 | 1 | 0 | 0 | 0 | - | 1 | 2.9 |
| Hap_10 | 0 | 0 | 1 | 0 | 0 | 0 | - | 1 | 2.9 |
| ITS2-based Haplotypes Distribution | |||||||||
| Haplotypes | Pakistan | Iran | India | China | Iraq | Saudi Arabia | Total | % Prevalence | |
| Hap_1 | 1 | 0 | 0 | 0 | 0 | 0 | - | 1 | 2.1 |
| Hap_2 | 1 | 0 | 0 | 0 | 0 | 0 | - | 1 | 2.1 |
| Hap_3 | 1 | 8 | 21 | 5 | 1 | 2 | - | 38 | 80.8 |
| Hap_4 | 0 | 0 | 0 | 1 | 0 | 0 | - | 1 | 2.1 |
| Hap_5 | 0 | 0 | 0 | 1 | 0 | 0 | - | 1 | 2.1 |
| Hap_6 | 0 | 1 | 0 | 0 | 0 | 0 | - | 1 | 2.1 |
| Hap_7 | 0 | 1 | 0 | 0 | 0 | 0 | - | 1 | 2.1 |
| Hap_8 | 0 | 0 | 0 | 0 | 1 | 0 | - | 1 | 2.1 |
| Hap_9 | 0 | 1 | 0 | 0 | 0 | 0 | - | 1 | 2.1 |
| Hap_10 | 0 | 0 | 1 | 0 | 0 | 0 | - | 1 | 2.1 |
| Cytochrome Oxidase I | |||||||
|---|---|---|---|---|---|---|---|
| Variable | Pakistan | Iran | India | China | Brazil | Sudan | Sri Lanka |
| Number of sequences (n) | 41 | 4 | 7 | 10 | 1 | 2 | 2 |
| Number of segregating sites | 43 | 0 | 47 | 0 | 0 | 0 | 0 |
| Number of haplotypes (h) | 4 | 1 | 4 | 1 | 1 | 1 | 1 |
| Haplotype diversity (Hd) | 0.49146 | 0 | 0.80 | 0 | 0 | 0 | 0 |
| Nucleotide diversity (Pi) | 0.16555 | 0 | 0.22587 | 0 | 0 | 0 | 0 |
| k | 18.70732 | 0 | 25.5238 | 0 | n.d. * | 0 | 0 |
| Tajima’s D | 3.05205 | n.d. | 2.00315 | n.d. | n.d. | n.d. | n.d. |
| Fu’s Fs | 43.549 | n.d. | 10.026 | n.d. | n.d. | n.d. | n.d. |
| Cytochrome Oxidase II | |||||||
| Variable | Pakistan | Iran | India | China | Brazil | UAE | - |
| Number of sequences (n) | 5 | 13 | 2 | 10 | 2 | 2 | - |
| Number of segregating sites | 1 | 7 | 3 | 4 | 0 | 5 | - |
| Number of haplotypes (h) | 2 | 4 | 2 | 4 | 1 | 2 | - |
| Haplotype diversity (Hd) | 0.6 | 0.526 | 1 | 0.533 | 0 | 1 | - |
| Nucleotide diversity (Pi) | 0.00107 | 0.00398 | 0.00536 | 0.00143 | 0 | 0.00893 | - |
| k | 0.6 | 2.231 | 3 | 0.8 | 0 | 5 | - |
| Tajima’s D | 1.22474 | −0.042 | n.d. | −1.6671 | n.d. | n.d. | - |
| Fu’s Fs | 0.626 | 1.343 | n.d. | −1.345 | n.d. | n.d. | - |
| Internal Transcribed Spacer 2 | |||||||
| Variable | Pakistan | Iran | India | China | Iraq | Saudi Arabia | - |
| Number of sequences (n) | 3 | 11 | 22 | 7 | 2 | 2 | - |
| Number of segregating sites | 4 | 3 | 71 | 6 | 1 | 0 | - |
| Number of haplotypes (h) | 3 | 4 | 2 | 3 | 2 | 1 | - |
| Haplotype diversity (Hd) | 1 | 0.491 | 0.091 | 0.524 | 1 | 0 | - |
| Nucleotide diversity (Pi) | 0.00572 | 0.00117 | 0.01385 | 0.00368 | 0.00215 | 0 | - |
| k | 2.667 | 0.545 | 6.455 | 1.714 | 1 | 0 | - |
| Tajima’s D | n.d. | −1.6 | −2.6719 | −1.5241 | n.d. | n.d. | - |
| Fu’s Fs | n.d. | −2.042 | 13.242 | 1.014 | n.d. | n.d. | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khan, J.; Zhang, D.; Gholizadeh, S.; Deng, Y.; Aziz, A.; Chen, J.; Shah, P.T.; Lv, Z.; Chen, T. Phylogeographic Patterns and Genetic Diversity of Anopheles stephensi: Implications for Global Malaria Transmission. Trop. Med. Infect. Dis. 2025, 10, 109. https://doi.org/10.3390/tropicalmed10040109
Khan J, Zhang D, Gholizadeh S, Deng Y, Aziz A, Chen J, Shah PT, Lv Z, Chen T. Phylogeographic Patterns and Genetic Diversity of Anopheles stephensi: Implications for Global Malaria Transmission. Tropical Medicine and Infectious Disease. 2025; 10(4):109. https://doi.org/10.3390/tropicalmed10040109
Chicago/Turabian StyleKhan, Jehangir, Dongjing Zhang, Saber Gholizadeh, Yidong Deng, Abdul Aziz, Jianhuang Chen, Pir Tariq Shah, Zhiyue Lv, and Tao Chen. 2025. "Phylogeographic Patterns and Genetic Diversity of Anopheles stephensi: Implications for Global Malaria Transmission" Tropical Medicine and Infectious Disease 10, no. 4: 109. https://doi.org/10.3390/tropicalmed10040109
APA StyleKhan, J., Zhang, D., Gholizadeh, S., Deng, Y., Aziz, A., Chen, J., Shah, P. T., Lv, Z., & Chen, T. (2025). Phylogeographic Patterns and Genetic Diversity of Anopheles stephensi: Implications for Global Malaria Transmission. Tropical Medicine and Infectious Disease, 10(4), 109. https://doi.org/10.3390/tropicalmed10040109