Abstract
In this study, nanocomposites based on polycaprolactone (PCL) and two types of mesoporous silicas, MCM-41 and SBA-15, were attained by melt extrusion. The effect of the silica incorporated within the PCL matrix was observed, firstly, in the morphological characteristics and degradation behavior of the resultant composites. DSC experiments provided information on the existence of confinement in the PCL–SBA-15 materials through the appearance of an additional small endotherm, located at about 25–50 °C, and attributed to the melting of constrained crystallites. Displacement to a slightly lower temperature of this endothermic event was observed in the first heating run of PCL–MCM-41 composites, attributed to the inferior pore size in the MCM-41 particles. Thus, this indicates variations in the inclusion of PCL chains within these two mesostructures with different pore sizes. Real-time variable-temperature small-angle X-ray scattering (SAXS) experiments with synchrotron radiation were crucial to confirm the presence of PCL within MCM-41 and SBA-15 pores. Accurate information was also deduced from these measurements regarding the influence of these two mesoporous MCM-41 and SBA-15 silicas on PCL long spacing. The differences found in these morphological and structural features were responsible for the ultimate mechanical response exhibited by the two sets of PCL nanocomposites, with a considerably higher increase of mechanical parameters in the SBA-15 family.
1. Introduction
The study of the crystalline characteristics of organic solids formed in small spaces of controlled pore glasses has been relevant for a long time [1]. Its understanding constitutes a fundamental bridge between molecular scale and the relationship to bulk scale. A melting point depression was found by differential scanning calorimetry (DSC), and the influence of pore diameter on the magnitude of the reduction in melting temperature (Tm) and in the apparent melting enthalpy ΔHm has been reported [2]. The formation of small crystals was described theoretically in a classical discussion by Gibbs [3] and results from the effect of surface curvature on the equilibrium state of a pure substance [4]. A related theory was derived by Thomson [5] for the effect of curvature on the vapor pressure of liquid droplets. This theory may be extended to small crystals to give the same equation as that of Gibbs, as noted by Defay et al. [4] and the equation for the temperature shift of melting in confined geometries is often called the Gibbs–Thomson equation. Research on the comprehension of physical properties of the molecular scale has become even more challenging since the arrival of nanotechnology. Phenomena like glass transition, phase separation crystallization and the subsequent melting under confinement, were analyzed to understand the influence of finite size constraints on bulk properties. This important topic is even more remarkable in polymeric-based materials, due to the long size of macromolecular chains and their inherent characteristics. The development of polymer-based nanocomposites, thin films and coatings, nanolithography in semiconductor manufacturing, etcetera, has allowed obtaining a basic knowledge of the involved molecular phenomena, which are considered fundamental to the success of confinement awareness.
An extensive variety of inorganic materials with nanodimensions are currently available. Ordered mesoporous silicas belong to that group [6,7,8]. MCM-41 and SBA-15 are two of the most known members, both with a stable 3D framework and a hexagonally arranged structure, possessing nano-sized pores along with high porosity and surface area [6,8]. The diameter of those pores is in MCM-41 particles smaller than in SBA-15: typically 3 nm for the former and 7–9 nm for the latter. These unique features make mesoporous silicas very interesting materials for the preparation of polymeric nanocomposites, either through in situ polymerization of organic monomers, or by melt extrusion. The presence of polymeric chains within these hollow nanometric long channels going out through the openings could significantly favor the interactions between mesoporous silica and polymer matrix. Furthermore, the confinement of macrochains can affect the ultimate performance of these polymeric-based materials. The appearance of a small endotherm showing a depression of Tm and a reduction of ΔHm has been observed by DSC for in situ polymerized composites, based on high density polyethylene (HDPE) with either pristine or decorated MCM-41 particles [9,10,11]. This endotherm exhibits similar features to those noted in organic solids constrained in the small pores of glasses [2]. In fact, a primary melting process was found at around 130 °C, corresponding with the HDPE chains that grew on the exterior surfaces of silica particles, together with the mentioned secondary melting event, which appeared at lower temperatures (from 65 to 85 °C) and involved a much smaller amount of enthalpy, that was ascribed to the chains embedded within the nanometric channels in the silica particles. This minor endothermic process took place at higher temperatures (100–115 °C) when HDPE or ultrahigh molecular weight polyethylene (UHMWPE) nanocomposites were synthesized by in situ polymerization [12,13,14] in the presence of mesoporous SBA-15 silica.
The confinement of isotactic polypropylene (iPP) chains within the ordered hollow spaces existing in SBA-15 mesoporous silica has also been proven recently by DSC, for their nanocomposites obtained from either in situ polymerization or by extrusion [15,16]. The secondary endothermic process appeared in the interval ranging from around 90 to 115 °C. Its location was dependent on iPP molecular weight [17]. Small-angle X-ray scattering (SAXS) measurements, using synchrotron radiation, have confirmed the existence of iPP confinement within the nanometric channels and have constituted a reliable and powerful tool [18] to achieve more complete information to that deduced from DSC. The presence of confinement is derived from the observation of a noticeable discontinuity in the intensity of the first order (100) diffraction characteristic of these mesoporous silicas, related to their hexagonal arrangement. Former investigations on SBA-15 composites [19,20,21] have described that the intensity of the main SBA-15 diffraction was derived from the eventual scattering contrast between the walls and the inside of the mesopores. In the iPP nanocomposites, changes in the electron density taking place in the iPP chains arranged inside the SBA-15 particles were also presumed, which were in a partial ordering—semicrystalline—at low temperatures, or fully amorphous after the melting of the iPP crystallites existing within the nanometric channels. Similar to that described in polyethylene, a small secondary melting process or location of the (100) reflection discontinuity (both ascribed to the iPP chains confined and located into the silica mesostructure) appears at lower temperatures if MCM-41 particles, instead of SBA-15 ones, are incorporated into an iPP matrix by extrusion [22].
The capability of polymers for crystallizing either within nanometric channels, or in the outer mesoporous silica surface is different to the multiple crystallization observed in other different systems, such as polytetrafluoroethylene-polymethylmethacrylate (PTFE/PMMA) core-shell nanoparticles [23,24]. This latest thermal behavior has been rationalized within the frame of a fractionated crystallization mechanism. The multiple crystallization transitions were noted in crystalline polymers when dispersed as small particles, or microphase-separated in polymer blends or in block copolymers.
The aim of this research is to compare the characteristics of confinement and the effect of the size of the initial hollow pores in (nano)composites prepared by extrusion from polycaprolactone (PCL) and mesoporous silica, either MCM-41 or SBA-15 particles. The PCL matrix is biodegradable and melts at relatively low temperatures (60–65 °C), differently to PE and iPP. Furthermore, the incorporation of stiff silica particles can contribute to significantly improve the PCL mechanical behavior, mainly at room temperature and temperatures above and close to its Tm. Due to the relevant interest of these PCL (nano)composites from an academic and practical standpoint, they will be analyzed in terms of differences in morphology, thermal stability, phase transitions, crystalline characteristics and confinement effects, together with their viscoelastic and mechanical responses. Several techniques have been used, including: scanning electron microscopy (SEM); thermogravimetry (TGA); differential scanning calorimetry (DSC); simultaneous wide and small angle X-ray Scattering (WAXS and SAXS, respectively) measurements with synchrotron radiation; dynamic mechanical analysis (DMA), and stress-strain tests.
2. Materials and Methods
2.1. Materials and Chemicals
A commercially available polycaprolactone (purchased from Sigma-Aldrich) with an average molecular weight (Mn) of 80,000 g/mol and a density of 1.1450 g/cm3 has been used in the current investigation as a polymeric matrix. The MCM-41 and SBA-15 particles were purchased from Sigma-Aldrich (specific surface area, SBETMCM-41 = 966 m2/g and SBETSBA-15 = 619 m2/g; average mesopore diameter [25], DpMCM-41 = 2.9 nm and DpSBA-15 = 8.0 nm) and were used as received.
2.2. (Nano)Composite and Film Preparation
Composites with different contents in particles of MCM-41 (2, 9, 12% in weight) or of SBA-15 silica (3, 6 and 9% in weight) were processed by melt extrusion. The materials containing MCM-41 silica were labeled as PCLMCM2, PCLMCM9 and PCLMCM12, whereas names were PCLSBA3, PCLSBA6 and PCLSBA9 for those incorporating SBA-15 particles. Previous to extrusion, both the polymer and mesoporous silica particles were dried. The PCL was placed in an oven at 50 °C for 20 min, followed by a drying under vacuum, also at 50 °C, for 20 h. The silica particles were dried under vacuum at 100 °C for 24 h. A corotating twin-screw microextruder (Rondol, model: Microlab) was used with a screw temperature profile of 100, 105, 110, 120 and 110 °C from the hopper to the die, with the length-to-diameter ratio 20:1. Then, films were prepared by compression molding at 120 °C and at 30 bar for 6 min, in a hot-plate press (Collin, model 200 × 200). After, a cooling process was applied to the different materials from their molten state to room temperature for 4 min, at the relatively fast rate of around 80 °C/min and at a pressure of 30 bar.
