2.1. Validations of Subtle Structural Characteristics and Attempts in Upscaling MD Simulations of Lignocellulosics
In this section, we will consider the following
The first attempts at lignocellulosic MD simulations, the various force fields that have been tried for this purpose and the validation methodologies used.
The types of structural/molecular characteristics of lignocellulosics that were uncovered using MD and the insights gained thereof.
Scales of the various initial simulations in terms of number of atoms, structural complexity and upscaling using high performance parallel computing systems.
Faulon et al. [
20] carried out one of the first successful simulations of lignocellulosics in gymnospermous wood. They used the DREIDING force field for most of the simulations. DREIDING is a 1st generation force field with the specifics reported in [
7]. The simulations were carried out at 300 K and for a relatively small length ranging from 120 ps to 240 ps owing to the capabilities of the time (ca 1994) but were still able to determine profound structural information about lignocellulosics within the cell wall of gymnospermous wood [
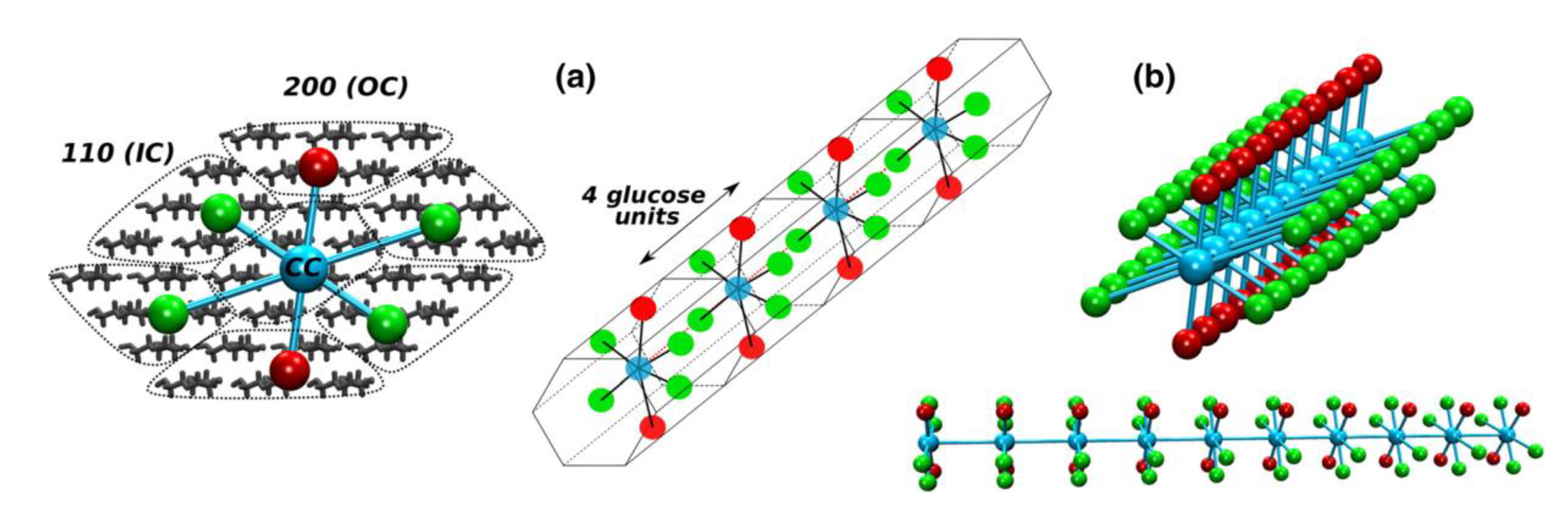
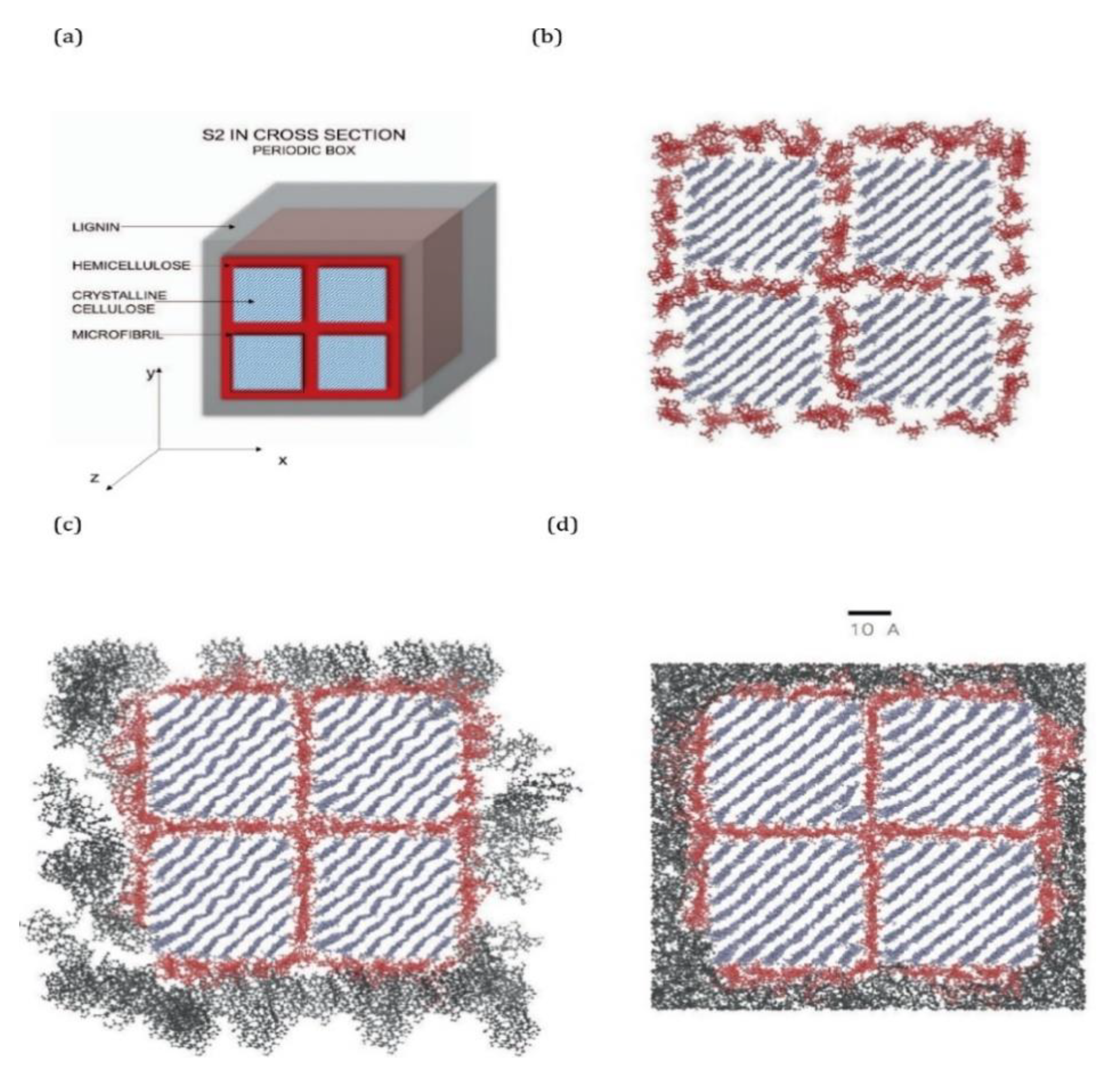
20]. They constructed the lignocellulosic model (
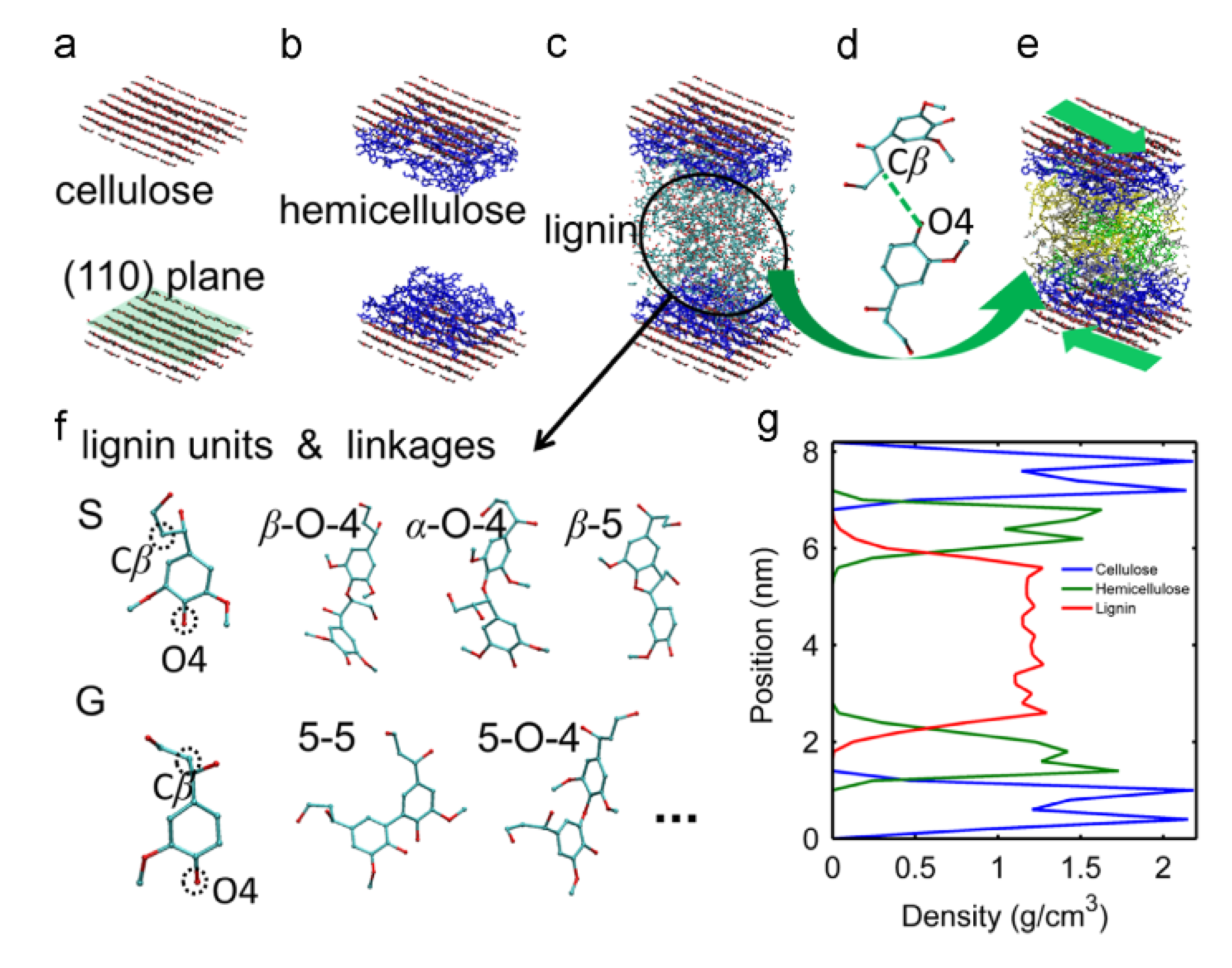
Figure 2) by incorporating 1 out of 4 separate lignin units within a polysaccharide unit comprised of cellulose chains and hemicellulose layers (
Figure 2).
