Abstract
Nonsteroidal anti-inflammatory drugs (NSAIDs) are commonly used as anti-inflammatory and analgesic agents. This family of drugs suppresses prostaglandin synthesis through inhibition of cyclooxygenase (COX) enzymes. Recent studies showed that anti-carcinogenic effects of these drugs are especially mediated by COX-2 enzyme. Etodolac is a COX-2 inhibitor and though not perfectly selective, it exhibits “preferential selectivity” for COX-2. In this study, the anti-proliferative and apoptotic effects of etodolac and its hydrazone or triazole derivatives (SGK 206 and SGK 242, respectively), were investigated on prostate cancer cell line PC-3 and human colorectal carcinoma cell line HT-29. Our data showed that SGK 206 and SGK 242 were more effective in the inhibition of proliferation and induction of apoptosis compared to etodolac in both cell lines.
1. Introduction
Nonsteroidal anti-inflammatory drugs (NSAIDs), regularly used for their anti-inflammatory and analgesic effects, were shown to have potency for cancer prevention as well [1]. This family of drugs suppresses prostaglandin synthesis through inhibition of cyclooxygenase (COX) enzymes. COX enzymes have two functional isoforms in human, COX-1 and COX-2. COX-1 is commonly expressed in body, showing constitutive activation. COX-2, on the other hand, is hardly detectable in normal conditions, but is induced upon stimulation by mitogenic agents, cytokines, growth factors etc.
Early reports about the role of NSAIDs in cancer were from the studies on colorectal cancer (CRC). Inducible form of COX enzymes, namely COX-2, was found to be elevated in CRC patients and early polyp formations were preceded by COX-2 induction [2]. Regular use of NSAIDs reportedly reduced CRC occurence 30–50%, mostly by COX inhibition, though later studies revealed that COX-2 inhibitors could also act on COX-independent pathways [3]. These studies were further confirmed by many others and in different types of cancers like prostate, breast or lung cancers.
Etodolac ((R,S) 2-[1,8-diethyl-1,3,4-tetrahydrapyrano(3,4-b)indole-1-yl] acetic acid), is an FDA-approved NSAID partially selective for COX-2. Etodolac has different structural characteristics than other coxibs in that it has no sulfonyl, sulfonamide or sulfone groups to facilitate COX-2 binding. Its toxicity and relative selectivity for COX-2 is low, and although there are numerous studies about its anti-carcinogenic activity, dose-response relationship on various cancer cell lines are not well established.
In this study, we focused on the anti-carcinogenic effects of etodolac and its hydrazone and triazole derivatives (SGK 206 and SGK 242, respectively) in two cancer cell lines. Both derivatives used in this study were synthesized and characterized by our collaborator .Dr.Küçükgüzel [4].
2. Materials and Methods
Both cell lines were maintained in DMEM medium, supplemented with 10% FBS, 1% glutamine and penicillin/streptomycin at 37 °C with 5% CO2 in atmosphere. Cell viability and growth inhibition were determined using MTT colorimetric assay, according to the instructions of manufacturer (Cell Proliferation Kit I (MTT), 11465007001, Roche, Indianapolis, USA). Briefly, cells were seeded into 96-well plates and left to grow in the presence and absence of agents for indicated time points (24, 48 and/or 72 h). At the end of incubation period, 10 μL of MTT was added to each well and incubated for 4 h, at 37 °C in 5% CO2 humidified incubator. The precipitated formazan, then, dissolved in 100 μL SDS and the absorbance was read at 570 nm using a multi-mode plate reader (Synergy H1, BioTek Instruments Inc., Winooski, VT, USA).
Apoptosis was evaluated by measuring mitochondrial membrane potential changes (MMP assay). Cells were maintained in similar conditions described above, except black opaque 96-well plates were used for culturing. After exposure to appropriate drug for indicated time points, cells were stained with 1 μL of JC-1 staining solution according to the protocol of kit, where JC-1, a lipophilic cationic dye with different absorbance wavelengths in healthy and apoptotic cells, was used to detect mitochondrial membrane depolarization (JC-1 Mitochondrial Membrane Potential Assay Kit, KA1324, Abnova, Taipei City, Taiwan). All tests were repeated at least three times (triplicate each) and statistical evaluation was performed using GraphPad Prism v5.0.
