Lipopolysaccharide-Induced Acute Kidney Injury Is Dependent on an IL-18 Receptor Signaling Pathway

 ,
,
Abstract
1. Introduction
2. Results
2.1. Interleukin (IL)-18Rα Deficiency Improves Survival after Lipopolysaccharide (LPS)-Induced Acute Kidney Injury (AKI)
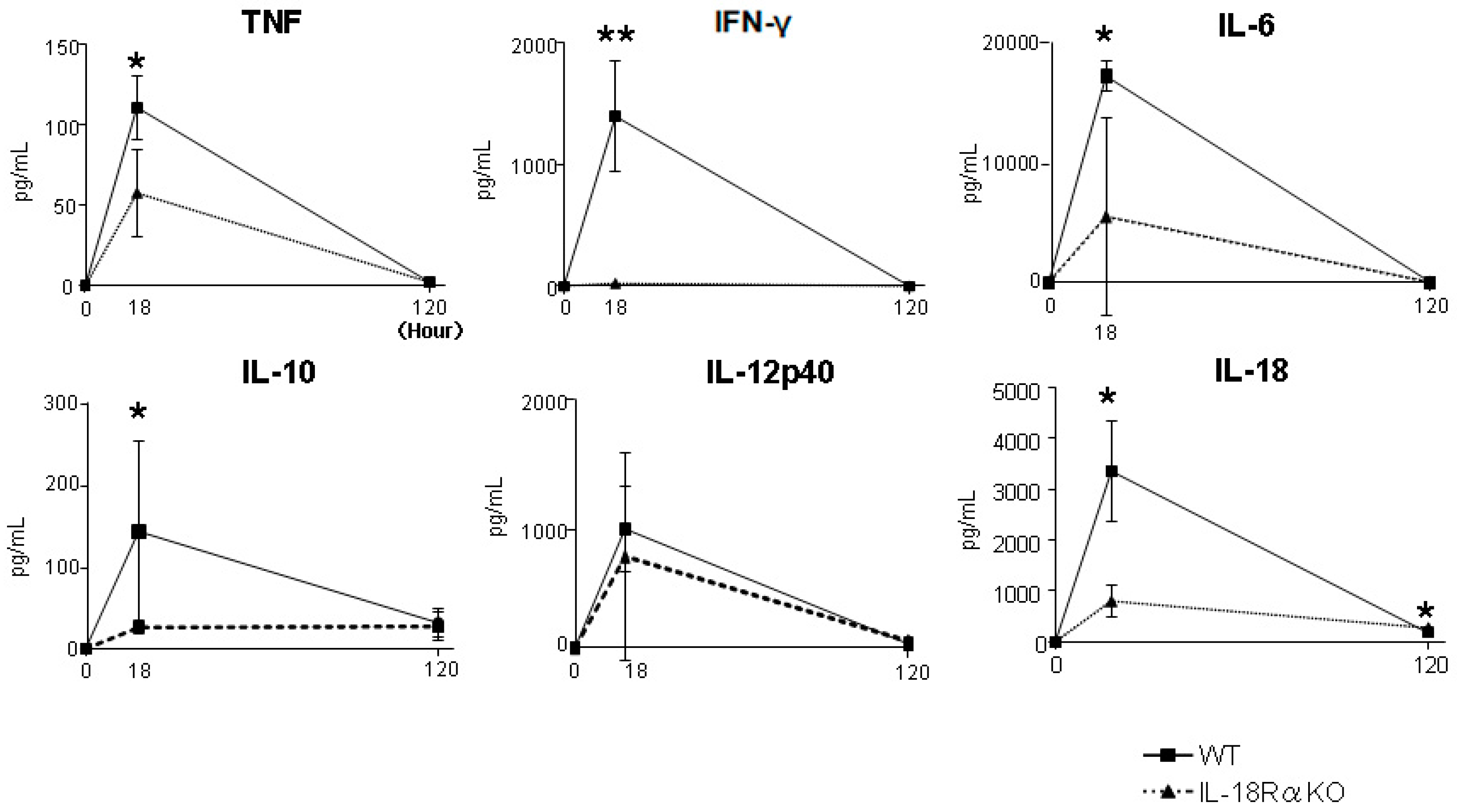
2.2. Serum Biomarkers in Acute Kidney Injury
2.3. The Infiltration of CD4+ T Cells and Antigen-Presenting Cells (APCs) in the Kidney
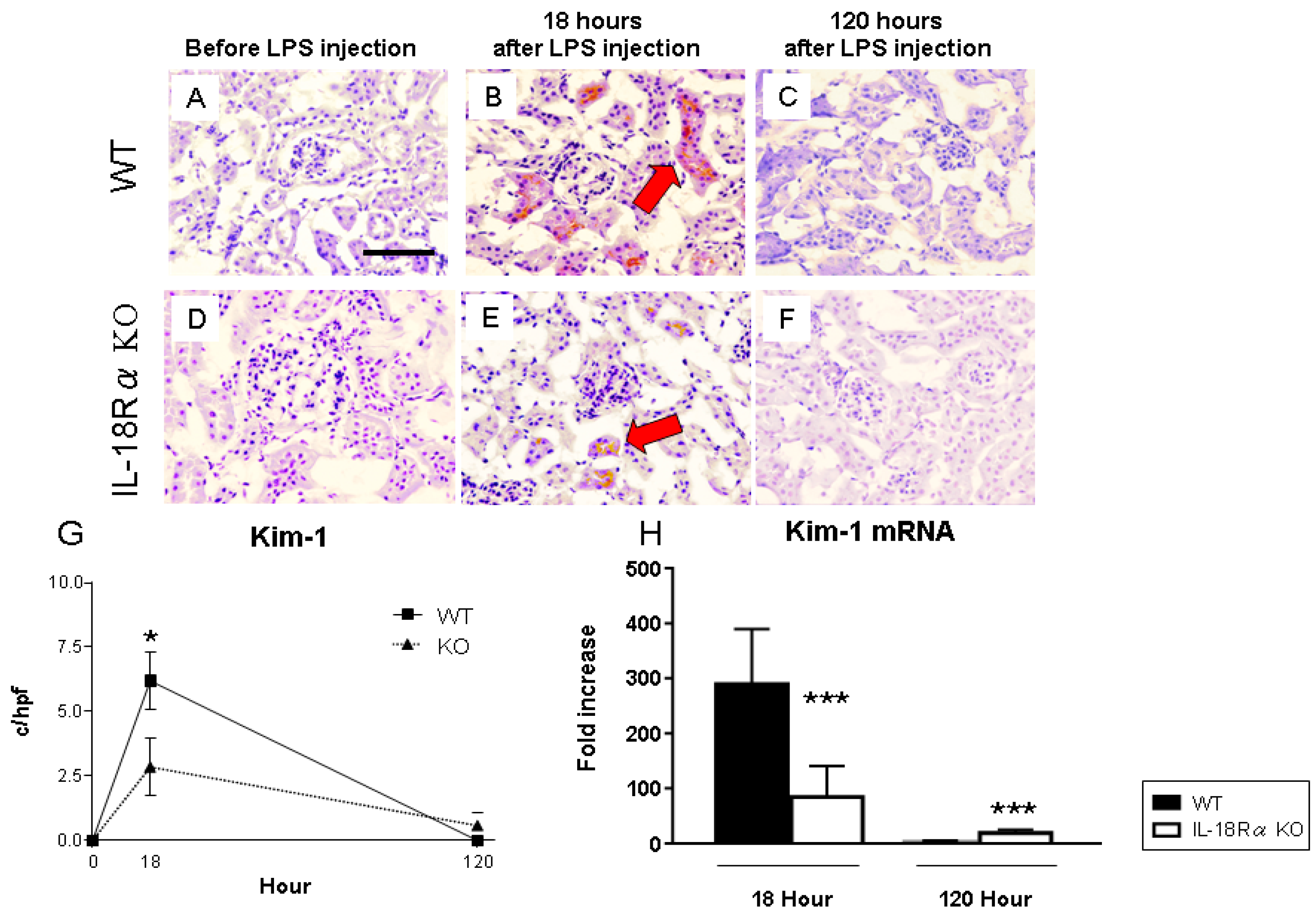
2.4. Renal Kim-1 Expression
2.5. Renal mRNA Expression in LPS-Induced AKI
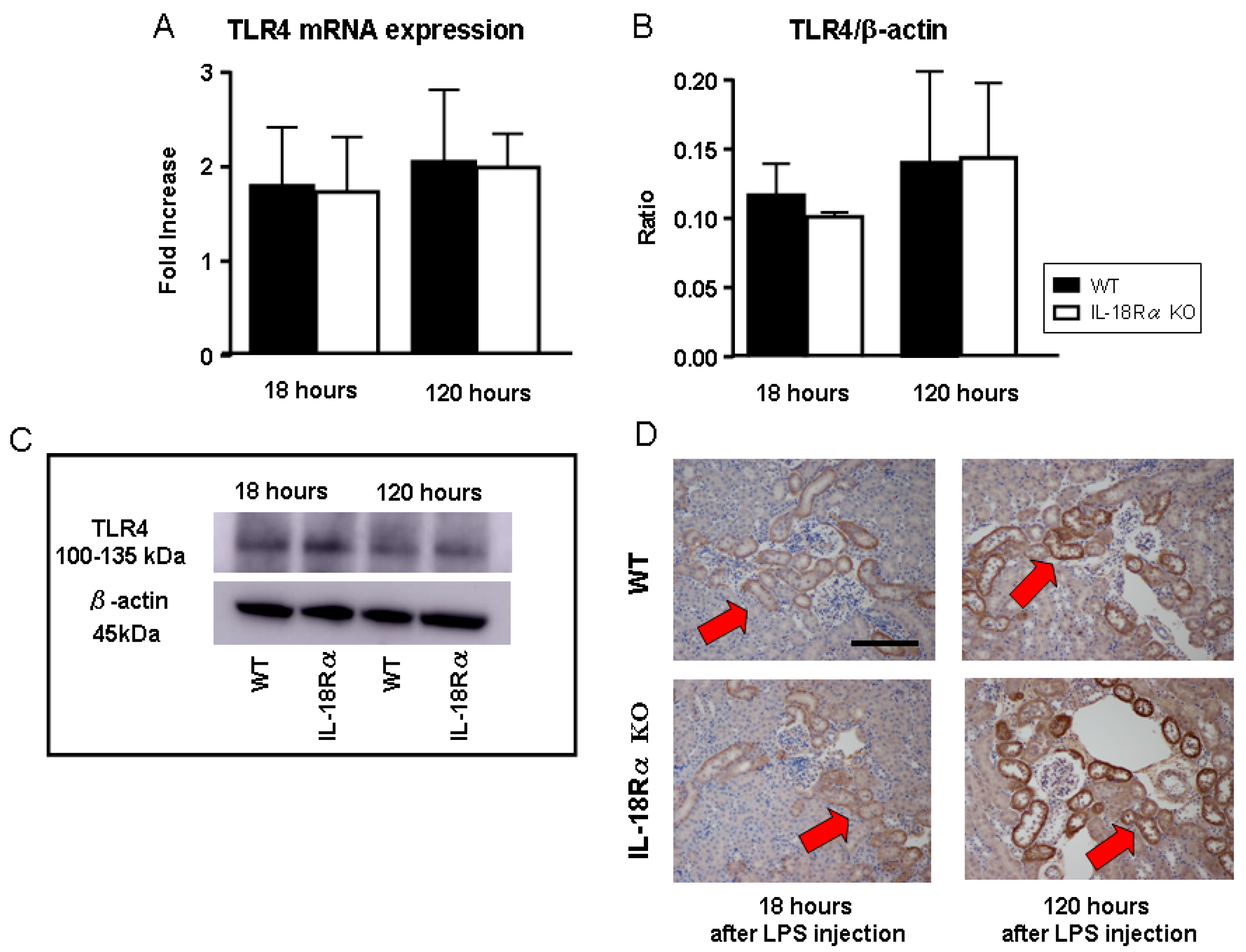
2.6. Toll-Like Receptor 4 (TLR4) Expression in LPS-Induced AKI
2.7. Intracellular Interferon-Gamma (IFN-γ) Staining in CD4+ T Cells and APCs after LPS-Induced AKI
2.8. Classically- and Alternatively-Activated Macrophages in LPS-Induced AKI
2.9. Splenocyte Adoptive Transfer Restored the Kidneys’ Susceptibility to LPS-Induced AKI
3. Discussion
4. Materials and Methods
4.1. Ethics Statement
4.2. Animals
4.3. Murine Model of Endotoxin-Induced Acute Kidney Injury
4.4. Assessment of Renal Injury
4.5. Measurements of mRNA Expression in the Kidney by Real-Time PCR
4.6. Serum Cytokines Quantitation by ELISA
4.7. Western blotting
4.8. FACS Analysis
4.9. Cell Sorting and Adoptive Transfer Experiments
4.10. Statistical Analysis
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| AKI | Acute Kidney Injury |
| APCs | Antigen-Presenting Cells |
| BUN | Blood Urea Nitrogen |
| c/gcs | Cell Numbers Per Glomerular Cross-Section |
| c/hpf | Cell Numbers Per High-Power Field |
| CCL2 | Chemokine (C-C Motif) Ligand 2 |
| CCR7 | C-C Chemokine Receptor Type 7 |
| DCs | Dendritic Cells |
| IFN | Interferon |
| IL-18Rα KO | Interluekin-18 Receptor α Knock-Out |
| IL-18Rβ | Interluekin-18 Receptor β |
| IL-4Rα | Interleukin-4 Receptor Alpha |
| iNOS | Inducible Nitric Oxidase Synthase |
| Kim-1 | Kidney Injury Molecule-1 |
| LPS | Lipopolysaccharide |
| MCP-1 | Monocyte Chemoattractant Chemokine-1 |
| MHC | Major Histocompatibility Complex |
| TLR4 | Toll-Like Receptor 4 |
| TNF | Tumor Necrosis Factor |
| WT | Wild-Type |
References
- Adachi, O.; Kawai, T.; Takeda, K.; Matsumoto, M.; Tsutsui, H.; Sakagami, M.; Nakanishi, K.; Akira, S. Targeted disruption of the MyD88 gene results in loss of IL-1- and IL-18-mediated function. Immunity 1998, 9, 143–150. [Google Scholar] [CrossRef]