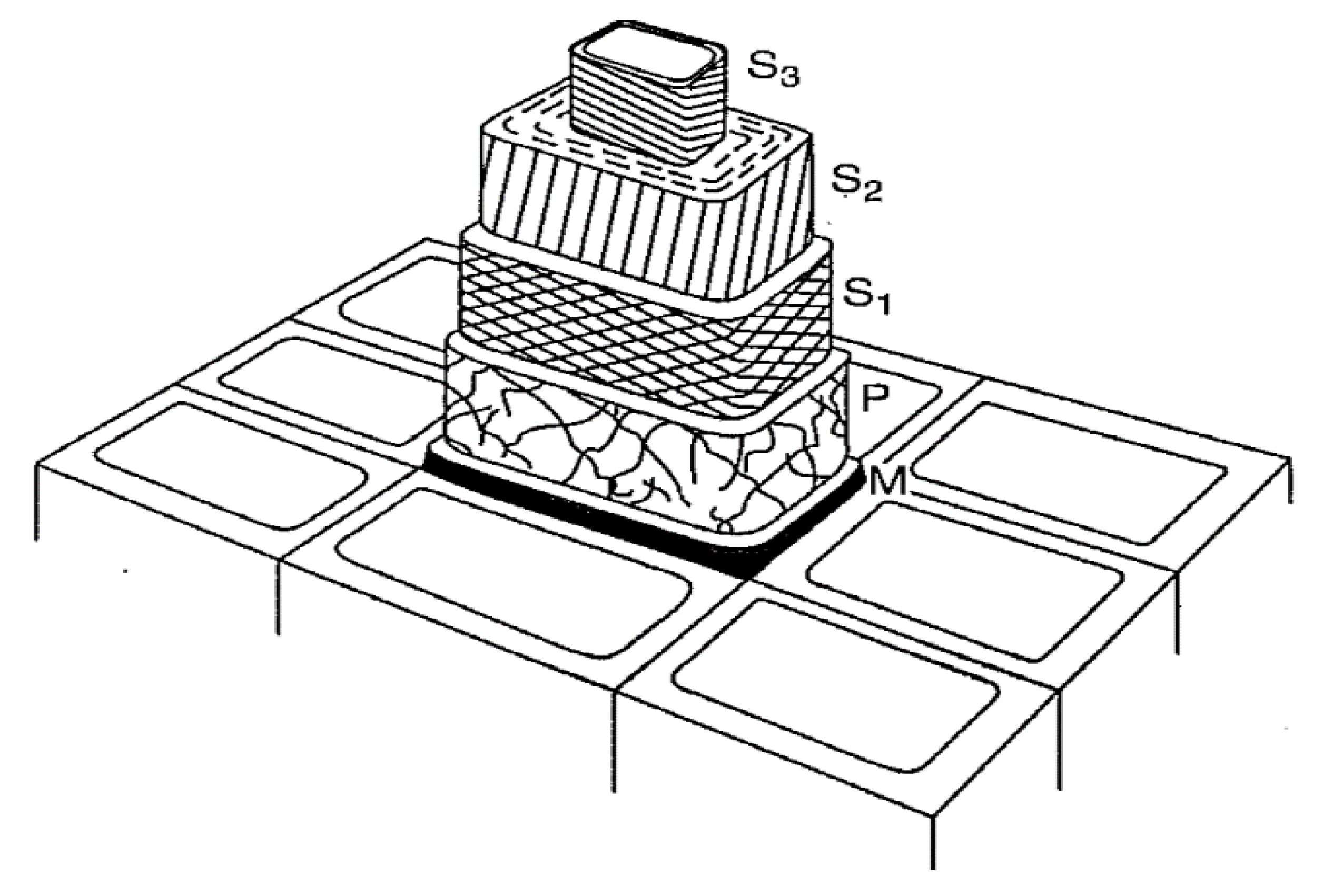
The lignin and hemicellulose were allowed to move during the MD simulation while the cellulose chains were fixed in place to approximate the crystalline I structure of cellulose within the cell walls of wood. The observations made by Faulon et al. [
20] showed that the lignin structures evolved to form hydrogen bonds with the hemicellulose chains and cellulose chains. Hydrogen bonds were present between the α and γ alcohols of the lignin and the alcohol functional groups of the different sugar units. The presence of hydrogen bonds between lignin and polysaccharides causes each lignin structure to retain its initial conformation. When lignin is associated with polysaccharides as in the secondary cell wall, the helical structure is probably elongated (as the first structure in
Figure 3) due to limited space between microfibrils and is also influenced by the crystallinity of the microfibrils. However, in portions of wood cells poor in polysaccharides such as the compound middle lamella, the structure is not supported by cellulose chains and is free to collapse to the most stable conformation-structure (as the last structure in
Figure 3).
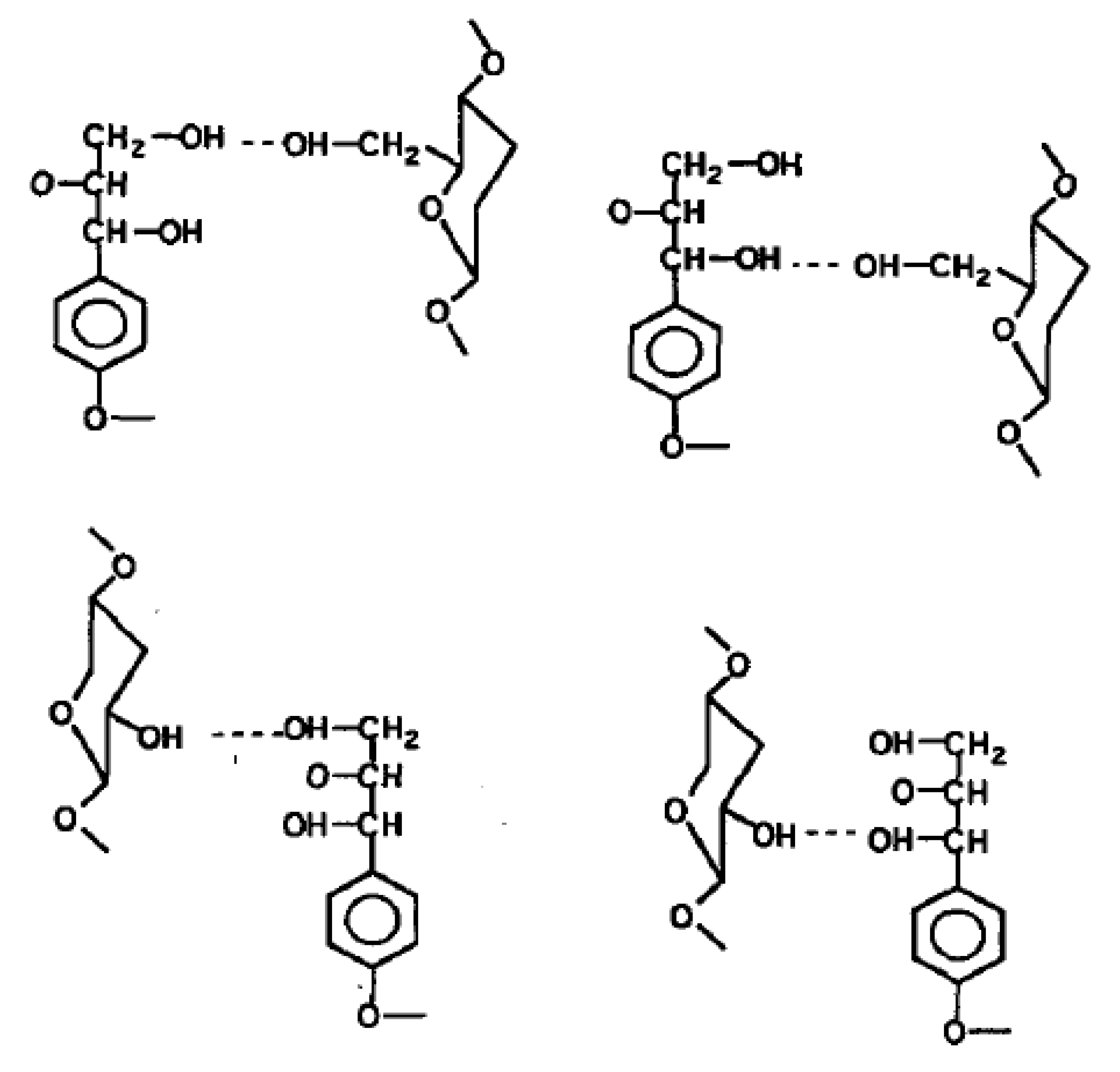
Faulon et al. [
20] also indicated the four possible sites for hydrogen bonds between lignin and polysaccharides in
Figure 4. While critical molecular information was obtained from their work, Faulon et al. [
20] did not determine any physical/mechanical properties of the lignocellulosics.
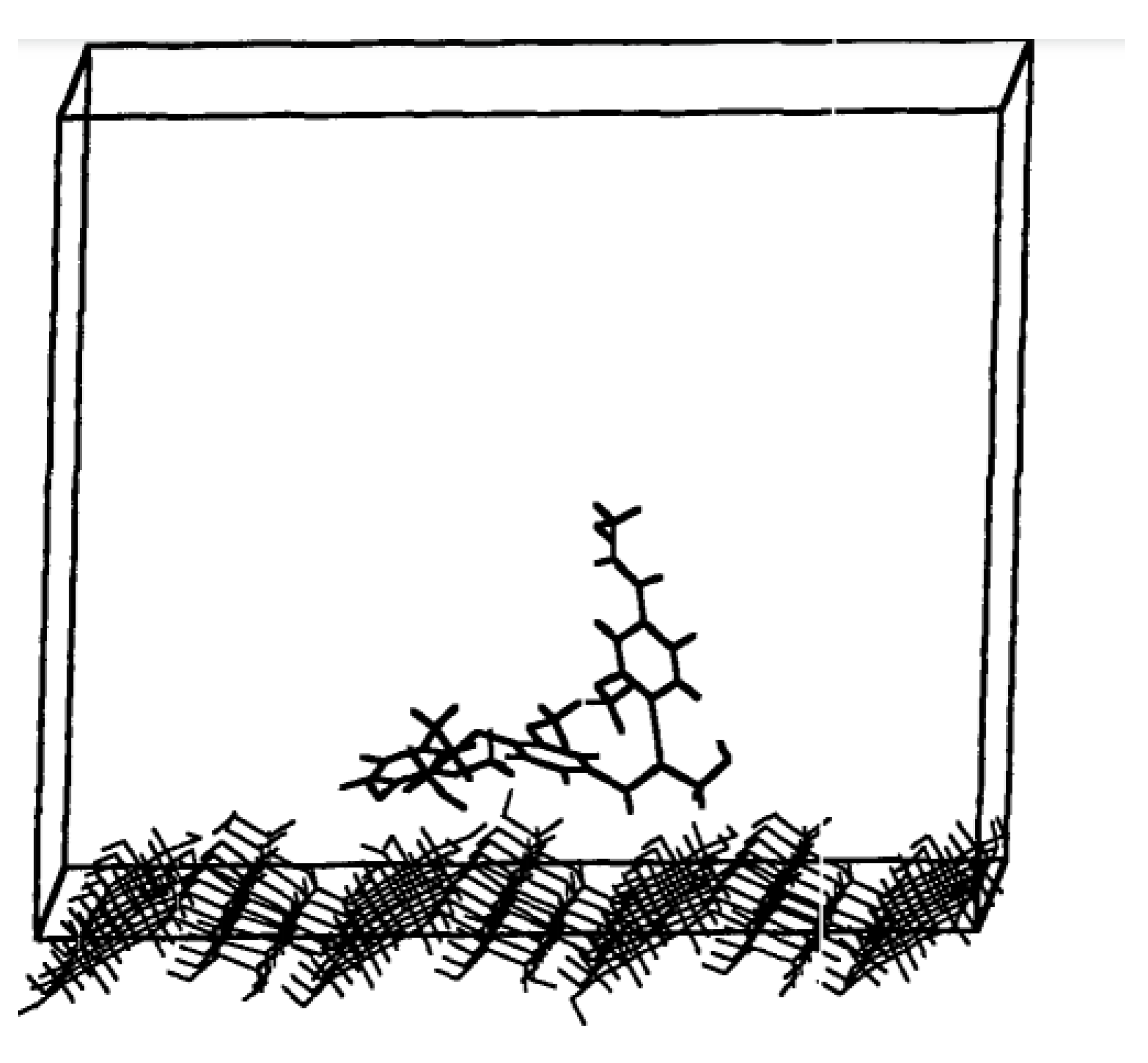
Houtman and Atalla [
21] expanded upon the work of Faulon et al [
20], using the consistent-valence forcefield (CVFF) for a similation time of 400 ps at 300 K and a cut off distance of 13 Å to model the interactions between a model lignin (coniferyl alcohol) compound with cellulose/polysaccharide surface. An illustration of the computational model used by Houtman and Atalla [
21] is shown in
Figure 5. It was found that two of the phenyl rings of the coniferyl alcohol are closely associated with the surface, but the third continued to remain free from strong associations with the surface. The adsorption of this third phenyl ring appears to be inhibited by the constraints of the β-O-4 linkage. Analysis of the cellulose surface showed that the lignin model was adsorbed over a region of high concentration of hydroxyl groups. This suggested that the dominant force holding the lignin model on the surface is electrostatic dipole–dipole interactions.