2.3. Scanning Electron Microscopy
Experiments of high-resolution field-emission scanning electron microscopy (FESEM) were carried out with S-8000 Hitachi equipment at room temperature, in different cryo-fractured sections of composites at distinct mesoporous contents. Thin sections of around 40 nm were cut by cryo-ultramicrotomy (Leica EM UC6) at −120 °C and deposited in a holder. Silica particles were dispersed in acetone in an ultrasonic bath for 5 min, and then deposited in a holder prior to observation.
2.4. Thermogravimetric Analysis
Thermogravimetric analysis (TGA) was performed with Q500 equipment of TA instruments, under either nitrogen or air atmosphere, at a heating rate of 10 °C/min. Degradation temperatures of the distinct materials were determined, as well as the exact mesoporous silica (either MCM-41 or SBA-15) amount incorporated into the composites prepared by extrusion, which was estimated as an average of the values obtained from the two environments.
2.5. Differential Scanning Calorimetry
Calorimetric analyses were carried out with a TA instruments Q100 calorimeter, connected to a cooling system and calibrated with different standards. The sample weights were around 3 mg. A temperature interval from −80 to 100 °C was studied, at a heating rate of 20 °C/min. For crystallinity determinations, a value of 135 J/g was considered for the melting enthalpy of the 100% crystalline PCL [26,27]. Errors in the determination of temperatures, the calculation of the enthalpy and the crystallinity were estimated at ±0.5 °C, ±4 J/g, and ±0.04 units, respectively.
2.6. X-ray Experiments with Synchrotron Radiation
Real-time variable-temperature simultaneous SAXS/WAXS experiments were carried out with synchrotron radiation with beamline BL11-NCD-SWEET at ALBA (Cerdanyola del Vallès, Barcelona, Spain), at a fixed wavelength of 0.1 nm. A PILATUS detector was used for SAXS (off beam, at a distance of 296 cm from sample) and a Rayonix for WAXS (at about 14.6 cm from sample, and at a tilt angle of around 29 degrees). A Linkam Unit, connected to a cooling system of liquid nitrogen, was employed for the temperature control. The calibration of spacings was obtained by means of silver behenate and Cr2O3 standards. The initial 2D X-ray images were converted into 1D diffractograms, as a function of the inverse scattering vector, s = 1/d = 2 sin θ/λ, by means of pyFAI python code (ESRF), modified by ALBA beamline staff. Film samples of around 5 × 5 × 0.2 mm were used in the synchrotron analysis.
2.7. Dynamic Mechanical Thermal Analysis (DMTA)
Viscoelastic relaxations were measured with a TA Q800 Dynamic Mechanical Thermal Analyzer, working in a tensile mode. The storage modulus, E’, loss modulus, E”, and the loss tangent, tan δ, of the different composites were determined as functions of temperature over the range from −150 to 125 °C, at fixed frequencies of 1, 3, 10 and 30 Hz, and at a heating rate of 1.5 °C/min. For this analysis, strips of 2.2 mm wide and 15 mm length were cut from the molded films.
2.8. Mechanical Behavior by Means of Stress-Strain Tests
Nominal stress-strain tests were performed at a temperature of 25 °C, and a stretching rate of 10 mm/min in MTS Q-Test Elite dynamometer with a load-cell of 100 N. Specimens for these experiments were punched out from the polymer films. The dimensions of these strips were 15 mm long, 1.9 mm wide and around 0.2 mm thick. At least, six different strips were stretched until fractured at a given specimen.
3. Results and Discussion
3.1. Morphological Characteristics
Details of the mesoporous MCM-41 and SBA-15 particles were obtained by high-resolution field-emission scanning electron microscopy (FESEM), as well as the dispersion of those silica particles within the PCL matrix. Figure 1a,b show FESEM pictures for pristine particles of the MCM-41 and SBA-15 silica used in this investigation.

Figure 1.
FESEM micrographs for the different materials. Pristine particles: (a) MCM-41 and (b) SBA-15; PCL (nano)composites with MCM-41: (c) PCLMCM2, (d) PCLMCM9 and (e) PCLMCM12; and PCL (nano)composites with SBA-15: (f) PCLSBA3, (g) PCLSBA6 and (h) PCLSBA9.
The MCM-41 particles exhibited their common irregular micrometric shape [25,28], whereas those for SBA-15 showed their vermicular elongated contour [13,25] with an average size of 350 nm wide and 0.9 μm long. The size found in these commercial mesoporous silicas was in agreement with those described previously in the literature for mesoporous MCM-41 and SBA-15 particles, synthesized in the laboratory [13,28]. The interior of the particle morphology consisted of a well-defined, uniform and ordered channel structure with a hexagonal arrangement. Both MCM-41 and SBA-15 silicas exhibit an analogous ordering, which displays their characteristic reflections, as will be discussed later on in the section of crystalline details. Although the type of arrangement is the same for MCM-41 and SBA-15, the position of their main (100) diffraction and the superior orders are different, since there are important differences between their hollow pore sizes, as aforementioned in the introduction.
Figure 1c–e show the FESEM micrographs for the set of distinct composites based on PCL and MCM-41, and Figure 1f–h show those materials incorporating SBA-15 particles. The distribution of MCM-41 silica was quite uniform at the two lower contents, but a certain agglomeration of particles can be seen in PCLMCM12. Despite the existence of aggregation, bulky inorganic domains were not observed along the specimens. The dispersion of SBA-15 seemed, however, to be better within the PCL matrix, independent of the content. The SBA-15 aggregates were of a smaller size than those formed from the MCM-41 particles. This fact could be associated with the irregular shape of the MCM-41 silica that makes more difficult to the PCL chains to be accommodated around their contour. Again, the agglomeration of SBA-15 was noted as its amounts rose in the material, clearly evident in the PCLSBA9 composite, as depicted in the Figure 1h.
3.2. Thermal Stability
Figure 2 shows the temperature dependence of the PCL weight loss under either inert (left) or oxidative (right) conditions in these composites with MCM-41 or SBA-15 particles. The inert TGA curve of pristine PCL (see in Figure 2a,c) indicates that its decomposition took place through a primary stage with an inflection point of around 360 °C (see data in Table 1), which involved most of the weight loss (around 95%). A detailed examination of this TGA curve highlights other minor consecutive mechanisms, located at higher temperatures, at around 438 °C. The study of the nature of the gases along the thermal degradation process, under the inert environment, allowed the proposal of a two-stage degradation mechanism [29,30]. The first process involved a statistical rupture of polyester chains via an ester pyrolysis reaction. The produced gases were identified as H2O, CO2, and 5-hexenoic acid. The second step leaded to the formation of ε-caprolactone (cyclic monomer) as result of an unzipping depolymerization process.
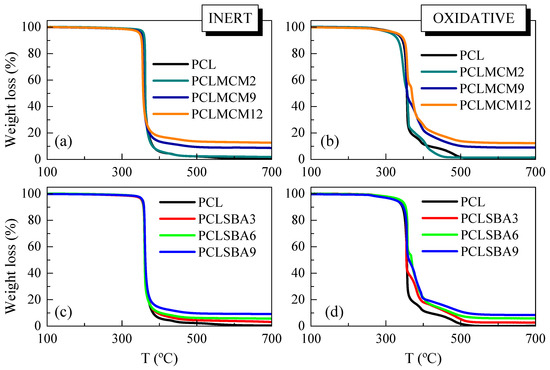
Figure 2.
TGA curves for the pristine PCL and its composites prepared by melt extrusion with MCM-41 or SBA-15, at different contents, under inert atmosphere (a,c) or oxidant environment (b,d), respectively.

Table 1.
TGA results under nitrogen and air atmospheres for neat PCL and its composites prepared by melt extrusion: temperature of a loss weight of 10% (T10%) and temperature at the maximum (Tmax) together with the SBA-15 wt.% at 600 °C and its average.