- O’Neill, L.A.; Dinarello, C.A. The IL-1 receptor/toll-like receptor superfamily: Crucial receptors for inflammation and host defense. Immunol. Today 2000, 21, 206–209. [Google Scholar] [CrossRef]
- Suzuki, N.; Chen, N.J.; Millar, D.G.; Suzuki, S.; Horacek, T.; Hara, H.; Bouchard, D.; Nakanishi, K.; Penninger, J.M.; Ohashi, P.S.; et al. IL-1 receptor-associated kinase 4 is essential for IL-18-mediated NK and Th1 cell responses. J. Immunol. 2003, 170, 4031–4035. [Google Scholar] [CrossRef] [PubMed]
- Raué, H.P.; Brien, J.D.; Hammarlund, E.; Slifka, M.K. Activation of virus-specific CD8+ T cells by lipopolysaccharide-induced IL-12 and IL-18. J. Immunol. 2004, 173, 6873–6881. [Google Scholar] [CrossRef] [PubMed]
- Parikh, C.R.; Abraham, E.; Ancukiewicz, M.; Edelstein, C.L. Urine IL-18 is an early diagnostic marker for acute kidney injury and predicts mortality in the intensive care unit. J. Am. Soc. Nephrol. 2005, 16, 3046–3052. [Google Scholar] [CrossRef] [PubMed]
- Bani-Hani, A.H.; Leslie, J.A.; Asanuma, H.; Dinarello, C.A.; Campbell, M.T.; Meldrum, D.R.; Zhang, H.; Hile, K.; Meldrum, K.K. IL-18 neutralization ameliorates obstruction-induced epithelial-mesenchymal transition and renal fibrosis. Kidney Int. 2009, 76, 500–511. [Google Scholar] [CrossRef] [PubMed]
- Yano, T.; Nozaki, Y.; Kinoshita, K.; Hino, S.; Hirooka, Y.; Niki, K.; Shimazu, H.; Kishimoto, K.; Funauch, M.; Matsumura, I. The pathological role of IL-18Rα in renal ischemia/reperfusion injury. Lab. Investig. 2015, 95, 78–91. [Google Scholar] [CrossRef] [PubMed]
- Ghose, P.; Ali, A.Q.; Fang, R.; Forbes, D.; Ballard, B.; Ismail, N. The interaction between IL-18 and IL-18 receptor limits the magnitude of protective immunity and enhances pathogenic responses following infection with intracellular bacteria. J. Immunol. 2011, 187, 1333–1346. [Google Scholar] [CrossRef] [PubMed]
- Devarajan, P. Emerging biomarkers of acute kidney injury. Contrib. Nephrol. 2007, 156, 203–212. [Google Scholar] [PubMed]
- Bajwa, A.; Kinsey, G.R.; Okusa, M.D. Immune mechanisms and novel pharmacological therapies of acute kidney injury. Curr. Drug Targets 2009, 10, 1196–1204. [Google Scholar] [CrossRef] [PubMed]