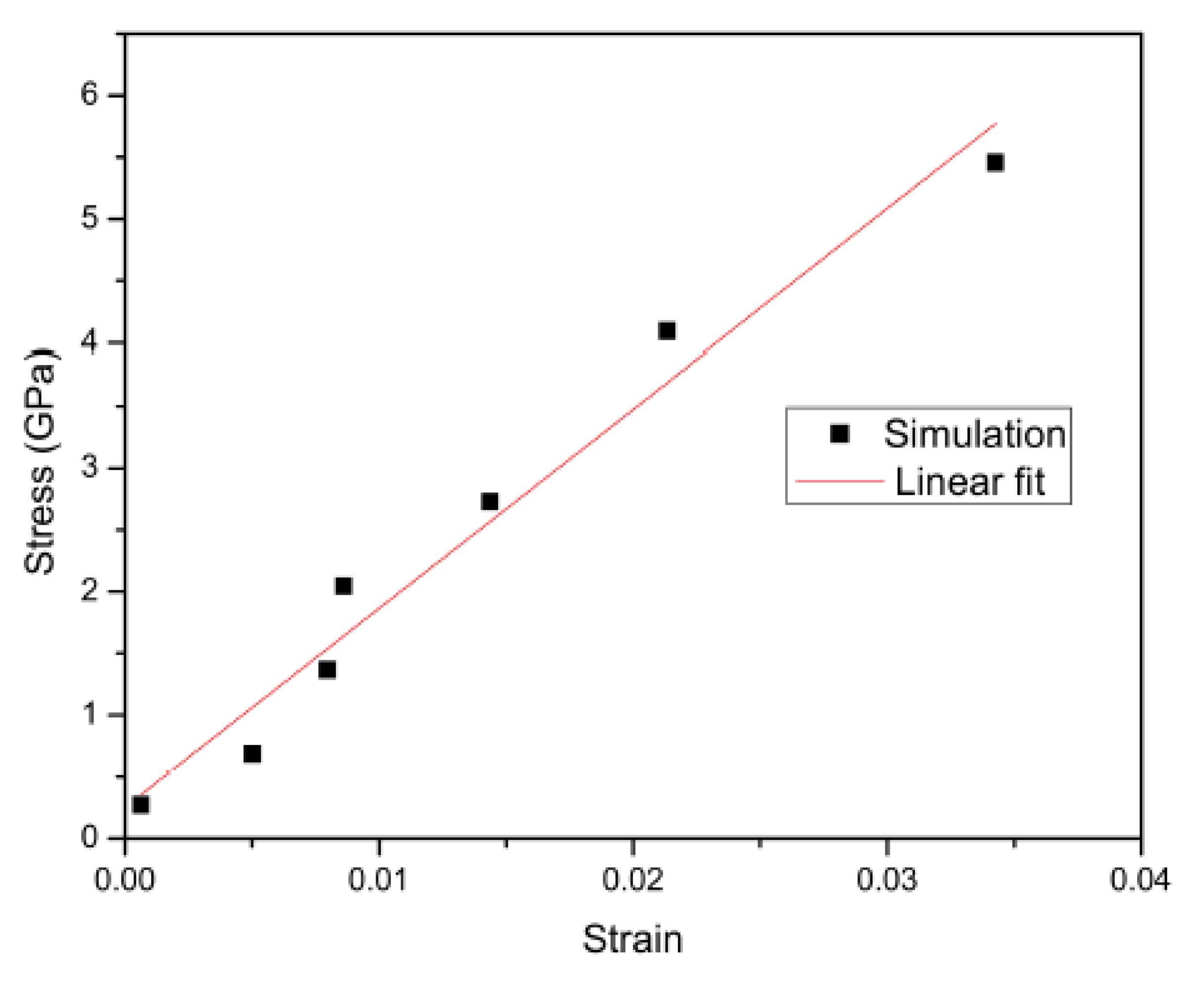
The CVFF was also used by Ganster and Blackwell [
22] in one of the first attempts to simulate crystalline polymorphs of cellulose (
Figure 6). The simulation was done for a period of 540 ps using the Verlet method [
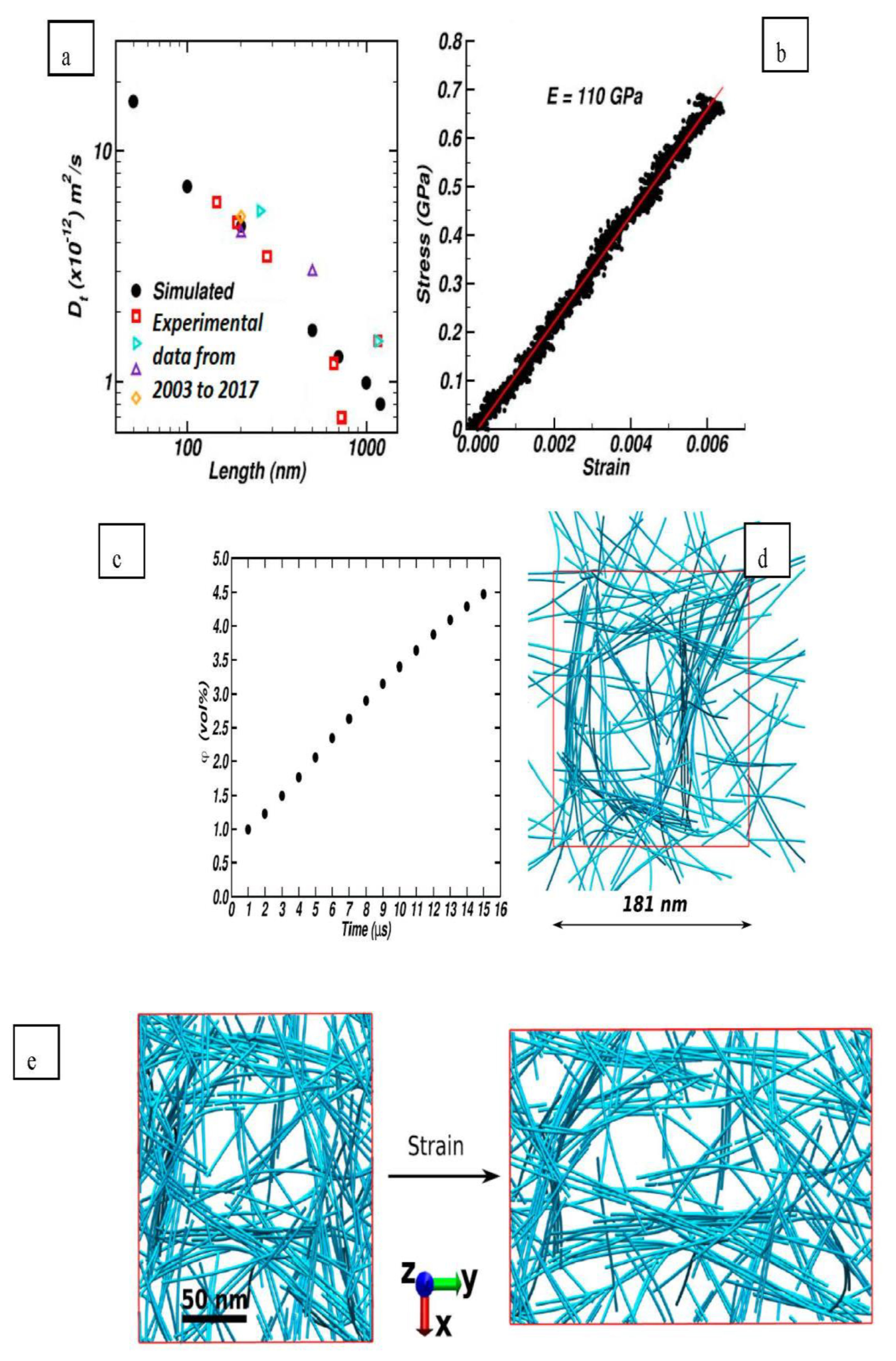
4] and a cut off of 9.5 Å at a starting temperature of 602 K which was slowly cooled to a target temperature of 298 K. No analysis was conducted regarding the thermal stability of the starting configuration at 602 K. Following the MD procedure, they found that there was a strong anisotropy for shear along the chains. In planes containing the chain axis, the shear modulus ranged from 5 to 20 GPa. Structural properties of the simulated cellulose were favourably comparable to real experimental data obtained through x-ray diffraction as shown in
Table 1.
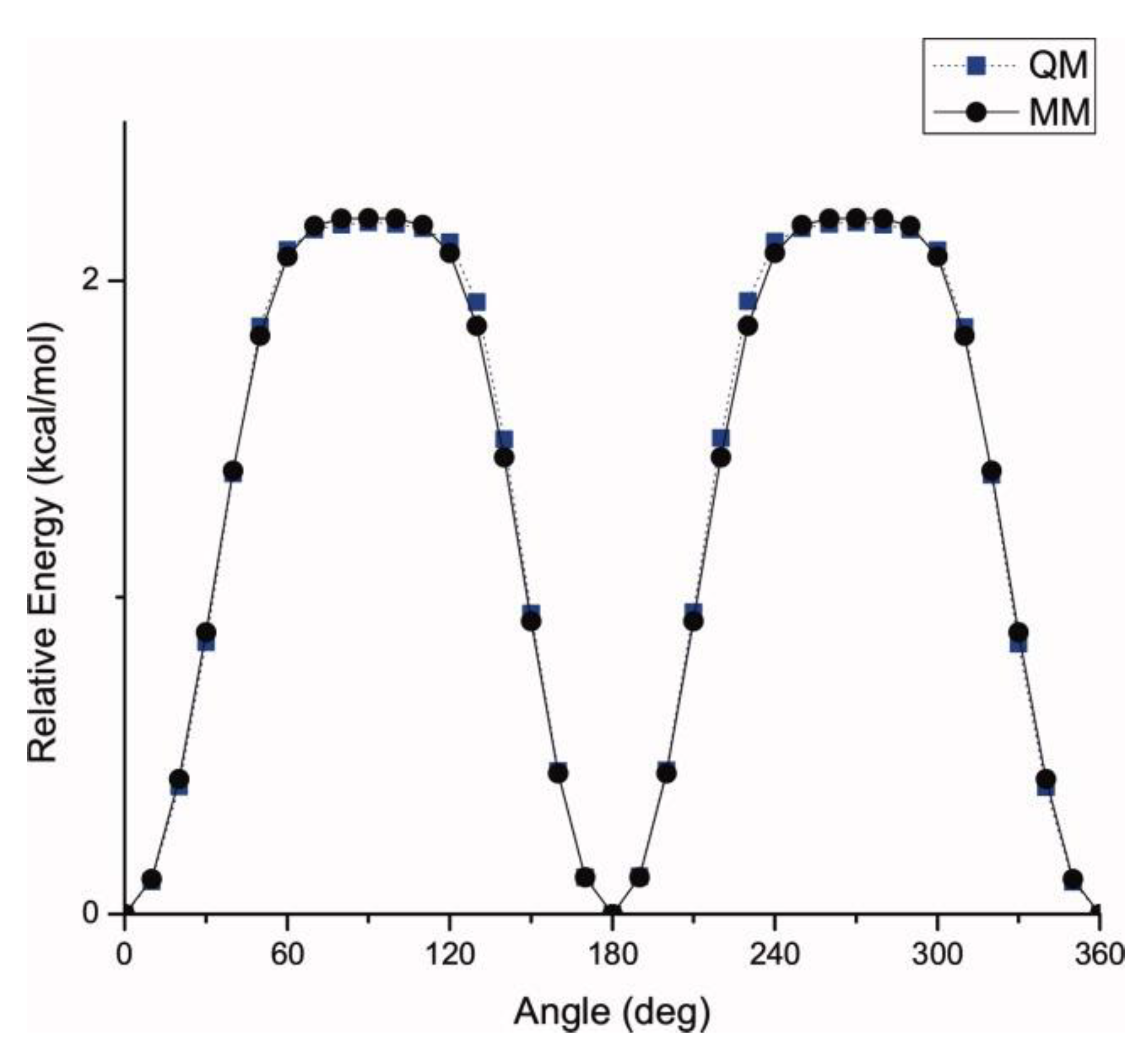
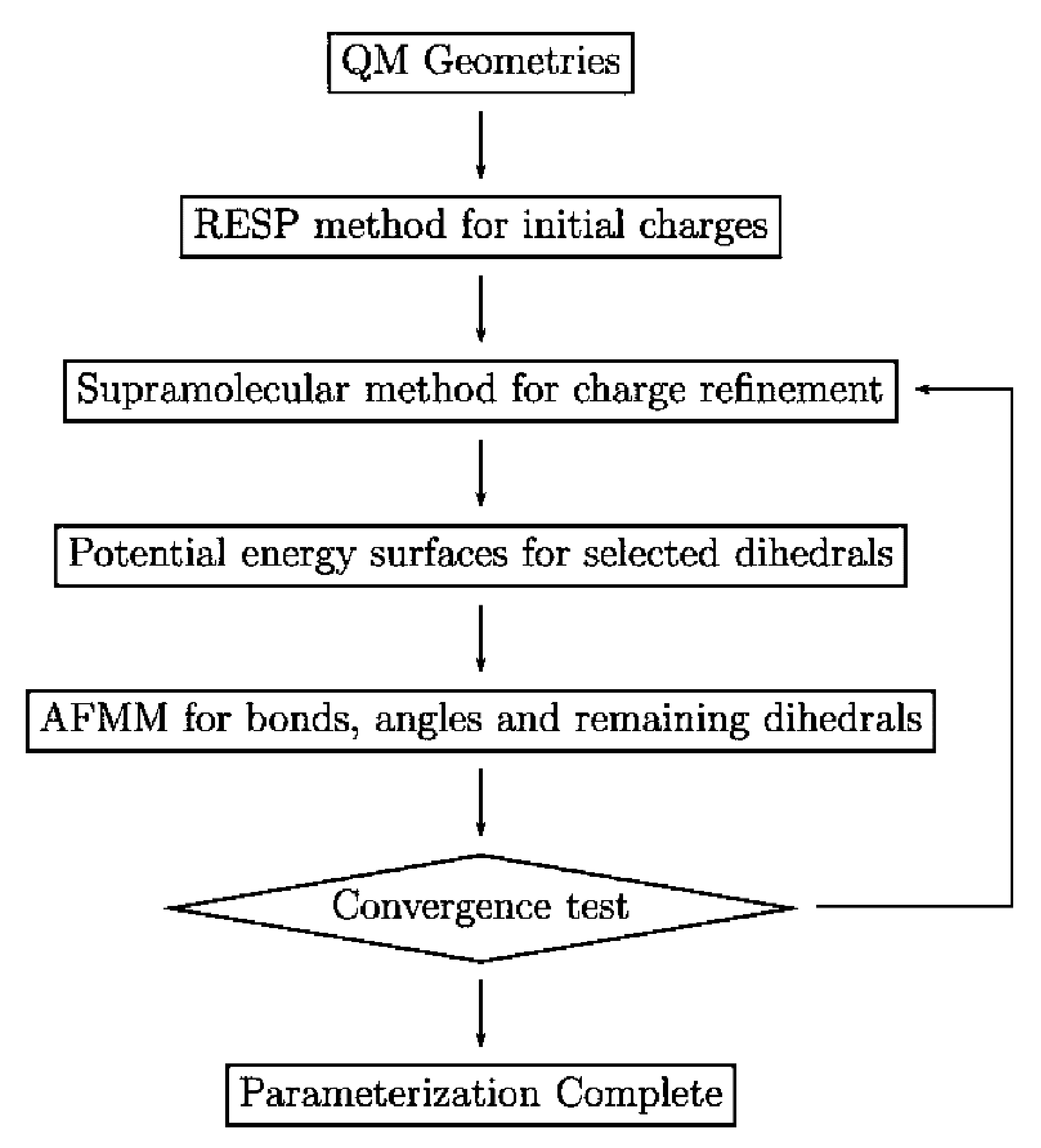
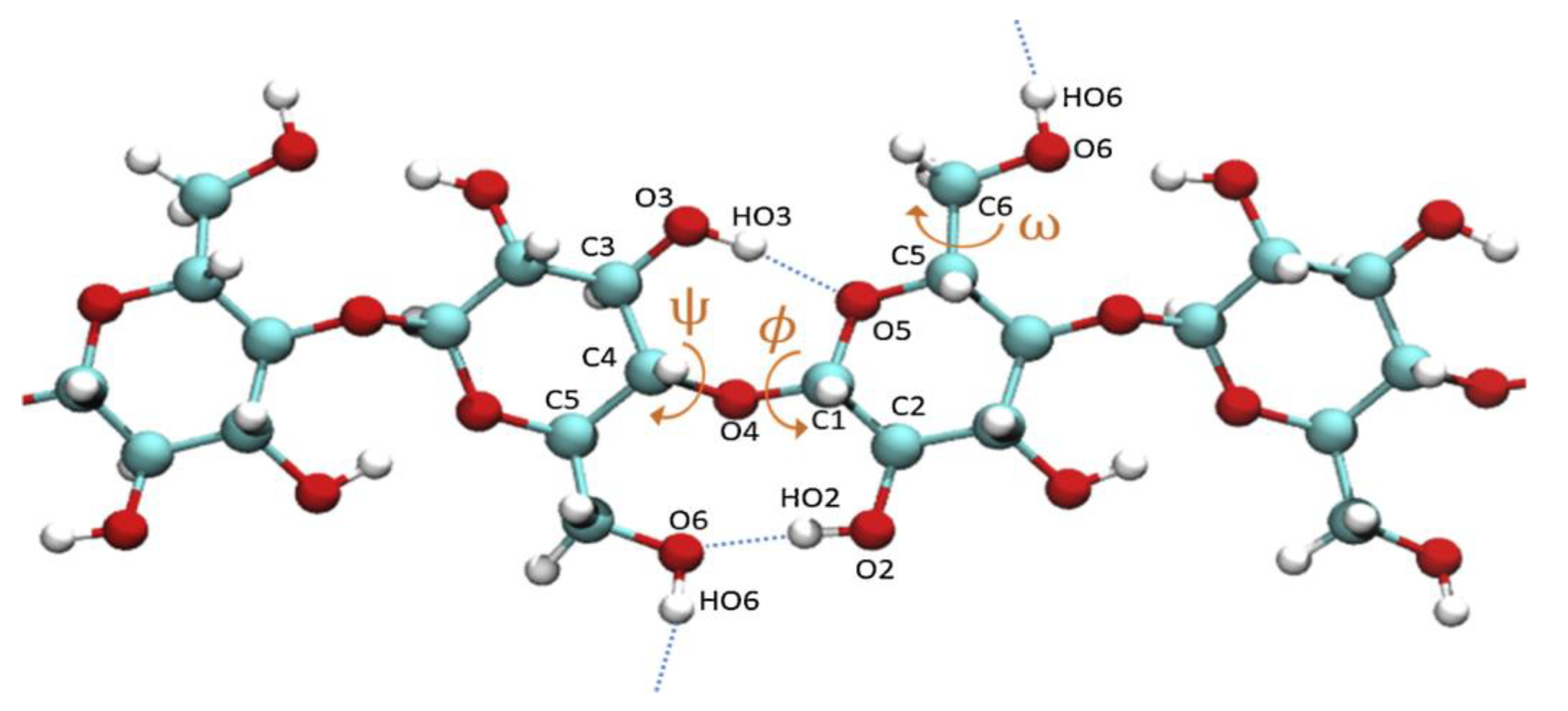
The MD algorithm developed by Petridis and Smith in [
23] involved optimization of the electrostatic interactions by assigning partial atomic charges so as to reproduce quantum chemical data. These charges were taken into account to ensure the presence of electronic polarization within the context of condensed phase simulations. Dihedral force constants were determined by examining potential energy surfaces. The interactions between bonded atoms were subsequently optimized with respect to quantum chemical vibrational data using the Automated Frequency Matching Method developed by Vaiana et al. [
24]. It was found that dihedral rotations around the β-O-4 linkage within the lignin structure played a significant role in determining the configuration of the lignin macromolecule [
25,
26]. Petridis et al. studied two equivalent dihedrals in lignin model system for showing the dihedral rotations [
23]. These are ω
1: C
2-C
1-O-C
α (or C
6-C
1-O-C
α equivalently) and ω