3. Results and Discussion
In our study, we investigated the effect of hydrazone and triazole derivatives of etodolac (SGK 206 and SGK 242) on prostate and colorectal cancer cell lines, PC-3 and HT-29. Novel hydrazone derivatives were found to be effective in the treatment of colon, ovarian, renal cancers [5,6]. One of the most benefical aspects of hydrazone groups is their preferential activity on malignant cells with respect to control cells. Triazoles are another candidate group with high biological activity. Anti-tumor activities for triazole derivatives were reported in colon adenocarcinoma, uterus carcinoma, cervical carcinoma, T cell leukemia and so on [7,8]. Based on these current evidences, hydrazone and triazole derivatives of etodolac were synthesized and tested for their anti-tumorigenic activities. All structures of the newly synthesized compounds were confirmed by spectral (FT-IR, 1H-NMR) methods.
Firstly, we studied low concentrations (0 to 100 µM) of etodolac on PC-3 cell line following 24 and 48 h incubation. No toxicity was observed in these low concentrations (data not shown). Therefore, PC-3 cells were treated with higher concentrations of etodolac (100 µM to 1000 µM) for 24, 48 and 72 h. Here, we observed that anti-proliferative and apoptotic effects of etodolac were only prominent after 500 µM (Figure 1).
On the other hand, derivatives of etodolac had cytotoxic effects in much lower doses at the end of 24 h incubation on PC-3 cells. Both drugs showed cytotoxic effects when concentrations were increased from 25 µM up to 100 µM. IC50 values for SGK 206 and SGK-242 were 40 µM and 25 µM, respectively. Similar to proliferation, SGK 206 and SGK 242 showed apoptotic effects in much lower doses (≥25 µM) (Figure 2). Apoptosis was 30% and 54% at 75 µM and 100 µM concentrations for SGK 206 and 28% and 89% at 25 µM and 50 µM concentrations for SGK 242.
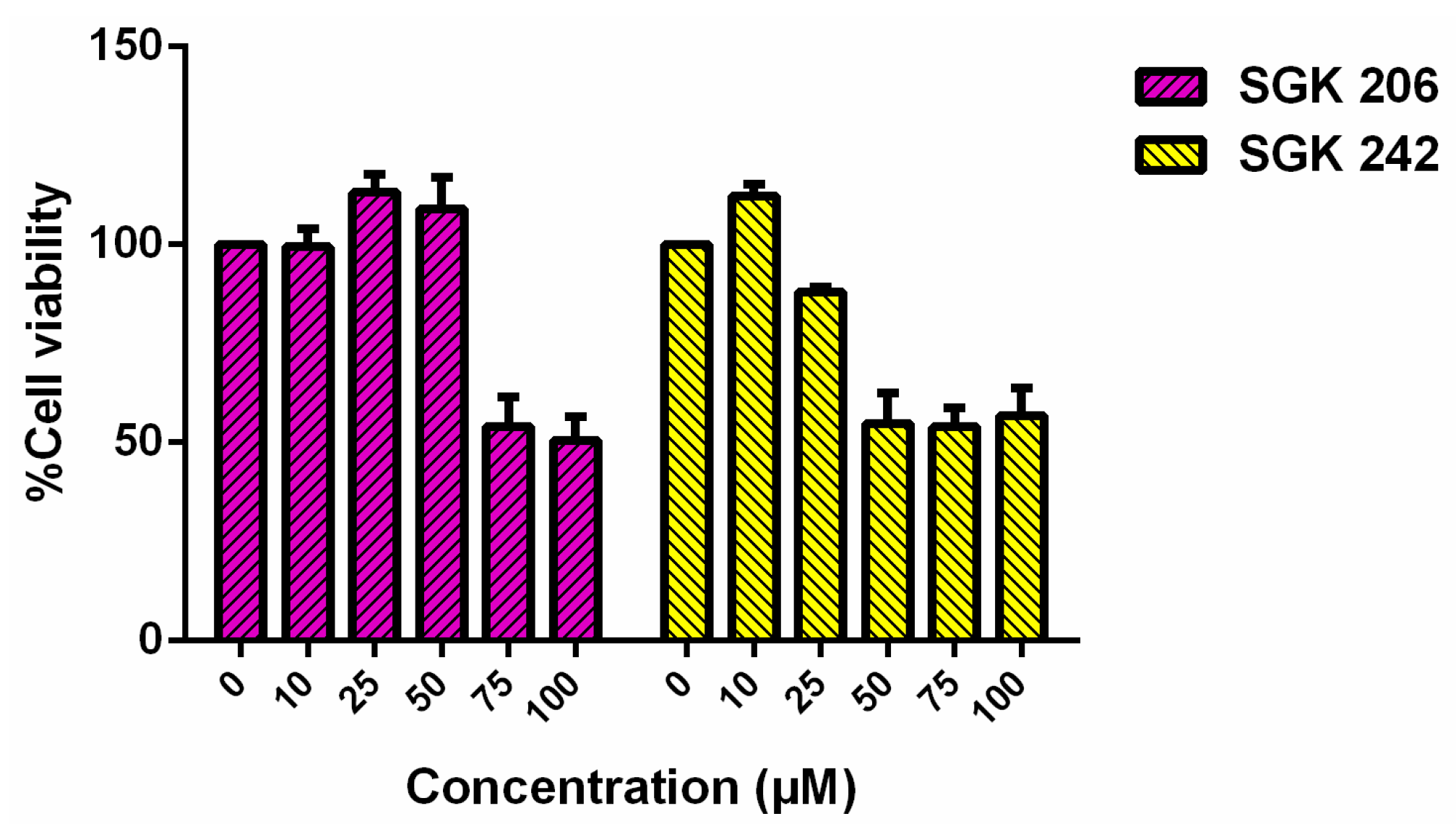
Derivatives of etodolac had cytotoxic effect on HT-29 cells as well (Figure 3). IC50 values for SGK 206 and SGK-242 were 70 µM and 26 µM, respectively.
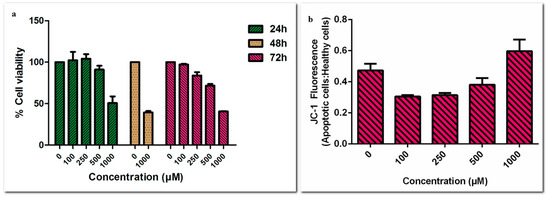
Figure 1.
(a) Cell viability assay results following 24, 48 and 72 h incubations and (b) MMP assay results following 24 h incubation at high concentrations of etodolac (0–1000 µM) in PC-3 cell line.
Figure 1.
(a) Cell viability assay results following 24, 48 and 72 h incubations and (b) MMP assay results following 24 h incubation at high concentrations of etodolac (0–1000 µM) in PC-3 cell line.
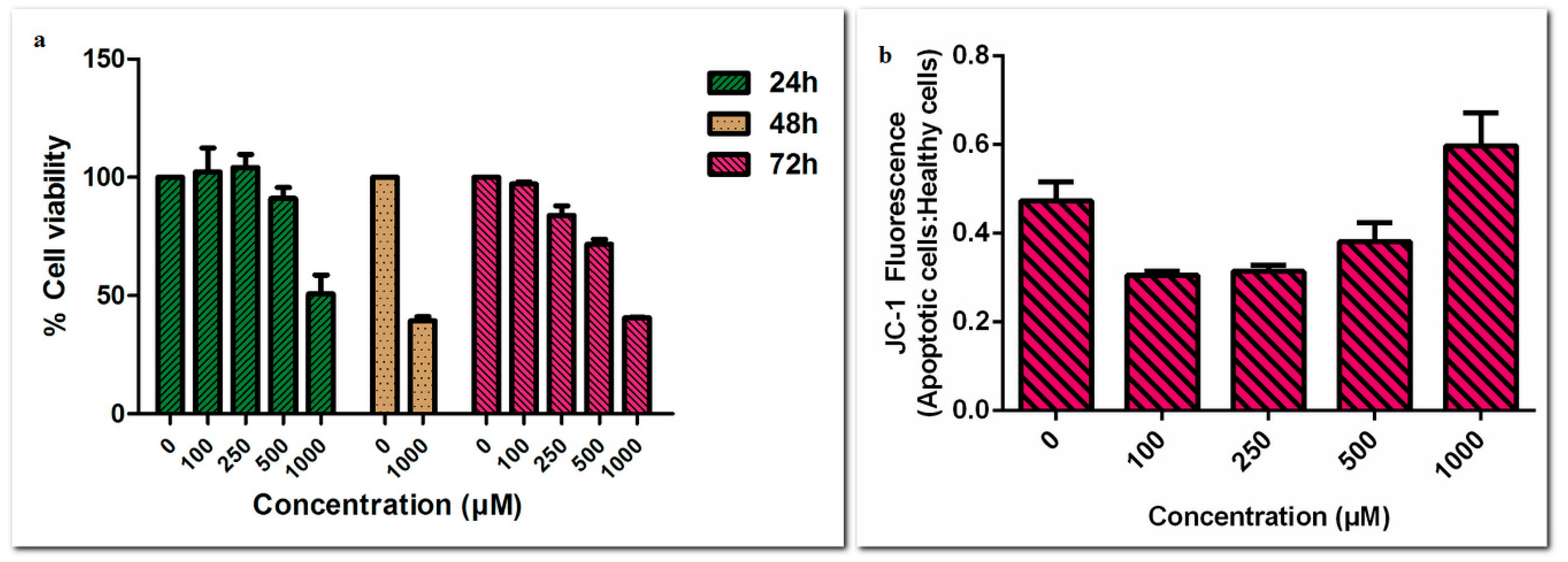
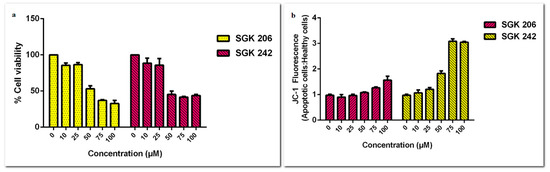
Figure 2.