- Day, Y.J.; Huang, L.; Ye, H.; Li, L.; Linden, J.; Okusa, M.D. Renal ischemia-reperfusion injury and adenosine 2A receptor-mediated tissue protection: The role of CD4+ T cells and IFN-gamma. J. Immunol. 2006, 176, 3108–3114. [Google Scholar] [CrossRef] [PubMed]
- Day, Y.J.; Huang, L.; Ye, H.; Linden, J.; Okusa, M.D. Renal ischemia-reperfusion injury and adenosine 2A receptor-mediated tissue protection: Role of macrophages. Am. J. Physiol. Ren. Physiol. 2005, 288, 722–731. [Google Scholar] [CrossRef] [PubMed]
- Cantaluppi, V.; Biancone, L.; Romanazzi, G.M.; Figureliolini, F.; Beltramo, S.; Galimi, F.; Camboni, M.G.; Deriu, E.; Conaldi, P.; Bottelli, A.; et al. Macrophage stimulating protein may promote tubular regeneration after acute injury. J. Am. Soc. Nephrol. 2008, 19, 1904–1918. [Google Scholar] [CrossRef] [PubMed]
- Humphreys, B.D.; Valerius, M.T.; Kobayashi, A.; Mugford, J.W.; Soeung, S.; Duffield, J.S.; McMahon, A.P.; Bonventre, J.V. Intrinsic epithelial cells repair the kidney after injury. Cell Stem Cell 2008, 2, 284–291. [Google Scholar] [CrossRef] [PubMed]
- Fremond, C.M.; Togbe, D.; Doz, E.; Rose, S.; Vasseur, V.; Maillet, I.; Jacobs, M.; Ryffel, B.; Quesniaux, V.F. IL-1 receptor-mediated signal is an essential component of MyD88-dependent innate response to Mycobacterium tuberculosis infection. J. Immunol. 2007, 179, 1178–1189. [Google Scholar] [CrossRef] [PubMed]
- Pedra, J.H.; Sutterwala, F.S.; Sukumaran, B.; Ogura, Y.; Qian, F.; Montgomery, R.R.; Flavell, R.A.; Fikrig, E. ASC/PYCARD and caspase-1 regulate the IL-18/IFN-gamma axis during Anaplasma phagocytophilum infection. J. Immunol. 2007, 179, 4783–4791. [Google Scholar] [CrossRef] [PubMed]
- Car, B.D.; Eng, V.M.; Schnyder, B.; Ozmen, L.; Huang, S.; Gallay, P.; Heumann, D.; Aguet, M.; Ryffel, B. Interferon gamma receptor deficient mice are resistant to endotoxic shock. J. Exp. Med. 1994, 179, 1437–1444. [Google Scholar] [CrossRef] [PubMed]
- Ogata, M.; Zhang, Y.; Wang, Y.; Itakura, M.; Zhang, Y.Y.; Harada, A.; Hashimoto, S.; Matsushima, K. Chemotactic response toward chemokines and its regulation by transforming growth factor-beta1 of murine bone marrow hematopoietic progenitor cell-derived different subset of dendritic cells. Blood 1999, 93, 3225–3232. [Google Scholar] [PubMed]