2: C
1-O-C
α-H. They observed that the molecular rotation around these dihedrals leads to severe steric hindrance between the two aromatic rings of guaiacyl and syringyl units connected with β-O-4 linkage. As an example, the comparison between the quantum mechanical and force field predicted dihedral rotations of anisole model are shown in
Figure 7. The predictions were obtained using a parameterization process shown in
Figure 8.
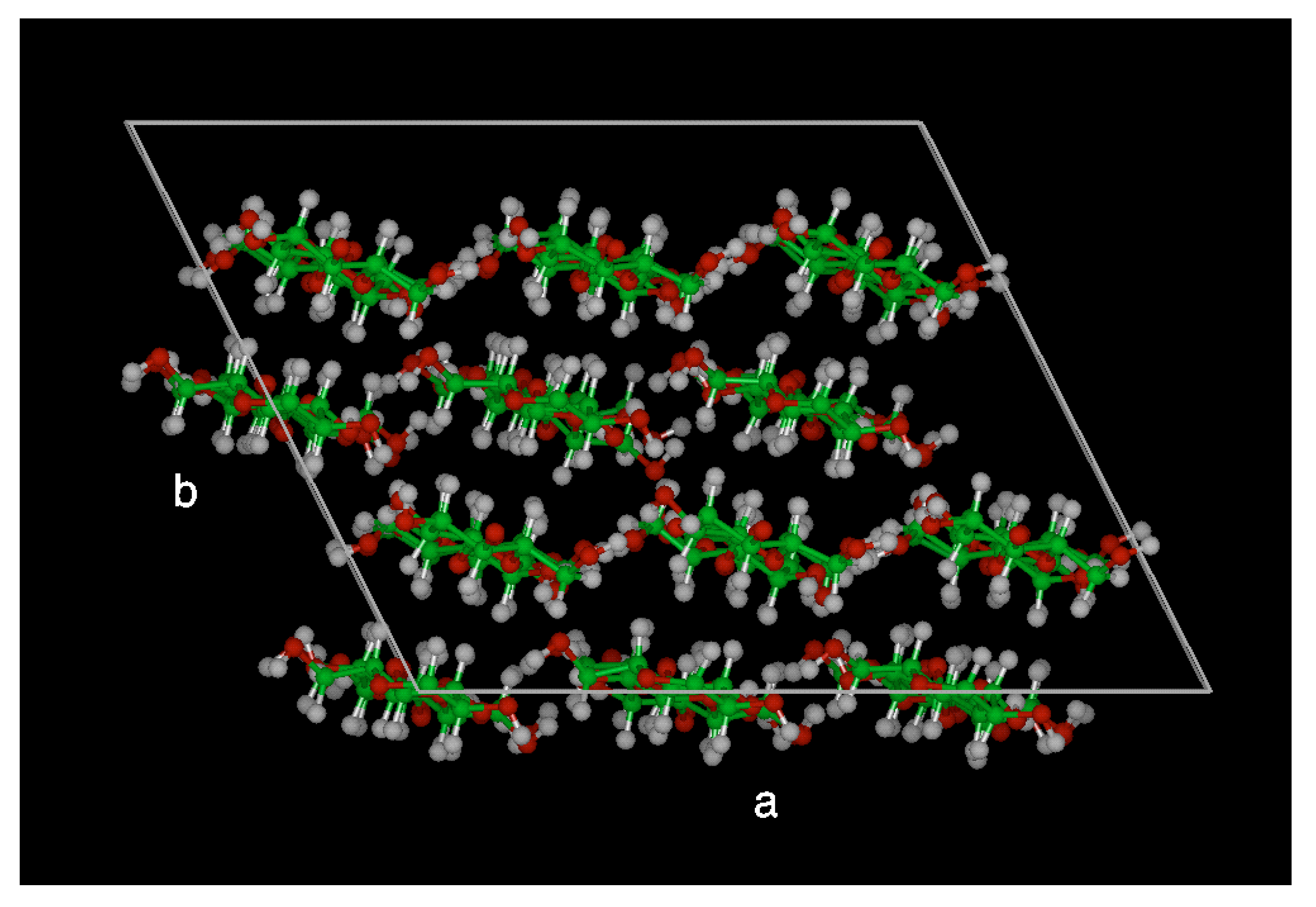
The dihedral rotations around the β-O-4 linkage of lignin structure confirmed the conjecture of Faulon et al. [
20]. In a separate study, the influence of intramolecular H-bonding on the dynamical conformational behaviour of the β-O-4 structure was studied to support the theoretical data. In this study, the experimental NMR data clearly indicated that the guaiacyl β-O-4 structure experiences conformational changes in solution and the intramolecular H- bonds to solvent molecules predominate. This study concluded that the guaiacyl β-O-4 structure is flexible and intramolecular H-bonds are not strong and persistent enough to confer rigidity to the molecule in solution. This concept was verified via both molecular modelling and NMR study [
28,
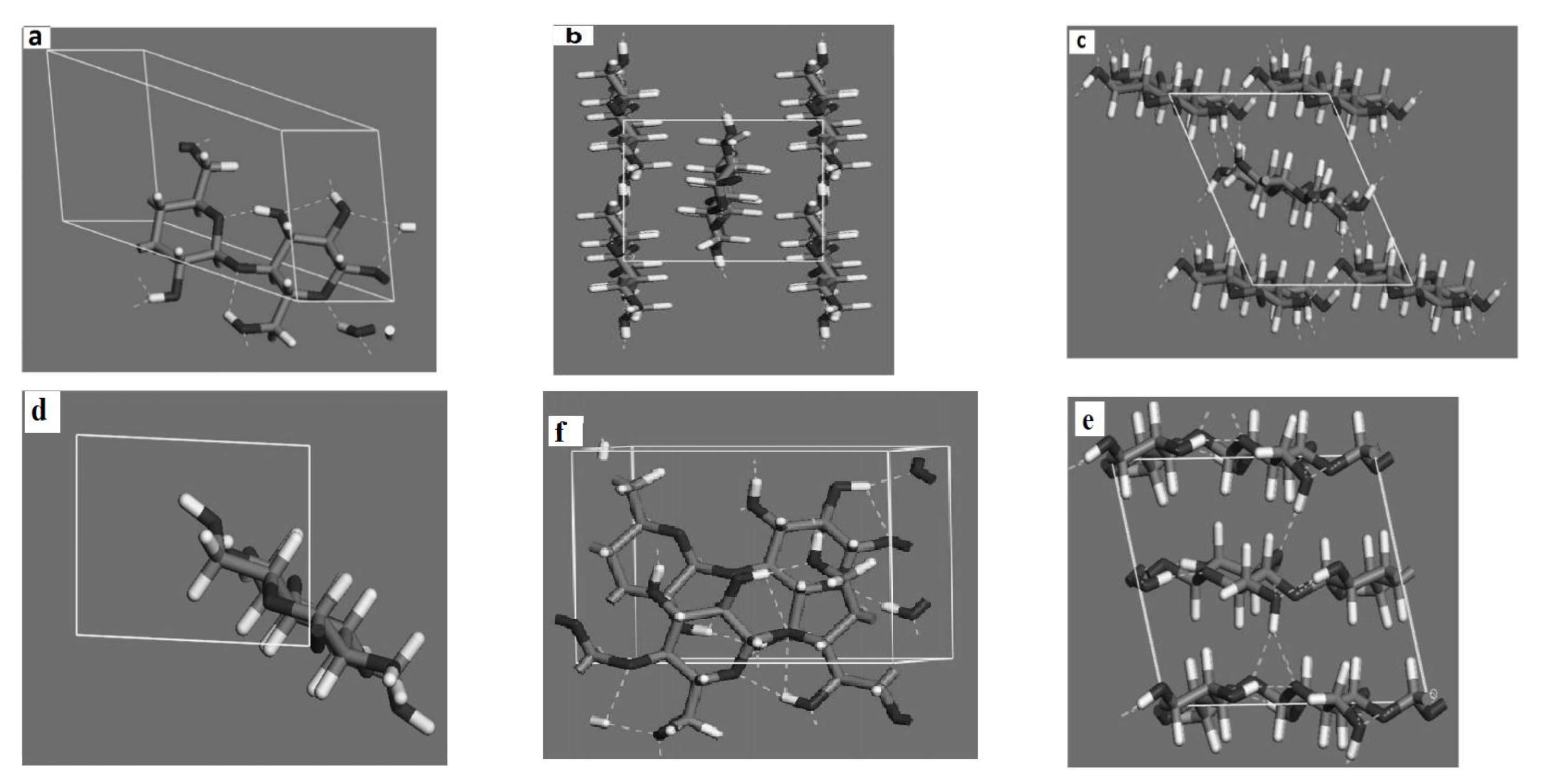
29]. The specific parameterization of the force field was done using two model compounds- anisole and 4-(1,2,3-trihydroxypropyl) phenol (PHP). To establish the validity of the developed molecular mechanics force field, the crystal structure of a lignin sub-unit dimer, erythro-2-(2,6-Dimethoxy-4-methylphenoxy)-1- (4-hydroxy-3,5-dimethoxyphenyl) propane-1,3-diol (EPD) was used. EPD is very similar to two syringyl units connected with a β-O-4 linkage, but with the hydroxy group of one of the phenol rings substituted by a methyl group. The excellent agreements between the MD and experimental data as regards the physical (in this case, crystalline) properties of EPD is shown in
Table 2.