(a) Cell viability assay and (b) MMP assay results for different concentrations of SGK 206 and SGK 242 (0–100 µM) following 24 h incubation in PC-3 cell line.
Figure 2.
(a) Cell viability assay and (b) MMP assay results for different concentrations of SGK 206 and SGK 242 (0–100 µM) following 24 h incubation in PC-3 cell line.
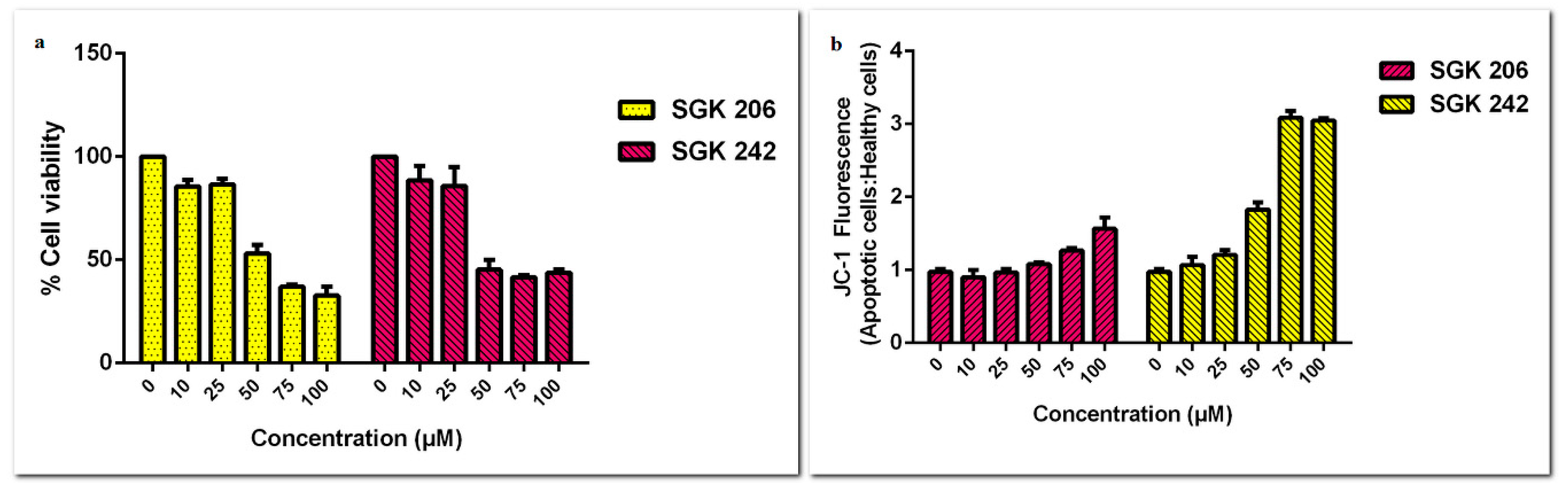
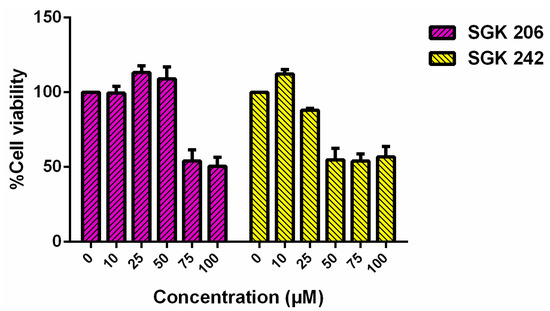
Figure 3.