- Brunialti, M.K.; Santos, M.C.; Rigato, O.; Machado, F.R.; Silva, E.; Salomao, R. Increased percentages of T helper cells producing IL-17 and monocytes expressing markers of alternative activation in patients with sepsis. PLoS ONE 2012, 7, e37393. [Google Scholar] [CrossRef] [PubMed]
- Kinoshita, K.; Yamagata, T.; Nozaki, Y.; Sugiyama, M.; Ikoma, S.; Funauchi, M.; Kanamaru, A. Blockade of IL-18 receptor signaling delays the onset of autoimmune disease in MRL-Faslpr mice. J. Immunol. 2004, 173, 5312–5318. [Google Scholar] [CrossRef] [PubMed]
- Nozaki, Y.; Kinoshita, K.; Yano, T.; Asato, K.; Shiga, T.; Hino, S.; Niki, K.; Nagare, Y.; Kishimoto, K.; Shimazu, H.; et al. Signaling through the interleukin-18 receptor α attenuates inflammation in cisplatin-induced acute kidney injury. Kidney Int. 2012, 82, 892–902. [Google Scholar] [CrossRef] [PubMed]
- Traylor, L.A.; Mayeux, P.R. Nitric oxide generation mediates lipid A-induced oxidant injury in renal proximal tubules. Arch. Biochem. Biophys. 1997, 338, 129–135. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Walker, L.M.; Mayeux, P.R. Role of nitric oxide in lipopolysaccharide-induced oxidant stress in the rat kidney. Biochem. Pharmacol. 2000, 59, 203–209. [Google Scholar] [CrossRef]
- Honda, T.; Egen, J.G.; Lämmermann, T.; Kastenmüller, W.; Torabi-Parizi, P.; Germain, R.N. Tuning of antigen sensitivity by T cell receptor-dependent negative feedback controls T cell effector function in inflamed tissues. Immunity 2014, 40, 235–247. [Google Scholar] [CrossRef] [PubMed]
- Crotty, S. Follicular helper CD4 T cells (TFH). Immunol. Rev. 2014, 260, 168–182. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Yamane, H.; Paul, W.E. Differentiation of effector CD4 T cell populations (*). Annu. Rev. Immunol. 2010, 28, 445–489. [Google Scholar] [CrossRef] [PubMed]
- Calvani, N.; Tucci, M.; Richards, H.B.; Tartaglia, P.; Silvestris, F. Th1 cytokines in the pathogenesis of lupus nephritis: The role of IL-18. Autoimmun. Rev. 2005, 4, 542–548. [Google Scholar] [CrossRef] [PubMed]
- McSorley, S.J. Immunity to intestinal pathogens: Lessons learned from Salmonella. Immunity 2009, 30, 277–288. [Google Scholar] [CrossRef] [PubMed]