Thermal PCL degradability under these inert conditions was not considerably affected by the incorporation of mesoporous particles. The presence of MCM-41 or SBA-15 silicas did not vastly change the location of main weight loss, although the MCM-41 particles seemed to slightly shift the maximum PCL degradation temperature to smaller values, compared with those found in the neat matrix, and the composites containing SBA-15, as clearly deduced from Figure 3a. MCM-41 silica has been employed, sometimes, as a catalyst for thermal decomposition. This effect was described for polyethylene (PE) degradation under N2, performed by TGA, in both presence and absence of mesoporous MCM-41 [31]. Other authors have shown the efficiency of mesoporous alumino-silicate MCM-41 as a promoter towards the degradation of polyolefins into liquid fuels [32]. This role as a promoter of decomposition was also found when MCM-41 silica was used in the double role of a catalyst carrier and filler for in situ polymerized PE-based composites employing either neat mesoporous particles or those decorated with undecenoic acid or silanes [9,10,33,34]. On the other hand, the process related to the PCL unzipping depolymerization became a shoulder and was broadened in both types of composites, those incorporating MCM-41 or SBA-15, in comparison with features observed in the pristine PCL. PCL degradation is known to also be affected by other parameters, such as polyester molecular weight and nature of the PCL end groups [29]. The increase of the former provokes a significant drop of degradation rate. This behavior is explained by statistical chain cleavage, triggered by the pyrolysis reaction at first degradation process.
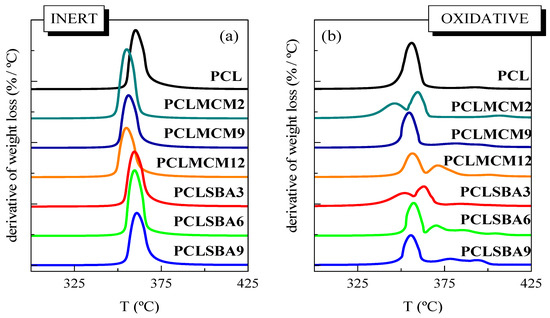
Figure 3.
DTGA curves for the pristine PCL and its composites attained by melt extrusion with MCM-41 or SBA-15, at different contents, under (a) inert atmosphere and (b) oxidant environment.
Figure 2b,d show the temperature dependence of neat PCL weight loss under air. Its behavior is more complex than that noted in inert atmosphere. Thus, it takes place in several stages, clearly noticeable in its TGA curve, since the PCL degradation occurs now via both thermolysis and oxidation, i.e., through thermo-oxidation. The presence of mesoporous MCM-41 or SBA-15 particles increased the complexity of the decomposition mechanism. At the lowest content, the main loss was split into two processes, as clearly noted from DTGA curves represented in Figure 3b, being that at the highest temperature of major intensity. As the silica content rose, independently of the addition of MCM-41 or SBA-15, other small decomposition stages were observed. All of these results indicate that under these oxidative conditions, the presence of mesoporous particles did not exert a catalytic effect in the PCL matrix of these composites. It is also interesting to remark that TGA curves obtained under both environments show that PCL degrades at a similar range of temperature, independently of the atmosphere employed, and when used as neat polymer or as a matrix in the composites.
3.3. Phase Transitions, Crystalline Characteristics and Confinement of PCL Chains
Pore size effect in the PCL thermal transitions is represented in Figure 4 for the PCL–MCM-41 and PCL–SBA-15 nanocomposites in the first heating run. Owing to the high flexibility of its chains, PCL glass transition appeared at very low temperature (around −64 °C). The addition of mesoporous silica, independent of the difference in the size of the pores between the MCM-41 and SBA-15 particles, did not greatly change the location of its glass transition temperature (Tg), as can be deduced from the data in Table 2. A displacement of Tg, which was ascribed to the amorphous regions within the PCL matrix, to a slightly higher temperature can be seen in PCLMCM12, as well as PCLSBA6 and PCLSBA9. This indicates a small increase in the hindrance of PCL mobility along transition from its glass to its rubbery amorphous state for the highest silica contents. It seems that SBA-15 particles induced more major restrictions than MCM-41. Thus, lower contents lead to that small change.
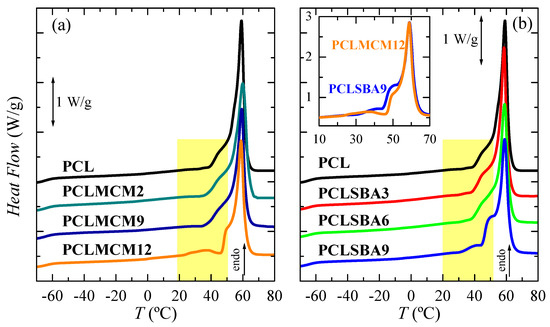
Figure 4.
DSC first melting curves for the neat PCL and its extruded composites with MCM-41 (a) and SBA-15 (b) particles. Curves have been shifted for the sake of clarity. The inset represents the heat flow (HF) comparison between the PCLMCM12 and PCLSBA9 materials.
Table 2.
DSC results for the different samples: glass transition (Tg)—calculated from the first heating run—first and second melting (Tm1 and Tm2) and crystallization (Tc) temperatures, and overall crystallinity (normalized to the actual PCL content in the material) for the first melting (fcm1NORM), crystallization (fcCNORM) and second melting (fcm2NORM) processes.
In the high temperature region, multiple small endothermic processes, together with a main melting peak are seen in Figure 4 for all the materials analyzed along this first heating scan. A similar complex melting process has been described in neat PCL and in some PCL composites or blends [27,35]. Thus, distinct endothermic events are noticeable, which are related to the existence of several melting-recrystallization stages. These steps, above around 40 °C, are associated with the initial thermal history imposed during processing, since a fast cooling from the melt is involved, leading to a formation of thin and imperfect crystals able to evolve toward thicker and more perfect ones by staying at room temperature. The main melting peak was rather similar in all these samples, establishing that size of the major PCL crystallites was not affected by the incorporation of mesoporous particles, regardless of being MCM-41 or SBA-15.
Some differences were, however, noticed with increasing the mesoporous amounts at a given family, and between the two highest contents for the distinct silicas evaluated. As mentioned, an intense shoulder was seen above 40 °C in PCL, PCLMCM2 and PCLMCM9, which overlapped with the main melting peak. In PCLMCM12, an additional small endothermic event was noticeable at the temperature interval ranging from 25 to 45 °C, and the shoulder merged with the main melting process then narrowed, and moved to slightly higher temperatures. A similar behavior was noted in the PCL–SBA-15 nanocomposites, PCLSBA9 being the sample with an extra process overlapped with the shoulder, previous to the primary melting peak. It was remarkable that the PCLMCM2 and PCLSBA3 curves, as well as the PCLMCM9 and PCLSBA6 ones, could be superimposed efficiently due to their similar shape and position. Nevertheless, this superposition did not materialize in the range from 25 to 55 °C for PCLMCM12 and PCLSBA9 (see inset in Figure 4b). Taking into account the previous results found for PE and iPP, the differences observed in these two materials with the highest MCM-41 and SBA-15 contents were associated with distinct features in the filling of their pores [10,11,12,13,22]. The small endotherm, located from 25 to 45 °C, was assigned to the melting of the PCL crystals located and constrained inside the mesoporous pores. These crystals were much thinner than those growing out of mesoporous particles and, therefore, the Tm of such small crystals was considerably lower than those for the PCL crystals outside the channels, which corresponded with the main melting temperature. Furthermore, the size of the pores was not the same in MCM-41 as in the SBA-15 particles, being smaller in the former. Accordingly, crystallites that can be formed within the nanometric MCM-41 spaces have to be thinner than those grown in the SBA-15 channels, and they will melt at lower temperatures.
In the PE-based materials achieved by in situ polymerization, the smaller crystals developed inside MCM-41 were reported to melt by DSC at the interval from 50 to 90 °C [10,11], whereas those crystallized within SBA-15 particles melted between 95 and 115 °C [12,13]. In extruded iPP nanocomposites, there was not observation by DSC of any secondary endothermic event in the iPP–MCM-41 materials [22], whereas this was located between 95 and 115 °C for those crystals confined within the nanometric pores of SBA-15 particles [16,17,18,22]. In the present extruded PCL-based materials, prepared using either MCM-41 or SBA-15, as clearly seen in the inset of Figure 4b, the small endotherm was found at 37.5 °C in PCLMCM12, and 42.5 °C in PCLSBA9. The temperature intervals for the two types of mesoporous silica overlapped to a significant extent, probably due to the low main melting temperature exhibited by this biodegradable polymer. Furthermore, the enthalpy involved in these secondary endotherms was also very small, this fact being associated with the approach used for the preparation of these materials. They were obtained by extrusion, and the PCL chains were able to fill out the nanometric spaces of these two mesoporous silica by the action of shear force during processing. A larger amount of PCL chains would probably be accommodated within the channels, if in situ polymerization had been used instead of melt extrusion. A more detailed analysis of the differences in confinement within MCM-41 and SBA-15 particles will be performed later, when real-time variable-temperature SAXS measurements with synchrotron radiation are further discussed.