Fine structural characteristics of lignocellulosic materials also entails understanding the interactions between the individual lignocellulosic phases and the water molecules they exist in association with. Faulon et al. [
20] were able to isolate the bonds/moieties in lignocellulosics that contribute most to the hydrogen bonding interactions but did not conduct any specific water-cellulose-lignin interaction simulations. The water molecules that form an intrinsic part of the lignocellulosic network. Therefore, when it comes to understanding lignocellulosic behaviour, it is fundamental to take into account the dynamics of these water molecules when they are in contact with the lignocellulosic material. To study the dynamics of the bound water with cellulose, O’Neill et al. [
30] utilised a MD based approach and validated the simulations using quasi-elastic neutron scattering (QENS). In a previous work, Petridis et al. [
31] had shown how cellulose was found to be more rigid in the hydrated state than in the dry state, and evidence was found that in hydrated cellulose the cellulose chains are more closely packed and water molecules bridge the chains through hydrogen bonding interactions.
The molecular model used in [
30] contained four aligned cellulose (cellulose Iα) microfibrils. Each microfibril contained 36 chains with a degree of polymerization of 80. The system was solvated at 0.20 g water/g cellulose to match the experiments. The simulations were performed employing the GROMACS software [
11] and a parameterised CHARMM C36 carbohydrate force field similar to [
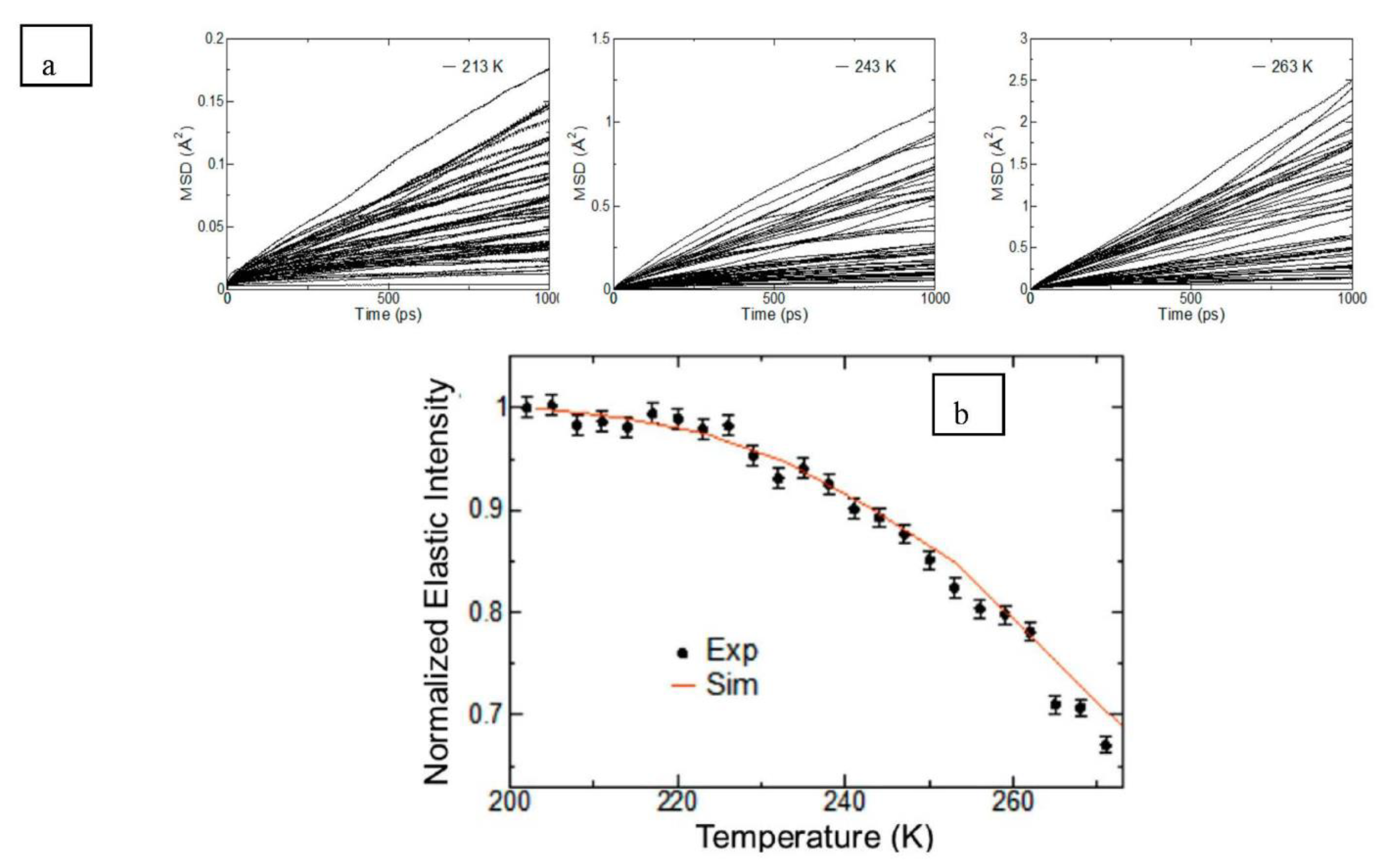
27]. The TIP4P water model was chosen for simulating the water molecules owing to the derived self-diffusion coefficient being in good agreement with experiments. Periodic boundary conditions were employed together with the Particle Mesh Ewald (PME) algorithm and an interaction cut-off distance of 12 Å for electrostatics and between 9 and 10 Å for the van der Waals interaction to reduce computational requirements. Each system was simulated for a total of 11 ns at three temperatures: 213 K, 243 K and 263 K and at atmospheric pressure. The data from the final 5 ns of each simulation were analysed, once the system had reached equilibration. This methodology, where the initial non equilibrium simulation data is discarded and the final few ns post equilibrium is only taken into account, was utilised for estimating diffusion coefficients in semicrystalline polymer-lignocelluosic composites by Prasad et al. [
32,
33]. In [
32,
33], semi empirical multiphase modelling was used to further increase the accuracy of the simulated data combined with the use of the PCFF (Polymer Consistent Force Field) for MD simulations. O’Neill et al. [
30] in
Figure 9 correlated the elastic intensity ‘S
el (T)’ at a temperature ‘T’ for the neutron scattering wave vector ‘Q’ to the mean squared displacement/MSD (
) of the bound water molecules using the Gaussian approximation given by Equation (18). Using the Einstein relationship also used by Prasad et al. [
32,
33] amongst others, the MSD could then be used to estimate the diffusion coefficient D.
As seen from
Figure 9b, an excellent agreement was found between the calculated and measured MSD showing that the simulated system was very accurate in terms of estimating the dynamics of water in real crystalline cellulose. O’Neill et al. [
30] stated that the knowledge of such dynamical properties of bound water in lignocellulosic systems and the partition behaviour of the bound water in the cellulose supramolecular structure could further enhance the understanding of water’s role in the change of biomass morphology during different pre-treatment regimes for biofuels production. This can potentially lead to more efficient pre-treatment approaches.
Bound water dynamics was then correlated to the molecular orientation of the cellulose molecules by Petridis et al. [
31]. Similar to [
30], the simulated system contains four aligned cellulose Iα fibres, the predominant allomorph of bacterial cellulose, each consisting of 36 chains with a chain length of 80 glucose monomers. The shape of the fibres is “diamond”, with the hydrophilic (100) and (010) crystallographic faces predominantly exposed to the solvent in this case, water and the interaction of water with cellulose hydrophilic and hydrophobic surfaces was studied. The simulated fibres were studied at dry and at hydrated conditions (0.20 g of water per g of cellulose). The parameterised CHARMM C36 carbohydrate force field was once again used for the cellulose and the TIP4P-EW model was used to simulate the bound water molecules. The simulations were performed with the program GROMACS [
11] using a time step of 2 fs. Bonds involving hydrogen atoms were constrained using the LINCS algorithm (fourth order with one iteration), and for water the SETTLE algorithm was used. Periodic boundary conditions were employed, and the PME algorithm [
31] was used for electrostatic interactions. A cut-off of 12 Å was used for the electrostatic interactions and 9−10 Å for the van der Waals interactions. Each system was simulated at a pressure of 105 Pa and at 16 temperatures: from T = 153 to 293 K in 10 K increments and at 298 K. Simulations were 11 ns long with data from the last 10 ns of simulation at each temperature being analysed similar to [
31,
32,
33].