- Hyodo, Y.; Matsui, K.; Hayashi, N.; Tsutsui, H.; Kashiwamura, S.; Yamauchi, H.; Hiroishi, K.; Takeda, K.; Tagawa, Y.; Iwakura, Y.; et al. IL-18 up-regulates perforin-mediated NK activity without increasing perforin messenger RNA expression by binding to constitutively expressed IL-18 receptor. J. Immunol. 1999, 162, 1662–1668. [Google Scholar] [PubMed]
- McLachlan, J.B.; Catron, D.M.; Moon, J.J.; Jenkins, M.K. Dendritic cell antigen presentation drives simultaneous cytokine production by effector and regulatory T cells in inflamed skin. Immunity 2014, 40, 235–247. [Google Scholar] [CrossRef] [PubMed]
- Muller, A.J.; Filipe-Santos, O.; Eberl, G.; Aebischer, T.; Spath, G.F.; Bousso, P. CD4+ T cells rely on a cytokine gradient to control intracellular pathogens beyond sites of antigen presentation. Immunity 2012, 37, 147–157. [Google Scholar] [CrossRef] [PubMed]
- Gracie, J.A.; Forsey, R.J.; Chan, W.L.; Gilmour, A.; Leung, B.P.; Greer, M.R.; Kennedy, K.; Carter, R.; Wei, X.Q.; Xu, D.; et al. A proinflammatory role for IL-18 in rheumatoid arthritis. J. Clin. Investig. 1999, 104, 1393–1401. [Google Scholar] [CrossRef] [PubMed]
- Nakahira, M.; Ahn, H.J.; Park, W.R.; Gao, P.; Tomura, M.; Park, C.S.; Hamaoka, T.; Ohta, T.; Kurimoto, M.; Fujiwara, H. Synergy of IL-12 and IL-18 for IFN-gamma gene expression: IL-12-induced STAT4 contributes to IFN-gamma promoter activation by up-regulating the binding activity of IL-18-induced activator protein 1. J. Immunol. 2002, 168, 1146–1153. [Google Scholar] [CrossRef] [PubMed]
- Yamamura, M.; Kawashima, M.; Taniai, M.; Yamauchi, H.; Tanimoto, T.; Kurimoto, M.; Morita, Y.; Ohmoto, Y.; Makino, H. Interferon-gamma-inducing activity of interleukin-18 in the joint with rheumatoid arthritis. Arthritis Rheumatol. 2001, 44, 275–285. [Google Scholar] [CrossRef]
- Gutierrez-Ramos, J.C.; Bluethmann, H. Molecules and mechanisms operating in septic shock: Lessons from knockout mice. Immunol. Today 1997, 18, 329–334. [Google Scholar] [CrossRef]
- Okamura, H.; Tsutsi, H.; Komatsu, T.; Yutsudo, M.; Hakura, A.; Tanimoto, T.; Torigoe, K.; Okura, T.; Nukada, Y.; Hattori, K.; et al. Cloning of a new cytokine that induces IFN-gamma production by T cells. Nature 1995, 378, 88–91. [Google Scholar] [CrossRef] [PubMed]
- Munder, M.; Mallo, M.; Eichmann, K.; Modolell, M. Murine macrophages secrete interferon gamma upon combined stimulation with interleukin (IL)-12 and IL-18: A novel pathway of autocrine macrophage activation. J. Exp. Med. 1998, 187, 2103–2108. [Google Scholar] [CrossRef] [PubMed]