The overall degree of PCL crystallinity can be determined from the DSC curves. The results obtained during the first melting process, after the normalization of enthalpies to the actual PCL amount at each composite, are reported in Table 2. Two different trends were observed by changing the mesoporous silica added. The incorporation of MCM-41 led to a reduction of PCL crystallization capability and, consequently, to a decrease in crystallinity. If SBA-15 particles were included, PCL crystallinity of the resultant nanocomposites remained constant in these films, processed by applying a fast cooling from the melt. As aforementioned, the MCM-41 particles seemed to be worse-distributed than the SBA-15 ones within the PCL matrix and, in addition, the formation of inorganic domains with a larger size were noticed (see Figure 1). These domains increased with the amount of MCM-41 particles in the nanocomposite. These features seem to impose difficulty on the PCL for the crystallization of the PCL–MCM-41 family; this hindrance was enlarged as MCM-41 content raised in the final material.
Figure 5a shows the curves attained for the subsequent DSC cooling process from the melt. An exothermic event is clearly noticeable in all of these materials based on PCL. Furthermore, it can be observed that the two mesoporous silicas exerted an evident nucleating effect in the PCL crystallization; then, its location shifted to a higher temperature for all the composites. Nevertheless, this influence was dependent on the type of silica, with the effect of SBA-15 particles being more important than that triggered by MCM-41. At a given set, this easier capability of PCL for crystallizing at higher temperatures than the pristine PCL matrix worsened as silica content rose, although Tc was always higher in the nanocomposites than in the neat PCL. This fact might also be associated with the increase in the size of inorganic domains.
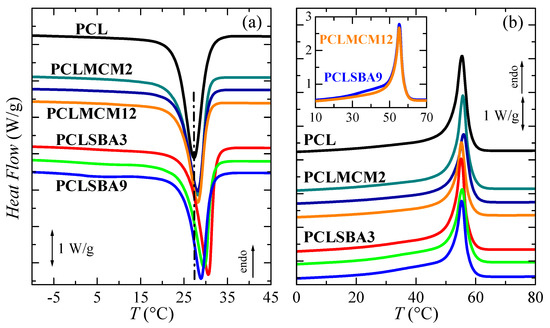
Figure 5.
DSC curves corresponding to: (a) cooling from the melt and (b) second heating run for: PCL, PCLMCM2, PCLMCM9, PCLMCM12, PCLSBA3, PCLSBA6 and PCLSBA9, from top to bottom. Curves have been shifted for the sake of clarity. Inset represents comparison between PCLMCM12 and PCLSBA9.
PCL crystallinity has also been determined from these cooling DSC curves as listed in Table 2. A reduction of crystallinity can clearly be observed, compared with that determined from the as-processed films. Despite the DSC cooling rate being slower than that used in the film preparation—20 °C/min instead of approximately 80 °C/min—which should lead to more and thicker crystals, the difference was associated with the fact that specimens from the as-processed films were maintained at room temperature for 2 days before their DSC testing. As mentioned, during that time thin crystallites were able to undergo several melting-recrystallization processes, allowing an increase in crystallinity. Under these conditions, the incorporation of mesoporous silica provoked a diminishment in PCL crystallinity, which was considerable when MCM-41 was added, and small when SBA-15 particles was added, within the experimental error. This decrease became larger as silica content rose.
Figure 5b depicts the DSC curves observed during the second heating run after PCL crystallization at 20 °C/min. The behavior was simpler than that found during the initial scan. Here, only an endothermic peak was noted, differently from the previous complex melting curves. Second heating was initiated just after cooling finished and then, the developed crystallites did not have sufficient time to evolve towards more perfect crystalline entities. Moreover, these crystals grown under 20 °C/min were thicker than those generated in the initial process. Consequently, melting–recrystallization processes were not seen in this second heating scan, and the Tm values were shifted to a slightly lower temperature. Location was independent of the incorporation of mesoporous particles (Figure 5b and Table 2). The absence of the melting–recrystallization process resulted in crystallinity being lower than that of the first melting process. As mesoporous silica content increased, crystallinity was significantly reduced in the nanocomposites containing MCM-41, and only slightly reduced in those with SBA-15 particles. Comparing PCLMCM12 and PCLSBA9—the two materials where evidence of confinement was observed during the first DSC heating run—shows a tiny increment of heat flow in the interval from 25 to 45 °C for the latter. This was again associated with the existence of PCL crystals confined in the nanometric SBA-15 mesostructure, although the amount of crystallites was reduced. Moreover, no evidence of confinement was found by DSC during this second melting process in PCLMCM12. Additional information will be discussed from real-time variable-temperature synchrotron experiments in the region of small angles. The presence of PCL chains within the silica channels indicates that there was a certain amount of PCL within the constrained nanometric spaces and, accordingly, these systems could be named as nanocomposites.
The crystalline nature of these PCL-based materials is also evident from Figure 6. It shows the WAXS profiles at room temperature, obtained with synchrotron radiation, for the pristine PCL, the composites with either MCM-41 (Figure 6a) or SBA-15 (Figure 6b) particles, and the two neat silicas. The PCL typically crystallized into an orthorhombic lattice [36]. There were not noticeable changes in the location of its characteristic (100), (110) and (200) diffraction peaks with the presence of mesoporous silicas. It was also deduced that the pristine MCM-41 and SBA-15 particles were amorphous at this short-range, showing a wide halo that was centered at an approximate s value of 2.54 nm−1. The amplified inset in Figure 6a shows that the PCL composites containing MCM-41 and the pure MCM-41 silica exhibited the higher orders of their hexagonal arrangement at the lowest s values in these diffractograms. The main order was in the s interval corresponding with the SAXS range, as displayed in Figure 7. The differences in location between the MCM-41 and SBA-15 reflections in their hexagonal assembly were ascribed to the distinct size of the pores of these two silicas, since their initial ordering was not affected by extrusion (see below).
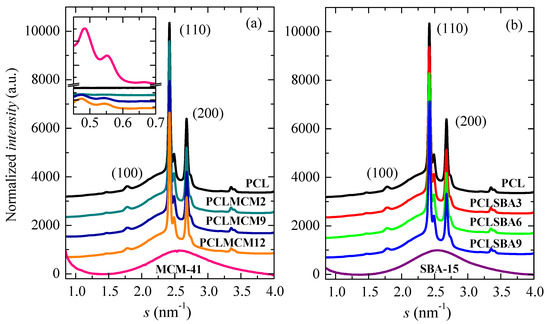
Figure 6.
WAXS profiles at room temperature, obtained with synchrotron radiation, for pristine PCL homopolymer, MCM-41 and SBA-15 particles, together with those for their nanocomposites prepared by extrusion: (a) PCL–MCM-41 and (b) PCL–SBA-15. Inset shows the higher orders of the MCM-41 hexagonal arrangement in the neat silica and in the distinct nanocomposites.
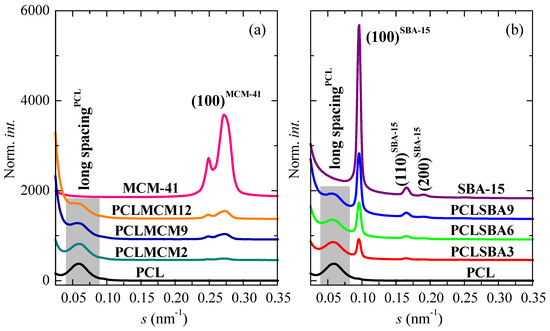
Figure 7.
SAXS profiles at room temperature for the pristine PCL, mesoporous MCM-41 and SBA-15 as well as for their composites: (a) PCL–MCM-41 and (b) PCL–SBA-15 sets.
Crystallinity degree can be also determined from X-ray diffraction at the wide-angle region, if a two-phase model is considered, by comparison with the area under the crystalline peaks to the total scattered intensity [37]. An estimation of PCL crystallinity in these materials implies the previous subtraction of amorphous halo from silica, since it appears at the same interval as the X-ray diffractions of neat PCL (as depicted in Figure 6). After that, the PCL profile was decomposed into its own amorphous halo and crystalline diffractions. The values are listed in Table 3. Very similar trends to those previously described from the first heating DSC experiments were found. Accordingly, a considerable reduction in crystallinity can be observed in the PCL–MCM-41 set with increasing MCM-41 content, whereas only a slight decrease was noticeable in those for the PCL–SBA-15 family with rising SBA-15 composition.
Table 3.
Characteristics of the PCL crystalline phase for the pristine polymer and the different composites: fcPCLWAXS (crystallinity degree determined by WAXS at room temperature); LPCLSAXS (long spacing estimated by SAXS at room temperature), and lc (most probable crystal size calculated assuming a two-phase model: lc = LPCLSAXS·fcPCLWAXS).