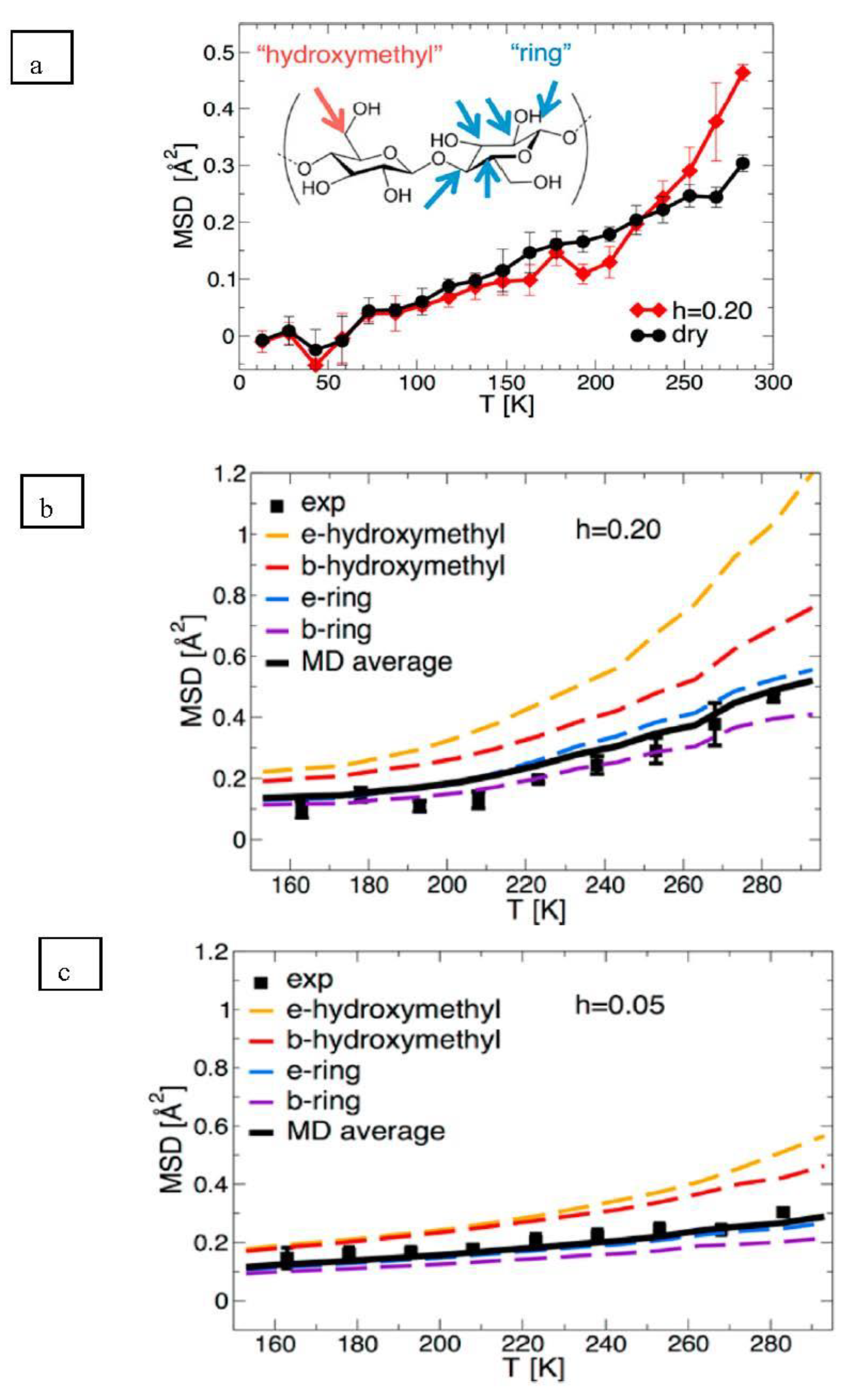
The experimental atomic MSD, a quantitative measure of the average magnitude of atomic motions, of the two cellulose samples is shown in
Figure 10. Up to ∼210 K both dry and hydrated cellulose display a relatively small increase in their MSD, indicating the underlying atomic fluctuations are not affected by hydration. However, a dynamical transition, indicated by a steep increase of MSD at T > 210 K is observed only in the hydrated sample (
Figure 10a). This transition arises from the onset of large-amplitude, anharmonic dynamics. The simulation-derived MSD was then decomposed by Petridis et al. [
31] into contributions from water-exposed or surface (“e-”) and water-buried or core (“b-”) hydroxymethyl and chain atoms in
Figure 10b,c for the hydrated and dry cellulose simulated systems respectively. In all simulations, the MSD of the hydroxymethyl hydrogens was found to be always higher than that of the ring hydrogen atoms. Furthermore, those rings and hydroxymethyl hydrogens exposed to the water molecules (i.e., in hydrated condition) had higher MSD values than those in the core (
Figure 10c). This indicated that there was a higher enhancement of molecular mobility upon hydration for the ring and hydroxymethyl hydrogens making up the surface groups relative to those groups in the core. A strong temperature dependence of the MSD is found only in the hydrated system and only for the exposed hydroxymethyl hydrogens, which account for only 1.5% of the total nonexchangeable hydrogen atoms in the entire system. In contrast, the average MSD of buried ring hydrogen atoms, which comprise 70% of the nonexchangeable H, is nearly independent of temperature. Therefore, the weaker experimental dynamical transition in cellulose relative to other biopolymers arises from the relatively small proportion of nonexchangeable hydrogens in the solvent-exposed side chain hydroxymethyl groups.
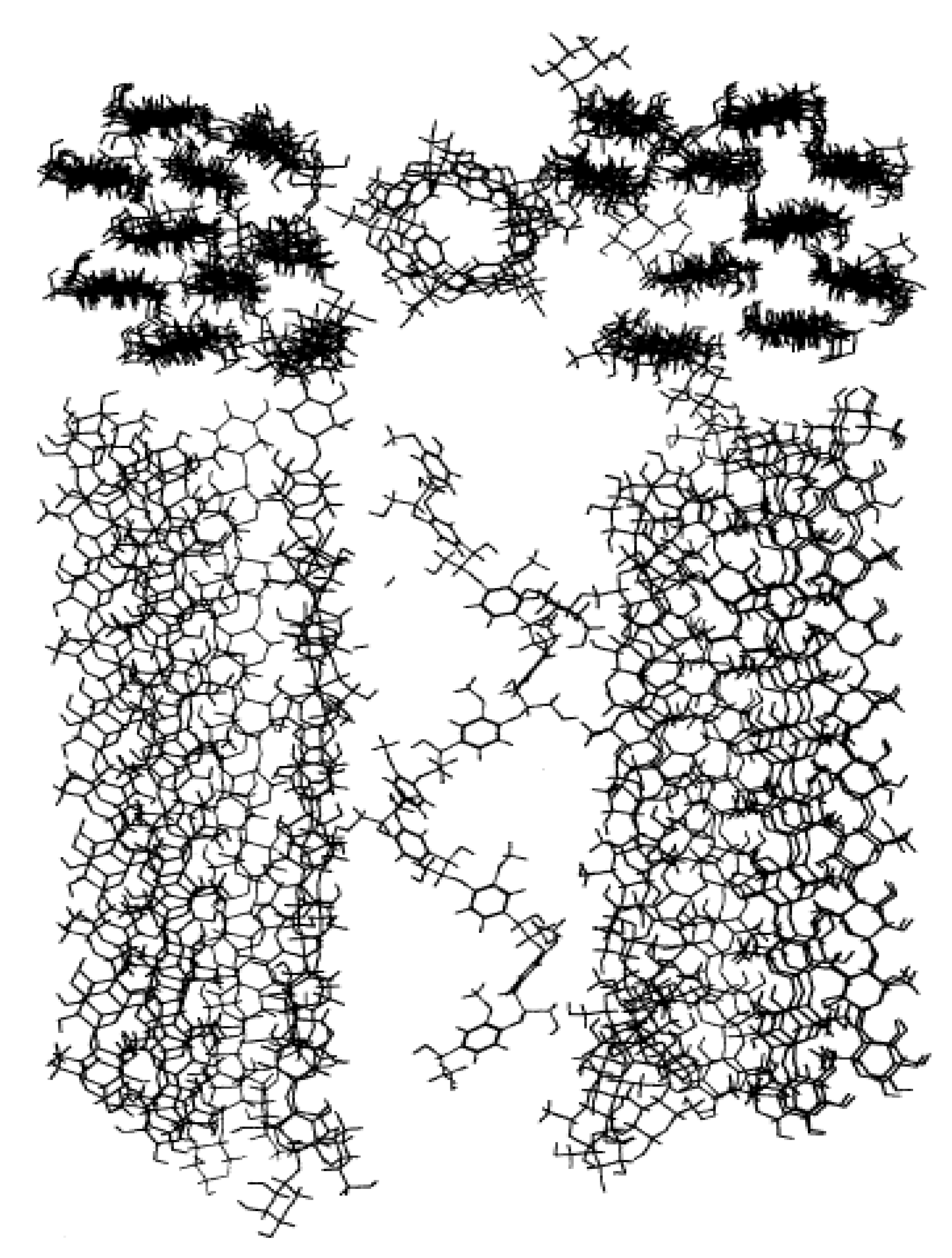
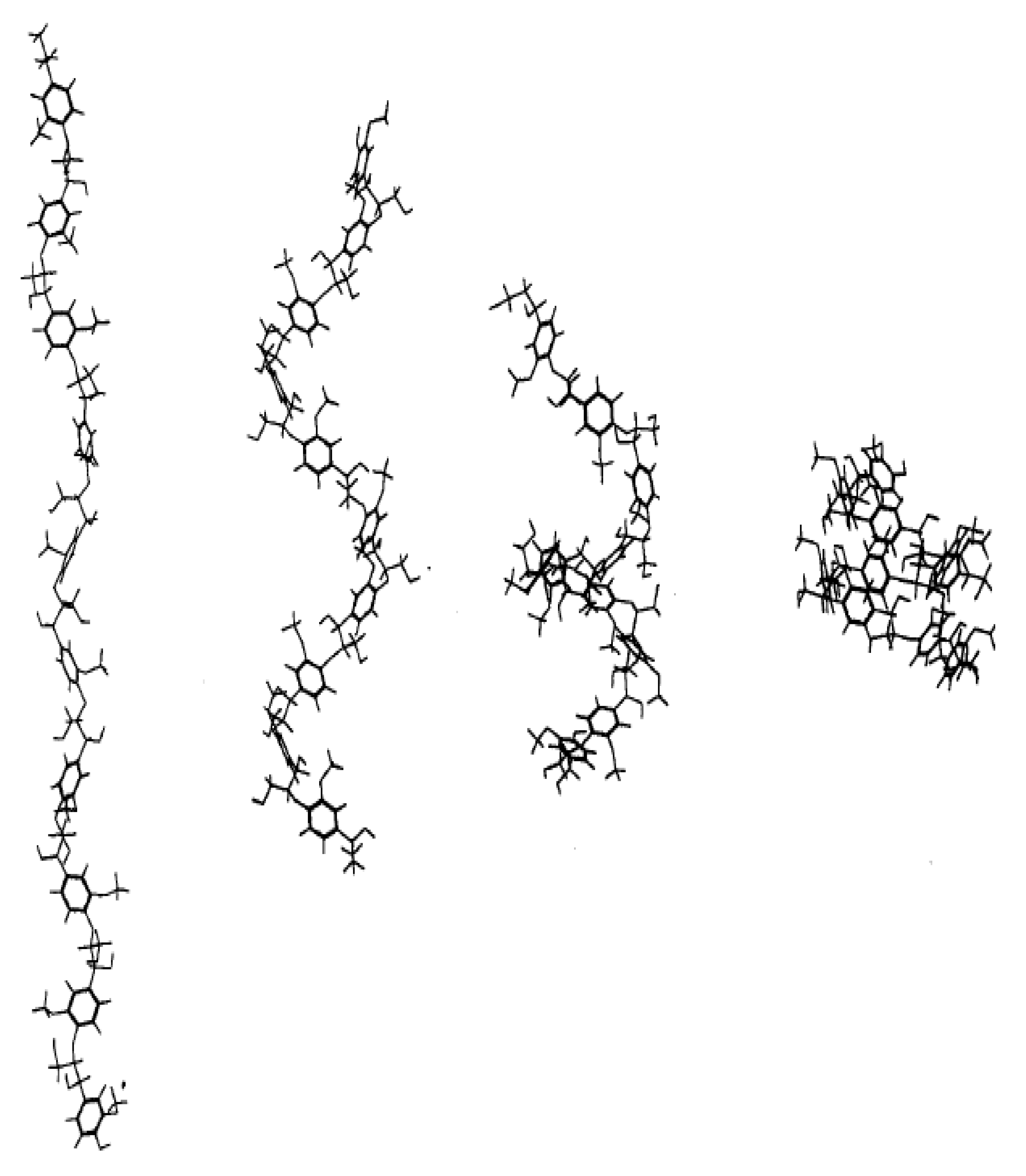
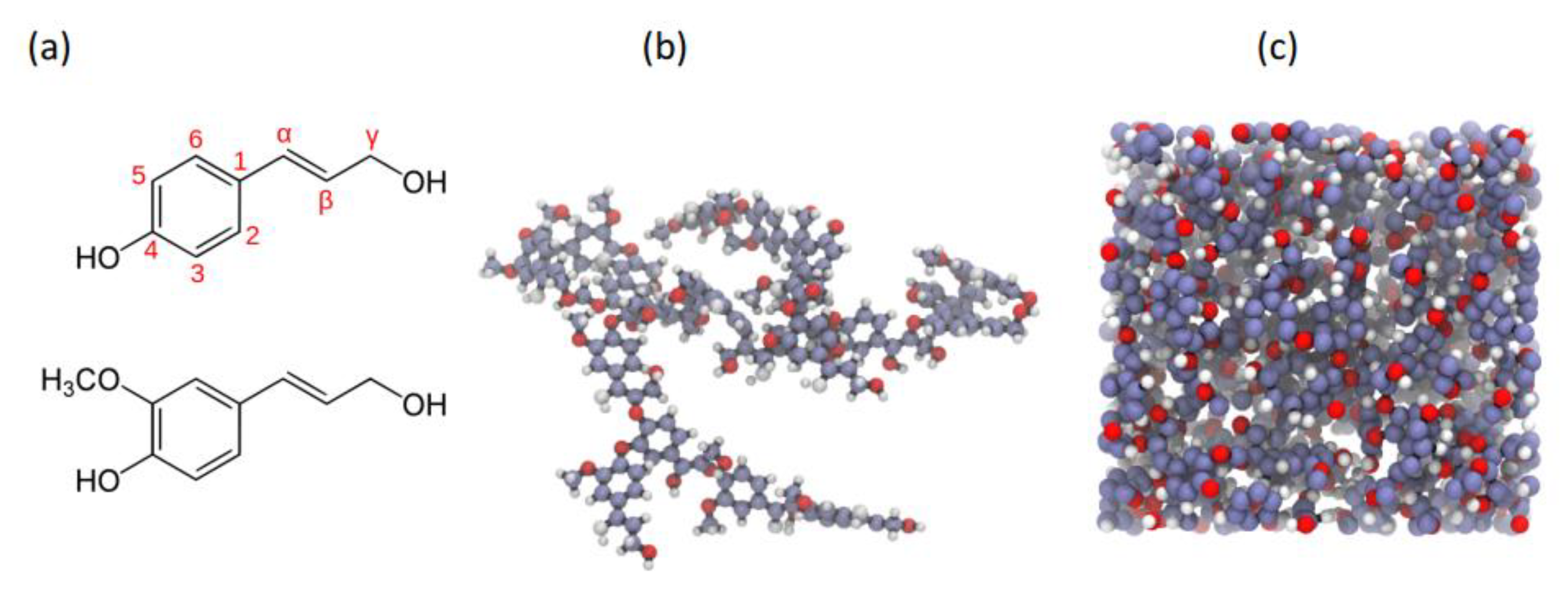
The higher enhancement of molecular mobility upon hydration of surface groups relative to those in the core was also found for lignin, another significant component of the lignocellulosic phase that comprises plant cell walls by Petridis et al. [
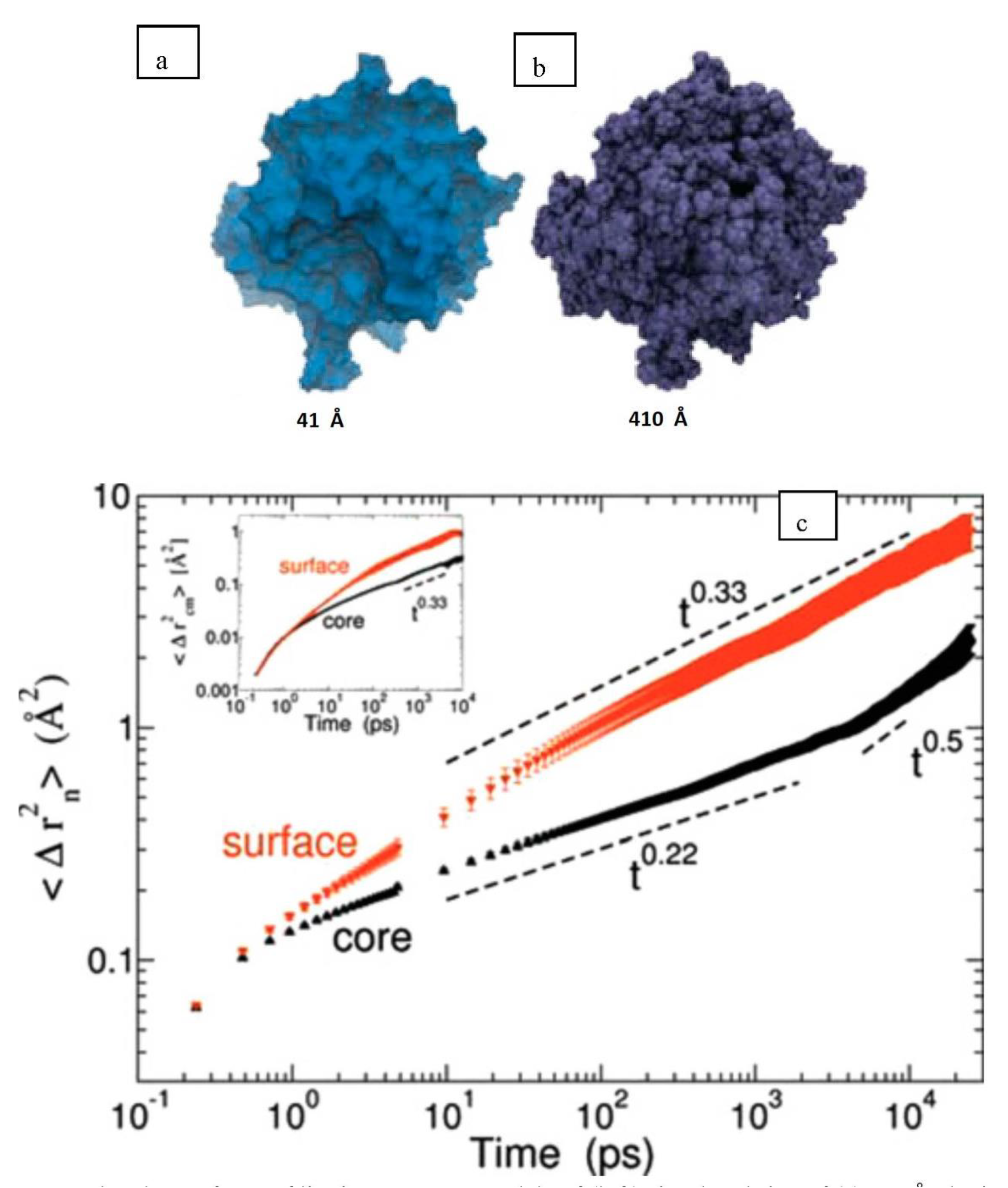
34]. Simulated version of lignin aggregates, each with 25 13-kDa lignin polymers, were constructed and were simulated in similar hydrated conditions to [
31] also using a parameterised CHARMM force field for lignin and the TIP3P water model. Extracted softwood (pine) lignin was first analysed using small-angle neutron scattering and an average aggregate size of up to 1300 Å was seen while the MD constructed models had a diameter of around 41 Å. To use MD to simulate true to life lignin models, Petridis et al. [
34] suggested that at a scale of 1–1000 Å there is a scale invariance in both structure and behaviour of lignocellulosics. Therefore, a 410 Å (
Figure 11b) aggregate was built using the 41 Å MD structure (
Figure 11a). The MSD behaviour of the lignin molecules is then shown in