- Fehniger, T.A.; Shah, M.H.; Turner, M.J.; VanDeusen, J.B.; Whitman, S.P.; Cooper, M.A.; Suzuki, K.; Wechser, M.; Goodsaid, F.; Caligiuri, M.A. Differential cytokine and chemokine gene expression by human NK cells following activation with IL-18 or IL-15 in combination with IL-12 implications for the innate immune response. J. Immunol. 1999, 162, 4511–4520. [Google Scholar] [PubMed]
- Yoshimoto, T.; Okamura, H.; Tagawa, Y.I.; Iwakura, Y.; Nakanishi, K. Interleukin 18 together with interleukin 12 inhibits IgE production by induction of interferon-gamma production from activated B cells. Proc. Natl. Acad. Sci. USA 1997, 94, 3948–3953. [Google Scholar] [CrossRef] [PubMed]
- Trinchieri, G. Interleukin-12: A proinflammatory cytokine with immunoregulatory functions that bridge innate resistance and antigen-specific adaptive immunity. Annu. Rev. Immunol. 1995, 13, 251–276. [Google Scholar] [CrossRef] [PubMed]
- Dinarello, C.A.; Kaplanski, G. Interleukin-18 treatment options for inflammatory diseases. Expert Rev. Clin. Immunol. 2005, 1, 619–632. [Google Scholar] [CrossRef] [PubMed]
- Mazodier, K.; Marin, V.; Novick, D.; Farnarier, C.; Robitail, S.; Schleinitz, N.; Veit, V.; Paul, P.; Rubinstein, M.; Dinarello, C.A.; et al. Severe imbalance of IL-18/IL-18BP in patients with secondary hemophagocytic syndrome. Blood 2005, 106, 3483–3489. [Google Scholar] [CrossRef] [PubMed]
- Paulukat, J.; Bosmann, M.; Nold, M.; Garkisch, S.; Kämpfer, H.; Frank, S.; Raedle, J.; Zeuzem, S.; Pfeilschifter, J.; Mühl, H. Expression and release of IL-18 binding protein in response to IFN-gamma. J. Immunol. 2001, 167, 7038–7043. [Google Scholar] [CrossRef] [PubMed]
- Chiossone, L.; Audonnet, S.; Chetaille, B.; Chasson, L.; Farnarier, C.; Berda-Haddad, Y.; Jordan, S.; Koszinowski, U.H.; Dalod, M.; Mazodier, K.; et al. Protection from inflammatory organ damage in a murine model of hemophagocytic lymphohistiocytosis using treatment with IL-18 binding protein. Front. Immunol. 2012, 3, 239. [Google Scholar] [CrossRef] [PubMed]
- Torigoe, K.; Ushio, S.; Okura, T.; Kobayashi, S.; Taniai, M.; Kunikata, T.; Murakami, T.; Sanou, O.; Kojima, H.; Fujii, M.; et al. Purification and characterization of the human interleukin-18 receptor. J. Biol. Chem. 1997, 272, 25737–25742. [Google Scholar] [CrossRef] [PubMed]