Figure 7 shows the SAXS profiles at room temperature for the neat PCL, mesoporous MCM-41 and SBA-15 silicas as well as their composites. On one hand, it is clearly noticeable that the hexagonal ordering of the mesoporous particles has been maintained throughout extrusion for the distinct composites, as mentioned from the FESEM pictures and from the inset of Figure 6a for those containing MCM-41 particles. Thus, the characteristic (100)hexagonal main reflections can be noticed in the two sets of nanocomposites. The high intensity and collimation of synchrotron radiation used proves that the first (100)MCM-41 diffraction of the MCM-41 hexagonal ordering, which is commonly asymmetric, was actually split into two contributions, appearing at 0.249 nm−1 and 0.274 nm−1, for this commercial MCM-41 silica. Moreover, Figure 7 also displays in the SAXS profiles the higher orders ((110)SBA-15 and (200)SBA-15) in the neat SBA-15 and in their materials [6,8,38]. These higher orders in the pristine MCM-41 and in their related composites are seen in the WAXS scale, as aforementioned.
In addition to the diffractions ascribed to mesoporous silicas, a broad peak was also evident in the region of the lowest values of s. This was ascribed to the variation of the electron density existing in the PCL matrix, as consequence of its semicrystalline nature and its lamellar crystallites, i.e., associated with its most probable long spacing. The results for the different samples are reported in Table 3. The presence of mesoporous silica leads to an increase in the values of long spacing, independent of their pore size. Nevertheless, the most probable PCL crystallite size (lc), determined assuming a two-phase model, showed two different trends: it decreased in the PCL–MCM-41 composites with increasing content of MCM-41 mesoporous particles, and it raised with SBA-15 composition in the PCL–SBA-15 materials. In spite of these differences in crystal sizes, similar Tm values during the first melting were observed for all of the specimens, a fact that can be associated with the existence of melting–recrystallization processes during the heating of the different materials.
As mentioned above, and contrary to the case of PCLSBA9, no evidence of confinement was found for PCLMCM12 by DSC in the cooling and in the second melting processes. Consequently, additional real-time variable-temperature synchrotron experiments in the region of small angles were performed, considering that this SAXS technique was valuable for confirming the existence of confinement. As previously reported, the confinement of polymeric chains into the channels of mesoporous silicas led to significant intensity variations of the silica SAXS diffraction peaks [17,18,21,39,40], which have been interpreted by taking into account that both pore-filling and scattering contrast between the inside and the walls of the mesoporous channels were affecting the intensity of the diffraction peaks [19,20].
Clear variations in intensity of the SBA-15 main SAXS peak were found for composite PCLSBA9 in the first melting, in the cooling from the melt, and in the subsequent second melting. Figure 8a clearly shows such variation for the second melting of this composite, with a discontinuity at around 39 °C, which agrees with the presumed DSC confinement (see Figure 5b). Figure 8b also shows a form of step for the PCLMCM12 at around 34 °C, even though no event was observed by DSC for this composite, as seen in Figure 5b. The variation was relatively smaller than that for PCLSBA9, and it was also found in the first melting, in agreement with the DSC results. On the contrary, no obvious differences were found during the cooling from the melt. Incidentally, note in Figure 8 that the SBA-15 peak appears at rather small angles, very close to the long spacing of PCL, which is clearly observed in Figure 8a. On the contrary, the MCM-41 peak appears at higher s values (see also Figure 7), corresponding with a significantly smaller size.
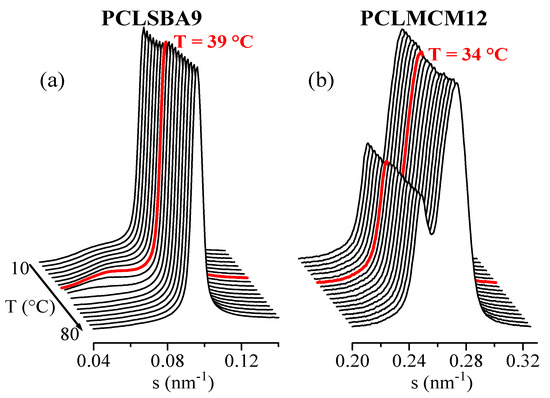
Figure 8.
Lorentz-corrected synchrotron SAXS profiles corresponding with the second melting, at 20 °C/min, of samples PCLSBA9 (a) and PCLMCM12 (b). Only 1 out of every 2 frames has been plotted, for clarity.
These intensity changes can be analyzed by representing the variation of the mesoporous SAXS peak intensity as a function of temperature, and the subsequent derivative of that intensity changes can be easily compared with the DSC curves. This has been performed for the composite PCLSBA9, and the results are shown in Figure 9a. As observed, such derivatives are very sensitive to the presence of confinement, much more than for the main melting or crystallization events, which were depleted in relation to the DSC curves. Importantly, the temperature regions for the confinement events were analogous from the two techniques, SAXS and DSC. Thus, confined melting appeared at around 48 and 38 °C for the first and second melting, respectively, whereas the confined crystallization appears centered at around 10 °C.
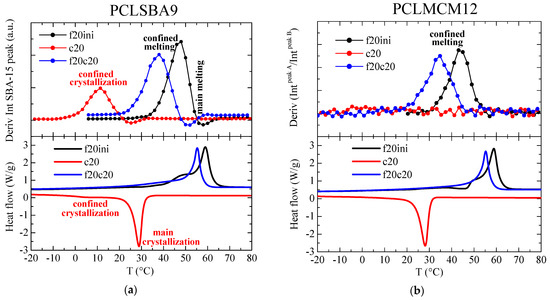
Figure 9.
(a) Variation with temperature of the intensity derivative for the SBA-15 diffraction in sample PCLSBA9 (upper) during the first melting (f20ini), cooling (c20) and second melting (f20c20) processes, compared with the corresponding DSC curves (lower); (b) Variation with temperature of the derivative of the intensities ratio between the two components in the MCM-41 peak for sample PCLMCM2 (upper) during the first melting (f20ini), cooling (c20) and second melting (f20c20) processes, compared with the corresponding DSC curves (lower).
These features were not so evident for PCLMCM12, since the area variations were very noisy. In the case of the MCM-41 composites, however, it has previously been shown that an appropriate alternative for quantifying confinement [22] was the analysis of the ratio of intensities between the two peaks arising from the bimodal hexagonal arrangement in MCM-41, which exhibited two different peaks, as seen in Figure 8b, at s values around 0.249 and 0.274 nm−1 (pore diameters of 4.02 and 3.65 nm). The corresponding derivative of the ratio of intensities between those two components is depicted in Figure 9b. Noise was only observed in the variation for the cooling from the melt. On the contrary, clear events were deduced for the first and second melting processes. Thus, the presumed confined melting appeared at around 43 and 34 °C for the first and second melting, respectively.
It can be concluded, therefore, that a considerably smaller confinement was deduced in the case of the MCM-41 composites, which can be expected because the diameter of the existing pores was much smaller than in the case of the SBA-15 composites. This difference in pore size was also responsible for the different melting temperatures of the confined crystals, which was around 5 °C less in the case of the PCLMCM12 composite, in comparison with PCLSBA9. These results are in agreement with those found by DSC during the first melting. This difference was not as large as in other cases [10,11,12,13], but it should be considered that the melting temperatures of regular PCL crystals were also rather low: only around 60 °C.
As a final aspect of this confinement analysis, it is not surprising that the DSC results derived from crystallization and second melting in these PCL–MCM-41 materials were not able to distinguish eventual exotherms or endotherms arising from confined crystallites, since the amount of PCL chains inside the pores of MCM-41 was significantly smaller than that within SBA-15, as the pore size was much smaller in the former. As mentioned above, there was no observation by DSC of any secondary exothermic or endothermic events in the extruded nanocomposites of iPP and MCM-41 [22].
3.4. Viscoelastic Behaviour and Mechanical Properties
Dynamic mechanical analysis was performed as a function of temperature to gain information about stiffness and relaxation processes exhibited by the different materials, and to evaluate the effect of amount and type of mesoporous silica. Figure 10 shows the dependence on temperature of storage modulus, E′, (Figure 10a,b) and relaxation processes in tan δ (Figure 10c,d) for the two families of composites. For a given family, an increase in E’ and a slight reduction in intensity of the most prominent relaxation were noticeable as silica content was raised. This effect was also widely described in the literature for different fillers [41,42,43,44] and can be explained in terms of the classical mechanical reinforcement exerted by the relatively rigid particles, which also lead to restricted mobility in the polymer matrix [45].