Figure 11c and similar to semi crystalline polymers, seems to exist in 3 regimes viz., the ballistic regime (t ~ 0.1 ps), the confined/caged regime (0.1 ps < t < 5 ns) and the sub-diffusive regime (t > 5 ns). The second insight is that there exists a clear difference in MSD behaviour between the surface and core lignin molecules. Lignin monomers at the surface of the aggregate displayed considerably faster dynamics beyond the ballistic region (a four-fold increase of their MSD at t = 10 ns compared to the core monomers). This increase is also much more pronounced for the lignin as compared to the core and surface cellulose moieties as analysed in [
31]. The higher crystallinities of the cellulose molecules as compared to the lignin molecules could be the reason for this, as crystalline polymers do show much lower MSDs than amorphous polymers at the same temperature. This was also established for diffusion of oxygen in crystalline and amorphous polyethylenes by Prasad et al. [
32].
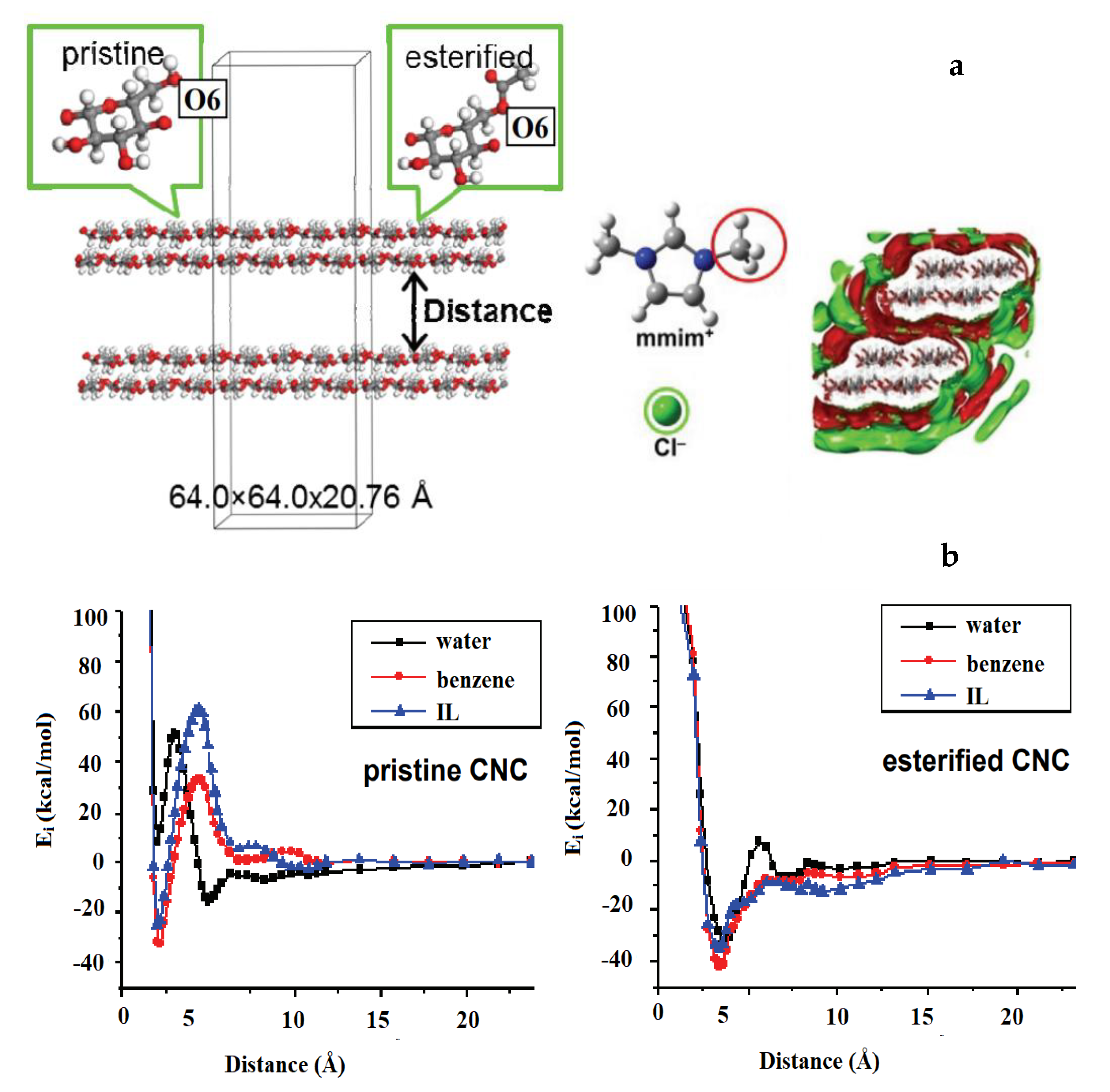
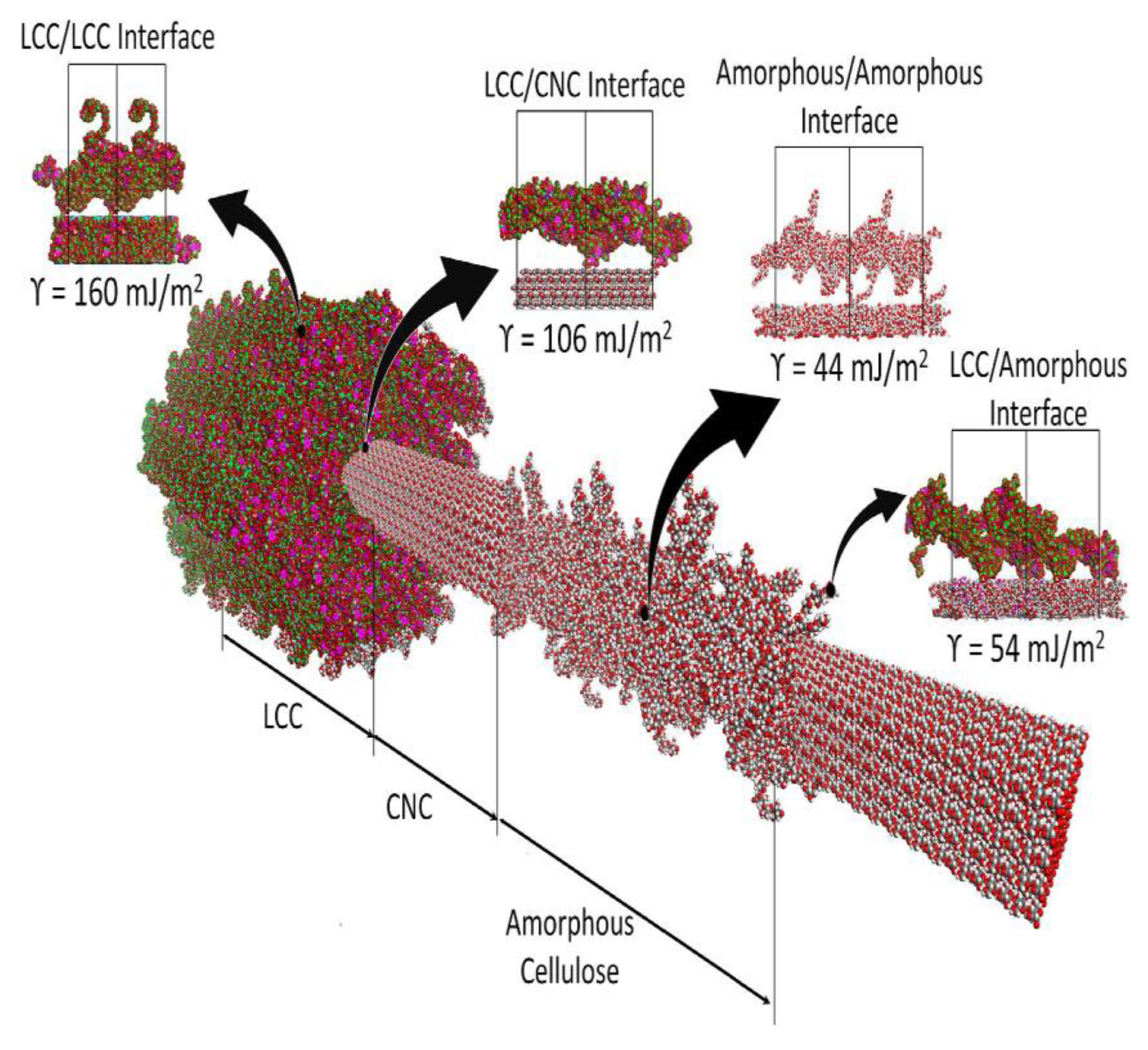
Behaviours of lignocellulosic materials when solvated or interacting with solvent molecules in addition to water were studied by Kovalenko [
35]. Three-layer Iα nanocrystalline cellulose fibrils (esterified and non-esterified) of ~9 nm in length with a DP of 34 were constructed using MD. Solvation with water, benzene and ionic liquid (IL) molecules (
Figure 12a) in neutral and ionic environments were studied using the 3-dimensional reference interaction site model with the Kovalenko–Hirata closure approximation (3d-RISM-KH). This method develops analytical summation of the free energy diagrams of the interaction between the macromolecule and the solvent beginning with molecular force field used. Thus, the 3D-RISM-KH integral equations for 3D site correlation functions can be obtained. Converging these site correlation functions, in turn, gives the solvation structure in terms of 3D distribution maps of solvent interaction sites around a solute macromolecule (supramolecule) of arbitrary shape. This also provides information about the solvation thermodynamics. Similar to the scale invariance of lignin structure over a limited length range. the 3d-RISM-KH method also bridges the gap between molecular structure and effective forces. However, the effectiveness of the 3d-RISM-KH is higher on multiple length scales. Specifically, in this work [
35], the effective interaction (potential of mean force) plots for pristine and esterified CNC in different solvents. Similar to Iα cellulose, the Iβ cellulose is stabilized by the hydration shell. The pristine CNC disaggregation barrier increases in the order of solvents: water < benzene < IL, which indicates that dispersion is favourable in water but is not favourable in benzene and IL. This is typical for hydrophilic surfaces. For esterified CNC, the potential of mean force in benzene and IL has a lower disaggregation barrier than in water, leading to facilitated dispersion in hydrophobic solvents. The lack of significant solvent expulsion barriers in benzene and IL suggests weak solvent-solute interactions. This example demonstrates how surface modifications can be employed to tune dispersion properties of CNC in various solvents [
36]. In addition to addressing the type of the surface modification, molecular simulation combined with the 3D-RISM-KH methodology allowed for the prediction of the degree of surface modification necessary to achieve a certain dispersion effect (
Figure 12b).