- Dinarello, C.A. Interleukin-18. Methods 1999, 19, 121–132. [Google Scholar] [CrossRef] [PubMed]
- Born, T.L.; Thomassen, E.; Bird, T.A.; Sims, J.E. Cloning of a novel receptor subunit, AcPL, required for interleukin-18 signaling. J. Biol. Chem. 1998, 6, 29445–29450. [Google Scholar] [CrossRef]
- Shimazu, H.; Kinoshita, K.; Hino, S.; Yano, T.; Kishimoto, K.; Nagare, Y.; Nozaki, Y.; Sugiyama, M.; Ikoma, S.; Funauchi, M. Effect of combining ACE inhibitor and statin in lupus-prone mice. Clin. Immunol. 2010, 136, 188–196. [Google Scholar] [CrossRef] [PubMed]
- Nozaki, Y.; Yamagata, T.; Yoo, B.S.; Sugiyama, M.; Ikoma, S.; Kinoshita, K.; Funauchi, M.; Kanamaru, A. The beneficial effects of treatment with all-trans-retinoic acid plus corticosteroid on autoimmune nephritis in NZB/WF mice. Clin. Exp. Immunol. 2005, 139, 74–83. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 18 h | 120 h | |
|---|---|---|
| Cytokines | WT vs. IL-18Rα KO | WT vs. IL-18Rα KO |
| IL-6 | 643.2 ± 201.5 vs. 72.1 ± 49.3 ** | 7.5 ± 4.6 vs. 2.4 ± 0.4 |
| IL-10 | 76.3 ± 10.3 vs. 33.3 ± 10.6 * | 8.4 ± 4.0 vs. 7.3 ± 1.8 |
| IL-12 | 3.2 ± 0.7 vs. 1.4 ± 0.8 | 0.3 ± 0.2 vs. 0.3 ± 0.1 |
| IL-18 | 2.7 ± 0.4 vs. 1.3 ± 0.2 ** | 1.3 ± 0.4 vs. 1.7 ± 0.1 |
| IFN-γ | 4.3 ± 2.5 vs. 1.8 ± 0.4 * | 3.4 ± 1.6 vs. 4.6 ± 0.6 |
| TNF | 27.5 ± 4.6 vs. 11.9 ± 4.2 * | 2.3 ± 0.5 vs. 2.9 ± 0.1 |
| Chemokines | ||
| CCL2/MCP-1 | 59.8 ± 15.7 vs. 28.7 ± 6.7 | 4.2 ± 1.3 vs. 4.7 ± 0.6 |
| Th Cell Subset Transcription Factors | ||
| GATA3 | 0.8 ± 0.4 vs. 0.9 ± 0.2 | 1.6 ± 0.7 vs. 1.8 ± 0.5 |
| T-bet | 2.5 ± 0.5 vs. 1.0 ± 0.2 * | 4.9 ± 1.8 vs. 5.8 ± 0.7 |
| IFN-γ+ | 18 h | 120 h |
|---|---|---|
| WT vs. IL-18Rα KO | WT vs. IL-18Rα KO | |
| CD4+ | 0.7 ± 0.1 vs. 0.3 ± 0.1 * | 0.2 ± 0.1 vs. 0.3 ± 0.1 |
| F4/80+ | 0.2 ± 0.0 vs. 0.1 ± 0.0 * | 1.4 ± 0.8 vs. 1.1 ± 0.5 |
| CD11b+ | 0.6 ± 0.1 vs. 0.2 ± 0.0 ** | 0.1 ± 0.0 vs. 0.2 ± 0.1 |
| CD11c+ | 0.3 ± 0.1 vs. 0.3 ± 0.1 | 0.6 ± 0.6 vs. 0.6 ± 0.2 |
| 18 h | 120 h | |
|---|---|---|
| M1 Macrophage | WT vs. IL-18Rα KO | WT vs. IL-18Rα KO |
| iNOS | 123.8 ± 12.8 vs. 12.9 ± 15.1 ** | 2.8 ± 3.2 vs. 2.2 ± 1.2 |
| M2 Macrophage | ||