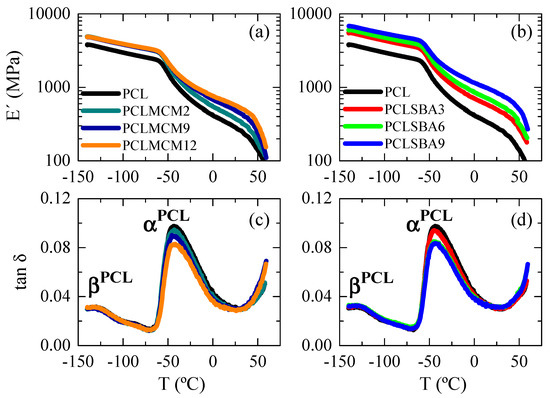
Figure 10.
Dependence of storage modulus and tan δ curves (at 3 Hz) on temperature for pristine PCL and composites PCL–MCM-41 ((a,c), respectively) and PCL–SBA-15 ((b,d), respectively).
The comparison of Figure 10a,b indicates the lower reinforcement that MCM-41 particles exert within PCL since storage moduli in the PCL–MCM-41 composites are inferior to those found in the PCL–SBA-15 materials at similar silica contents. This was also easily deduced from the E′ data reported in Table 4 at different temperatures. This lower filler effect found in the PCL–MCM-41 hybrids can be associated with their particular morphological and structural details: a worse dispersion of mesoporous particles, formation of a greater amount of aggregates, lower degree of PCL crystallinity, thinner crystallites and a reduced content of PCL chains confined within MCM-41 channels.
Table 4.
Values of storage modulus at different representative temperatures for the pristine PCL and its composites with either MCM-41 or SBA-15 particles.
These values of storage moduli could be compared with those calculated using the Halpin–Tsai and Mori–Tanaka models [41], although this aspect is out of the objective of this investigation, and will be analyzed in a further study.
Figure 10c,d show the relaxations observed in the tan δ representations for the different specimens. Two mechanisms are noted: βPCL and αPCL in order of increasing temperatures. As is clearly noticeable, the location of the two processes is independent of either silica content at a specific family, or the type of mesoporous at a given similar content. The βPCL relaxation was related to local mobility, with a similar molecular origin to the γ relaxation in polyethylene [46], although this took place at lower temperature. This fact is attributed to presence of thinner crystals that impose fewer restrictions in mobility to these amorphous regions. In both polymers, three or more CH2 units were involved in this relaxation [47].
The αPCL process was, however, ascribed to generalized movements involving long segments of polymeric chains located in the amorphous regions of PCL, and this relaxation took place during the corresponding glass transition of PCL [48,49]. The large decrease observed in the E′ values (Figure 10a,b) in the region of temperatures for those motions can be explained by the cooperative character of this relaxation. Additionally, the maximum value of the tan δ intensity also showed a slight decrease when the PCL content of the composites diminished, considering the restrictions of mobility because of the presence of mesoporous silica particles.
The mechanical response was also evaluated by tensile stress-strain measurements. Figure 11 shows the stretching curves for pure PCL and the different composites. All of them exhibited nominal curves that were characteristic for ductile polymeric materials. Three distinct regions are noted: initially, the stress rises on strain in a linear dependence (see insets), which allows determining Young’s modulus, E. Subsequently to this stage, an evident yield point is observed (see also insets) followed by a narrow section where stress is maintained as a constant. Finally, a third region, where an increase of the stress is again noticed as a strain becomes larger, that is related to a strain-hardening promoted by the stress-induced orientation of the PCL chains. The formation of a neck was observed during the deformation process performed under these conditions for all of specimens, independent of the absence or presence of silica and of type of the mesoporous particles incorporated. The necking propagation stage occurred in a short strain range because these materials were quite soft and exhibited a large deformation capability, as previously commented.
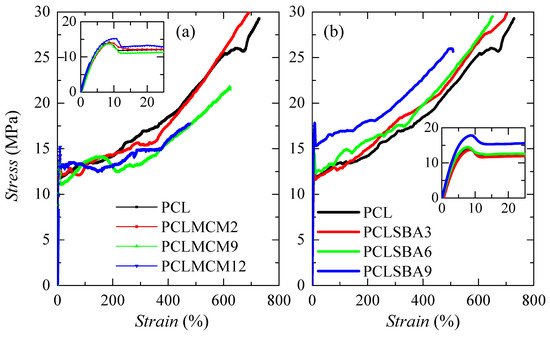
Figure 11.
Stress-strain behavior of pristine PCL and its composites prepared by extrusion with either MCM-41 (a) or SBA-15 (b) particles. Insets show the scale for the linear region, where Young’s modulus is estimated, and the yielding zone.
The main parameters achieved from these curves, Young’s modulus (E) and yield stress (σY), are represented in Figure 12. As depicted in Figure 12a, rigidity is enlarged and, consequently, E increases, as silica content is raised in the two sets of materials, as expected, since silica particles are harder and more rigid than PCL chains. Nevertheless, stiffness increased differently in the two families, with a much smaller increase in those composites containing MCM-41 particles, compared with those that incorporated SBA-15 silica. This feature corroborates the results deduced from the storage modulus at different temperatures (see Table 4), which show a similar trend.
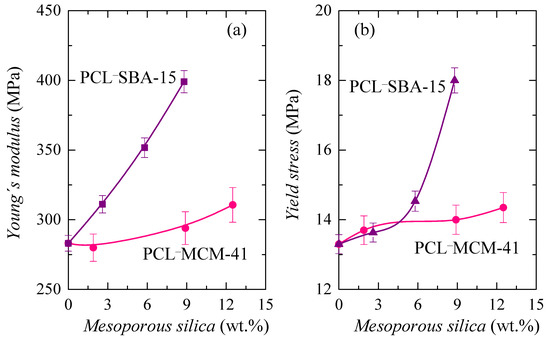
Figure 12.
(a) Values of Young’s modulus, E; and (b) yield stress, σY, deduced both from stress-strain tests for the PCL homopolymer and its composites with different MCM-41 and SBA-15 contents.
As aforementioned, there were several aspects that played an important role in the mechanical response. The worse distribution of MCM-41 particles and the formation of a greater amount of aggregates, together with the lower degree of PCL crystallinity, the thinner PCL crystallites and the reduced content of the PCL chains confined in MCM-41 channels of the PCL–MCM-41 composites led to an inferior, and less satisfactory, reinforcement effect, when compared with that exhibited by the PCL–SBA-15 materials. This was also deduced from the dependence of yield stress on silica content, as shown in Figure 12b.
Other attractive features found in these materials were related to the strain at break and the tensile strength. PCL shows the highest value in the former, since it was the softest material studied. The values decreased with increasing MCM-41 or SBA-15 content, but PCLMCM12, which was less deformable, was able to be strained up to almost 500% and showed a tensile strength of around 18 MPa. Accordingly, rigidity was improved more in the PCL–SBA-15 family than in the PCL–MCM-41 family, but the characteristics at rupture did not notably decrease. Moreover, a good reproducibility was found during the deformation process, either in the shape of the stress-strain curve or in the mechanical magnitudes derived from these measurements, for the different strips stretched in a specific material.
4. Conclusions
Composites based on PCL and different amounts of mesoporous MCM-41 or SBA-15 particles were achieved by melt extrusion. The SAXS results showed that the well-defined hexagonal arrangement exhibited by both of the pristine silicas was maintained by the mesoporous particles embedded within the PCL matrix in the resultant composites. Good dispersions were observed at different contents by FESEM, although the SBA-15 particles showed a more suitable distribution and led to an inferior formation of aggregates within the PCL matrix.
The presence of an endothermic event in the DSC first melting curve, which took place in the temperature interval ranging from 30 to 45 °C, indicated the development of crystals of a small size that were accommodated in the interior space of the nanometric MCM-41 and SBA-15 channels, meaning that there were confined PCL chains in both mesostructures. Confinement was confirmed by SAXS measurements.
The incorporation of mesoporous silica particles did not largely affect the PCL thermal degradation processes under either inert or air atmospheres, nor the location of Tg and main Tm. The variation of crystallinity and crystallite size was, however, dependent on silica content and on the type of mesoporous particle incorporated. Both parameters decreased as the MCM-41 amount increased in the PCL–MCM-41 composites, whereas crystallinity was constant, and crystal size rose with SBA-15 content in the PCL–SBA-15 family. This opposite dependence implies important differences in the parameters related to stiffness for the set of composites. Despite the incorporation of silica increasing the rigidity of the resultant materials, the improvement in the PCL–SBA-15 hybrids was much more significant than in the PCL–MCM-41 composites. Nevertheless, the behavior at break in terms of rupture strain and tensile strength were relatively maintained.
Mesoporous silica incorporation seems to be an appropriate and useful strategy for the improvement of the PCL mechanical performance, mainly with SBA-15 particles. Furthermore, mesoporous silica could provide capability for improving cell adhesion to PCL-based materials through the existing hydroxyl groups. These features may contribute to the increase of its bioapplications, such as in tissue engineering that requires stiffer materials with an enhanced mechanical response, or in other technological fields.