Cell viability assay results of HT-29 cell line for different concentrations of SGK 206 and SGK 242 (0–100 µM) following 24 h incubation.
Figure 3.
Cell viability assay results of HT-29 cell line for different concentrations of SGK 206 and SGK 242 (0–100 µM) following 24 h incubation.
Our preliminary tests also showed that effects of both agents on control primary dermal fibroblasts are lower, especially in the case of SGK-242 (data not shown). Concentration dependent increase in apoptosis was in agreement with MTT viability tests as verified by higher apoptotic levels induced by SGK-242 and SGK-206 after 50 µM concentrations. Detailed molecular mechanisms underlying these apoptotic responses are yet to be defined.
Findings of our study indicate that SGK 242 had substantial effects in inhibition of proliferation on PC-3 cells and HT-29 cells with respect to SGK-206 and had lower effect in control cells. While SGK-242 had same level of inhibition in both cell lines, SGK-206 was more efficient in PC-3 cells. Therefore both agents could be promising therapeutic targets in cancer treatments.
Acknowledgments
This study was supported by Marmara University Research Fund SAG-C-DRP-100914-0321.
References
- Thun, M.J.; Henley, S.J.; Patrono, C. Nonsteroidal anti-inflammatory drugs as anticancer agents: Mechanistic, pharmacologic, and clinical issues. J. Natl. Cancer Inst. 2002, 94, 252–266. [Google Scholar] [CrossRef]
- Hsi, L.C.; Joon Baek, S.; Eling, T.E. Lack of Cyclooxygenase-2 Activity in HT-29 Human Colorectal Carcinoma Cells. Exp. Cell Res. 2000, 256, 563–570. [Google Scholar] [CrossRef]
- Kobayashi, M.; Nakamura, S.; Shibata, K.; Sahara, N.; Shigeno, K.; Shinjo, K.; Naito, K.; Ohnishi, K. Etodolac inhibits EBER expression and induces Bcl-2-regulated apoptosis in Burkitt’s lymphoma cells. Eur. J. Haematol. 2005, 75, 212–220. [Google Scholar] [CrossRef] [PubMed]
- Çıkla-Süzgün, P.; Özsavcı, D.; Bingöl-Özakpınar, Ö.; Şener, A.; Çevik, Ö.; Özbaş-Turan, S.; Akbuğa, J.; Şahin, F.; Küçükgüzel, Ş.G. Synthesis, cytotoxicity, and pro-apoptosis activity of etodolac hydrazide derivatives as anticancer agents. Arch. Pharm. Chem. Life Sci. 2013, 346, 367–379. [Google Scholar] [CrossRef] [PubMed]
- Easmon, J.; Pürstinger, G.; Thies, K.S.; Heinisch, G.; Hofmann, J. Synthesis, structure-activity relationships, and antitumor studies of 2-benzoxazolyl hydrazones derived from alpha-(N)-acyl heteroaromatics. J. Med. Chem. 2006, 49, 6343–6350. [Google Scholar] [CrossRef] [PubMed]
- Küçükgüzel, Ş.G.; Koç, D.; Çıkla-Süzgün, P.; Özsavcı, D.; Bingöl-Özakpınar, Ö.; Mega-Tiber, P.; Orun, O.; Erzincan, P.; Sağ-Erdem, S.; Şahin, F. Synthesis of Tolmetin Hydrazide-Hydrazones and Discovery of a Potent Apoptosis Inducer in Colon Cancer Cells. Arch. Pharm. 2015, 348, 730–742. [Google Scholar] [CrossRef] [PubMed]
- Romagnoli, R.; Baraldi, P.G.; Cruz-Lopez, O.; Lopez Cara, C.; Carrion, M.D.; Brancale, A.; Hamel, E.; Chen, L.; Bortolozzi, R.; Basso, G.; et al. Synthesis and antitumor activity of 1,5-disubstituted 1,2,4-triazoles as cis-restricted combretastatin analogues. J. Med. Chem. 2010, 53, 4248–4258. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.K.; Kim, J.; Park, H.J. Design, synthesis, and biological evaluation of novel 2-pyridinyl-[1,2,4]triazoles as inhibitors of transforming growth factor beta1 type 1 receptor. Bioorg. Med. Chem. 2004, 12, 2013–2020. [Google Scholar] [CrossRef] [PubMed]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).