| IL-4Rα | 6.4 ± 1.7 vs. 2.4 ± 1.2 ** | 4.2 ± 1.3 vs. 4.7 ± 0.6 |
| M1/M2 ratio | 19.0 ± 4.0 vs. 4.2 ± 1.6 ** | 1.3 ± 0.5 vs. 1.3 ± 0.2 |
| Dendritic Cell | ||
| CCR7 | 9.8 ± 0.8 vs. 2.1 ± 0.7 *** | 1.9 ± 0.8 vs. 1.0 ± 0.2 |
| Forward Primer | Reverse Primer | |
|---|---|---|
| 18SrRNA | GTAACCCGTTGAACCCCATTC | GCCTCACTAAACCATCCAATCG |
| TNF | CGATCACCCCGAAGTTCAGTA | GGTGCCTATGTCTCAGCCTCTT |
| CCL2/MCP-1 | AAAAACCTGGATCGGAACCAA | CGGGTCAACTTCACATTCAAAG |
| GATA3 | AGGGACATCCTGCGCGAACTGT | CATCTTCCGGTTTCGGGTCTGG |
| RORγt | AGATTGCCCTCTACACG | GGCTTGGACCACGATG |
| T-bet | CCTGGACCCAACTGTCAACT | AACTGTGTTCCCGAGGTGTC |
| Gene Database No | |
|---|---|
| 18SrRNA | NM_026744.3 |
| IFN-γ | NM_008337.3 |
| IL-4 | NM_021283.2 |
| IL-6 | Mm00446190 |
| IL-10 | NM_010548.1 |
| IL-12p40 | NM_008352.2 |
| IL-18 | NM_008360.1 |
| IL-18R1 (IL-18Rα) | Mm00515180_m1 |
| IL-18R2 (IL-18Rβ) | Mm00516053_m1 |
| Kim-1 | NM_134248.1 |
| TLR4 | Mm00445273_m1 |
| CCR7 | Mm99999130_s1 |
| iNOS | Mm00440502 |
| IL-4Rα | Mm01275139 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nozaki, Y.; Hino, S.; Ri, J.; Sakai, K.; Nagare, Y.; Kawanishi, M.; Niki, K.; Funauchi, M.; Matsumura, I. Lipopolysaccharide-Induced Acute Kidney Injury Is Dependent on an IL-18 Receptor Signaling Pathway. Int. J. Mol. Sci. 2017, 18, 2777. https://doi.org/10.3390/ijms18122777
Nozaki Y, Hino S, Ri J, Sakai K, Nagare Y, Kawanishi M, Niki K, Funauchi M, Matsumura I. Lipopolysaccharide-Induced Acute Kidney Injury Is Dependent on an IL-18 Receptor Signaling Pathway. International Journal of Molecular Sciences. 2017; 18(12):2777. https://doi.org/10.3390/ijms18122777
Chicago/Turabian StyleNozaki, Yuji, Shoichi Hino, Jinhai Ri, Kenji Sakai, Yasuaki Nagare, Mai Kawanishi, Kaoru Niki, Masanori Funauchi, and Itaru Matsumura. 2017. "Lipopolysaccharide-Induced Acute Kidney Injury Is Dependent on an IL-18 Receptor Signaling Pathway" International Journal of Molecular Sciences 18, no. 12: 2777. https://doi.org/10.3390/ijms18122777
APA StyleNozaki, Y., Hino, S., Ri, J., Sakai, K., Nagare, Y., Kawanishi, M., Niki, K., Funauchi, M., & Matsumura, I. (2017). Lipopolysaccharide-Induced Acute Kidney Injury Is Dependent on an IL-18 Receptor Signaling Pathway. International Journal of Molecular Sciences, 18(12), 2777. https://doi.org/10.3390/ijms18122777