Author Contributions
Conceptualization, M.L.C. and M.d.R.R.; methodology, T.M.D.-R., E.B.-B., N.L.C.A., M.d.R.R., E.P. and M.L.C.; software, E.P.; validation, T.M.D.-R., E.B.-B. and N.L.C.A.; formal analysis, T.M.D.-R., E.B.-B., N.L.C.A., E.P. and M.L.C.; investigation, T.M.D.-R., E.B.-B., N.L.C.A., M.d.R.R., E.P. and M.L.C.; resources, E.P. and M.L.C.; writing—original draft preparation, E.P. and M.L.C.; writing—review and editing, T.M.D.-R., E.B.-B., N.L.C.A., M.d.R.R., E.P. and M.L.C.; supervision, M.L.C.; project administration, E.P. and M.L.C.; funding acquisition, E.P. and M.L.C. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by the Agencia Estatal de Investigación (AEI, Spain) together with the European Regional Development Fund (FEDER, UE) (grant numbers MAT2016-79869-C2-1-P and PID2020-114930GB-I00), by CSIC (grant number 2020AEP129) and ALBA Synchrotron Light Facility. TMDR is also grateful for her pre-doctoral funding (contract number BES-2017-082284).
Acknowledgments
Authors are grateful to the Characterization Service at ICTP-CSIC for FESEM, TGA and mechanical facilities as well as to its personnel for support. The synchrotron experiments were carried out at beamline BL11-NCD-SWEET at ALBA Synchrotron Light Facility with the collaboration of ALBA staff.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Haller, W. Chromatography on Glass of Controlled Pore Size. Nature 1965, 206, 693–696. [Google Scholar] [CrossRef]
- Jackson, C.L.; McKenna, G.B. The melting behavior of organic materials confined in porous solids. J. Chem. Phys. 1990, 93, 9002–9011. [Google Scholar] [CrossRef]
- Gibbs, J.W. On the Equilibrium of Heterogeneous Substances. In Collected Works, Volume I: Thermodynamics, Chapter III; Longmans: Green, NY, USA, 1928; pp. 55–353. [Google Scholar]
- Defay, R.; Prigogine, I.; Bellemans, A.; Everett, D.H. Surface Tension and Adsorption; Wiley: New York, NY, USA, 1966. [Google Scholar]
- Thomson, W. On the equilibrium of vapor at a curved surface of liquid. Philos. Mag. 1871, 42, 448–452. [Google Scholar] [CrossRef]
- Beck, J.S.; Vartuli, J.C.; Roth, W.J.; Leonowicz, M.E.; Kresge, C.T.; Schmitt, K.D.; Chu, C.T.-W.; Olson, D.H.; Sheppard, E.W.; McCullen, S.B.; et al. A new family of mesoporous molecular sieves prepared with liquid crystal templates. J. Am. Chem. Soc. 1992, 114, 10834–10843. [Google Scholar] [CrossRef]
- Roth, W.J.; Vartuli, J.C. Synthesis of mesoporous molecular sieves. Stud. Surf. Sci. Catal. 2005, 157, 91–110. [Google Scholar] [CrossRef]
- Zhao, D.; Feng, J.; Huo, Q.; Melosh, N.; Fredrickson, G.H.; Chmelka, B.F.; Stucky, G.D. Triblock copolymer syntheses of mesoporous silica with periodic 50 to 300 angstrom pores. Science 1998, 279, 548–552. [Google Scholar] [CrossRef] [PubMed]
- Cerrada, M.L.; Pérez, E.; Lourenço, J.P.; Campos, J.M.; Ribeiro, M.R. Hybrid HDPE/MCM-41 nanocomposites: Crystalline structure and viscoelastic behaviour. Microporous Mesoporous Mater. 2010, 130, 215–223. [Google Scholar] [CrossRef]
- Cerrada, M.L.; Pérez, E.; Lourenço, J.P.; Bento, A.; Ribeiro, M.R. Decorated MCM-41/polyethylene hybrids: Crystalline Details and Viscoelastic Behavior. Polymer 2013, 54, 2611–2620. [Google Scholar] [CrossRef]
- Cerrada, M.L.; Bento, A.; Pérez, E.; Lorenzo, V.; Lourenço, J.P.; Ribeiro, M.R. Hybrid Materials Based on Polyethylene and MCM-41 Particles Functionalized with Silanes: Catalytic Aspects of In Situ Polymerization, Crystalline Features and Mechanical Properties. Microporous Mesoporous Mater. 2016, 232, 86–96. [Google Scholar] [CrossRef]
- Ferreira, A.E.; Cerrada, M.L.; Pérez, E.; Lorenzo, V.; Cramail, H.; Lourenço, J.P.; Quijada, R.; Ribeiro, M.R. Hafnocene catalyst for polyethylene and its nanocomposites with SBA-15 by in situ polymerization: Immobilization approaches, catalytic behavior and properties evaluation. Eur. Polym. J. 2016, 85, 298–312. [Google Scholar] [CrossRef]
- Ferreira, A.E.; Cerrada, M.L.; Pérez, E.; Lorenzo, V.; Cramail, H.; Lourenço, J.P.; Ribeiro, M.R. UHMWPE/SBA-15 nanocomposites synthesized by in situ polymerization. Microporous Mesoporous Mater. 2016, 232, 13–25. [Google Scholar] [CrossRef]
- Ferreira, A.E.; Cerrada, M.L.; Perez, E.; Lorenzo, V.; Valles, E.; Ressia, J.; Cramail, H.; Lourenço, J.P.; Ribeiro, M.R. UHMWPE/HDPE in-reactor blends, prepared by in situ polymerization: Synthetic aspects and characterization. Express Polym. Lett. 2017, 11, 344–361. [Google Scholar] [CrossRef][Green Version]
- Barranco-García, R.; Ferreira, A.E.; Ribeiro, M.R.; Lorenzo, V.; García-Peñas, A.; Gómez-Elvira, J.M.; Pérez, E.; Cerrada, M.L. Hybrid materials obtained by in situ polymerization based on polypropylene and mesoporous SBA-15 silica particles: Catalytic aspects, crystalline details and mechanical behavior. Polymer 2018, 151, 218–230. [Google Scholar] [CrossRef]
- Barranco-García, R.; López-Majada, J.M.; Lorenzo, V.; Gómez-Elvira, J.M.; Pérez, E.; Cerrada, M.L. Confinement of iPP chains in the interior of SBA-15 mesostructure ascertained by gas transport properties in iPP-SBA-15 nanocomposites prepared by extrusion. J. Membr. Sci. 2018, 569, 137–148. [Google Scholar] [CrossRef]
- Barranco-García, R.; Gómez-Elvira, J.M.; Ressia, J.A.; Quinzani, L.; Vallés, E.M.; Pérez, E.; Cerrada, M.L. Effect of iPP molecular weight on its confinement within mesoporous SBA-15 silica in extruded iPP−SBA-15 nanocomposites. Microporous Mesoporous Mater. 2019, 294, 109945. [Google Scholar] [CrossRef]
- Barranco-García, R.; López-Majada, J.M.; Martínez, J.C.; Gómez-Elvira, J.M.; Pérez, E.; Cerrada, M.L. Confinement of iPP crystallites within mesoporous SBA-15 channels in extruded iPP-SBA-15 nanocomposites studied by Small Angle X-ray scattering. Microporous Mesoporous Mater. 2018, 272, 209–216. [Google Scholar] [CrossRef]
- Hammond, W.; Prouzet, E.; Mahanti, S.; Pinnavaia, T.J. Structure factor for the periodic walls of mesoporous MCM-41 molecular sieves. Microporous Mesoporous Mater. 1999, 27, 19–25. [Google Scholar] [CrossRef]
- Sauer, J.; Marlow, F.; Schüth, F. Simulation of powder diffraction patterns of modified ordered mesoporous materials. Phys. Chem. Chem. Phys. 2001, 3, 5579–5584. [Google Scholar] [CrossRef]
- Wang, X.; Ma, X.; Song, C.; Locke, D.R.; Siefert, S.; Winans, R.E.; Möllmer, J.; Lange, M.; Möller, A.; Gläser, R. Molecular basket sorbents polyethylenimine–SBA-15 for CO2 capture from flue gas: Characterization and sorption properties. Microporous Mesoporous Mater. 2013, 169, 103–111. [Google Scholar] [CrossRef]
- Barranco-García, R.; Gómez-Elvira, J.M.; Ressia, J.A.; Quinzani, L.; Vallés, E.M.; Pérez, E.; Cerrada, M.L. Variation of Ultimate Properties in Extruded iPP-Mesoporous Silica Nanocomposites by Effect of iPP Confinement within the Mesostructures. Polymers 2020, 12, 70. [Google Scholar] [CrossRef] [PubMed]
- Kapeliouchko, V.; Palamone, G.; Poggio, T.; Zuccheri, G.; Passeri, R.; Sparnacci, K.; Antonioli, D.; Deregibus, S.; Laus, M. PMMA-based core-shell nanoparticles with various PTFE cores. J. Polym. Sci. Part A Polym. Chem. 2009, 47, 2928–2937. [Google Scholar] [CrossRef]
- Laus, M.; Sparnacci, K.; Antonioli, D.; Deregibus, S.; Kapeliouchko, V.; Palamone, G.; Poggio, T.; Zuccheri, G.; Passeri, R. On the multiple crystallization behavior of PTFE in PMMA/PTFE nanocomposites from core-shell nanoparticles. J. Polym. Sci. Part B Polym. Phys. 2010, 48, 548–554. [Google Scholar] [CrossRef]
- Watanabe, R.; Hagihara, H.; Sato, H. Structure-property relationship of polypropylene-based nanocomposites by dispersing mesoporous silica in functionalized polypropylene containing hydroxyl groups. Part 1: Toughness, stiffness and transparency. Polym. J. 2018, 50, 1057–1065. [Google Scholar] [CrossRef]
- Nakagawa, S.; Kadena, K.-I.; Ishizone, T.; Nojima, S.; Shimizu, T.; Yamaguchi, K.; Nakahama, S. Crystallization Behavior and Crystal Orientation of Poly(ε-caprolactone) Homopolymers Confined in Nanocylinders: Effects of Nanocylinder Dimension. Macromolecules 2012, 45, 1892–1900. [Google Scholar] [CrossRef]
- Muñoz-Bonilla, A.; Cerrada, M.L.; Fernández-García, M.; Kubacka, A.; Ferrer, M.; Fernández-García, M. Biodegradable Polycaprolactone-Titania Nanocomposites: Preparation, Characterization and Antimicrobial Properties. Int. J. Mol. Sci. 2013, 14, 9249–9266. [Google Scholar] [CrossRef] [PubMed]
- Bento, A.; Lourenço, J.P.; Fernandes, A.; Cerrada, M.L.; Ribeiro, M.R. Functionalization of Mesoporous MCM-41 (Nano)particles: Preparation Methodologies, Role on Catalytic Features, and Dispersion Within Polyethylene Nanocomposites. ChemCatChem 2013, 5, 966–976. [Google Scholar] [CrossRef]
- Persenaire, O.; Alexandre, M.; Degée, A.P.; Dubois, P. Mechanisms and Kinetics of Thermal Degradation of Poly(ε-caprolactone). Biomacromolecules 2001, 2, 288–294. [Google Scholar] [CrossRef] [PubMed]
- Mofokeng, J.; Kelnar, I.; Luyt, A. Effect of layered silicates on the thermal stability of PCL/PLA microfibrillar composites. Polym. Test. 2016, 50, 9–14. [Google Scholar] [CrossRef]
- Marcilla, A.; Gómez-Siurana, A.; Menargues, S.; Ruiz-Femenia, R.; García-Quesada, J. Oxidative degradation of EVA copolymers in the presence of MCM-41. J. Anal. Appl. Pyrolysis 2006, 76, 138–143. [Google Scholar] [CrossRef]
- Aguado, J.; Serrano, D.P.; Romero, M.D.; Escola, J.M. Catalytic conversion of polyethylene into fuels over mesoporous MCM-41. Chem. Commun. 1996, 725–726. [Google Scholar] [CrossRef]
- Campos, J.; Lourenço, J.P.; Perez, E.; Cerrada, M.L.; Ribeiro, M.D.R. Self-Reinforced Hybrid Polyethylene/MCM-41 Nanocomposites: In-Situ Polymerisation and Effect of MCM-41 Content on Rigidity. J. Nanosci. Nanotechnol. 2009, 9, 3966–3974. [Google Scholar] [CrossRef] [PubMed]
- Bento, A.; Lourenço, J.P.; Fernandes, A.; Ribeiro, M.R.; Arranz-Andrés, J.; Lorenzo, V.; Cerrada, M.L. Gas permeability properties of decorated MCM 41/polyethylene hybrids prepared by in situ polymerization. J. Membr. Sci. 2012, 415–416, 702–711. [Google Scholar] [CrossRef]
- Blázquez-Blázquez, E.; Pérez, E.; Lorenzo, V.; Cerrada, M.L. Crystalline characteristics and their influence in the Mechanical Performance in Poly(ε-caprolactone)/High Density Polyethylene Blends. Polymers 2019, 11, 1874. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.; Dorset, D.L. Crystal Structure of Poly(ε-capro1actone). Macromolecules 1990, 23, 4604–4607. [Google Scholar] [CrossRef]
- Alexander, L.E. X-ray Diffraction Methods in Polymer Science; Wiley-Interscience: New York, NY, USA, 1969. [Google Scholar]
- Zhao, D.; Huo, Q.; Feng, J.; Chmelka, B.F.; Stucky, G.D. Nonionic Triblock and Star Diblock Copolymer and Oligomeric Surfactant Syntheses of Highly Ordered, Hydrothermally Stable, Mesoporous Silica Structures. J. Am. Chem. Soc. 1998, 120, 6024–6036. [Google Scholar] [CrossRef]
- Xu, X.; Song, C.; Andrésen, J.M.; Miller, B.G.; Scaroni, A.W. Preparation and characterization of novel CO2 “molecular basket” adsorbents based on polymer-modified mesoporous molecular sieve MCM-41. Microporous Mesoporous Mater. 2003, 62, 29–45. [Google Scholar] [CrossRef]
- Son, W.-J.; Choi, J.-S.; Ahn, W.-S. Adsorptive removal of carbon dioxide using polyethyleneimine-loaded mesoporous silica materials. Microporous Mesoporous Mater. 2008, 113, 31–40. [Google Scholar] [CrossRef]
- Ruggerone, R.; Plummer, C.J.; Herrera, N.N.; Bourgeat-Lami, E.; Månson, J.-A.E. Highly filled polystyrene–laponite nanocomposites prepared by emulsion polymerization. Eur. Polym. J. 2009, 45, 621–629. [Google Scholar] [CrossRef]
- Antonioli, D.; Laus, M.; Sparnacci, K.; Deregibus, S.; Kapeliouchko, V.; Palamone, G.; Poggio, T.; Zuccheri, G.; Passeri, R. Thermal and DMA Characterization of PTFE-PMMA Nanocomposites from Core-Shell Nanoparticles. Macromol. Symp. 2010, 296, 197–202. [Google Scholar] [CrossRef]
- Antonioli, D.; Laus, M.; Zuccheri, G.; Kapeliouchko, V.; Righetti, M.C.; Boarino, L.; Sparnacci, K. Preparation and Properties of PTFE-PMMA Core-Shell Nanoparticles and Nanocomposites. J. Nanotechnol. 2012, 2012, 875815. [Google Scholar] [CrossRef]
- Saladino, M.; Motaung, T.; Luyt, A.; Spinella, A.; Nasillo, G.; Caponetti, E. The effect of silica nanoparticles on the morphology, mechanical properties and thermal degradation kinetics of PMMA. Polym. Degrad. Stab. 2011, 97, 452–459. [Google Scholar] [CrossRef]
- Nielsen, L.E.; Landel, R.W. Mechanical Properties of Polymers and Composite; Marcel Dekker: New York, NY, USA, 1994. [Google Scholar]
- Sabater i Serra, R.; Escobar Ivirico, J.L.; Meseguer Dueñas, J.M.; Andrio Balado, A.; Gómez Ribelles, J.L.; Salmerón Sánchez, M. Dielectric relaxation spectrum of poly(ε-caprolactone) networks hydrophilized by copolymerization with 2-hydroxyethyl acrylate. Eur. Phys. J. E 2007, 22, 293–302. [Google Scholar] [CrossRef] [PubMed]
- Cerrada, M.L.; Pereña, J.M.; Benavente, R.; Pérez, E. Viscoelastic processes in vinyl alcohol-ethylene copolymers. Influence of composition and thermal treatment. Polymer 2000, 41, 6655–6661. [Google Scholar] [CrossRef]
- Harrison, K.L.; Jenkins, M.J. The effect of crystallinity and water absorption on the dynamic mechanical relaxation behaviour of polycaprolactone. Polym. Int. 2004, 53, 1298–1304. [Google Scholar] [CrossRef]
- Nikolic, M.S.; Mitric, M.; Dapcevic, A.; Djonlagic, J. Viscoelastic properties of poly(ε-caprolactone)/clay nanocomposites in solid and in melt state. J. Appl. Polym. Sci. 2016, 133, 42896. [Google Scholar] [CrossRef]












Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).