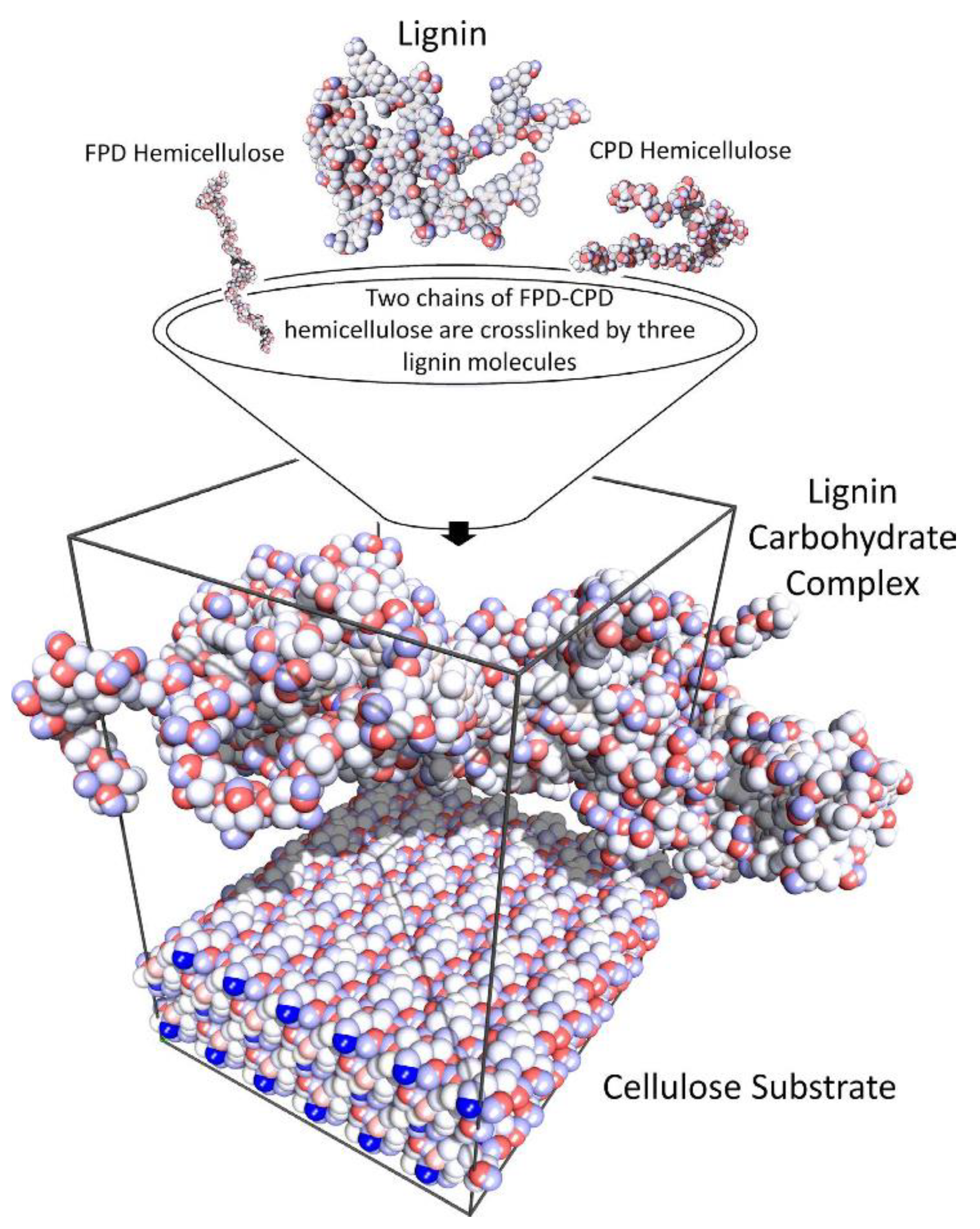
The accurate recreation of molecular mechanics is critical for the estimation of the physical properties of any simulated molecule. To this end a molecular mechanics force field was created for lignin by Petridis and Smith [
23] by using the CHARMM empirical force [
9] upon suitably parameterizing it. The lignocellulosic model of cellulose surrounded by lignin molecules in solution using the parameterised model in [
23] built by Petridis et al. [
27] is shown in
Figure 13. This model was then used to probe the interactions between lignin and cellulose at the atomic level, as well as provide a way to parameterize coarse-grained mesoscale models. The constructed lignin molecules contain equal numbers of left- and right-hand linkages. The molecular weight of lignins was of the order of 10,000 or greater, and the models had a molecular weight of 13,000. Crosslinks were formed when one unit participates in more than one linkage. In total, 26 lignins were built with varying degrees of crosslinking, but the average crosslink density was chosen to be consistent with the experimental value of 0.052 obtained from spruce wood (
Figure 13).
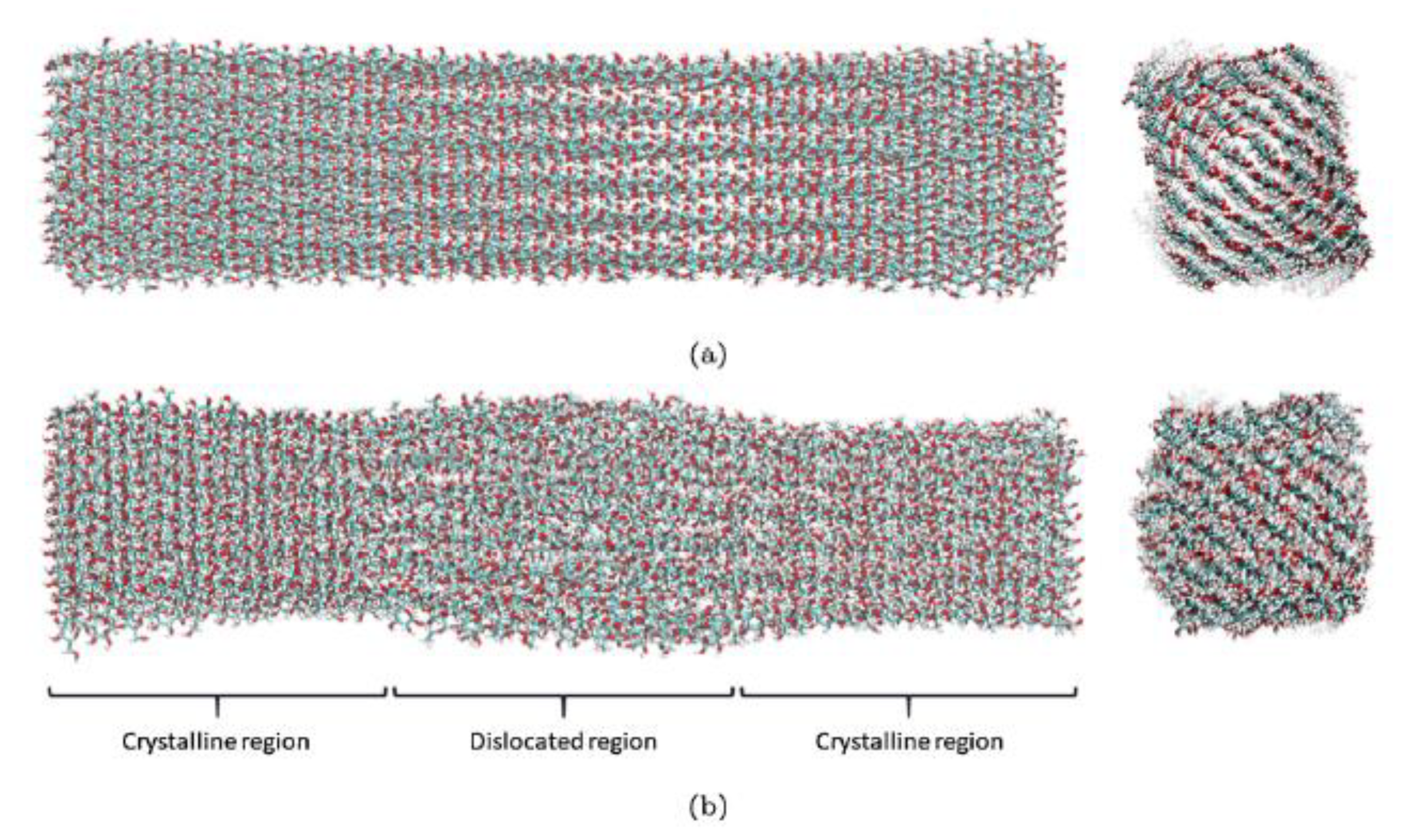
A similar parameterized CHARMM force field was applied for a multimillion atom system for lignocellulosics by Schulz et al. [
37]. The main difference between the generic CHARMM force field [
9] in this work and the parameterised CHARMM force field in [
37] is in the expression for electrostatic potential. The general CHARMM field uses the Coulombic expression shown in Equation (20) where ε and ε
0 are the dielectric constants of the system and vacuum, q
i and q
j are the charges on atoms i and j. In [
29] electrostatic potential was determined using the reaction field potential (V
rf) shown in Equation (19) where ε is the dielectric constant outside the radius rc, and r is the distance separating two charges. The reaction field potential was developed by Onsager [
38] and used within the context of molecular simulation (specifically, Monte Carlo simulations by Barker and Watts [
39])
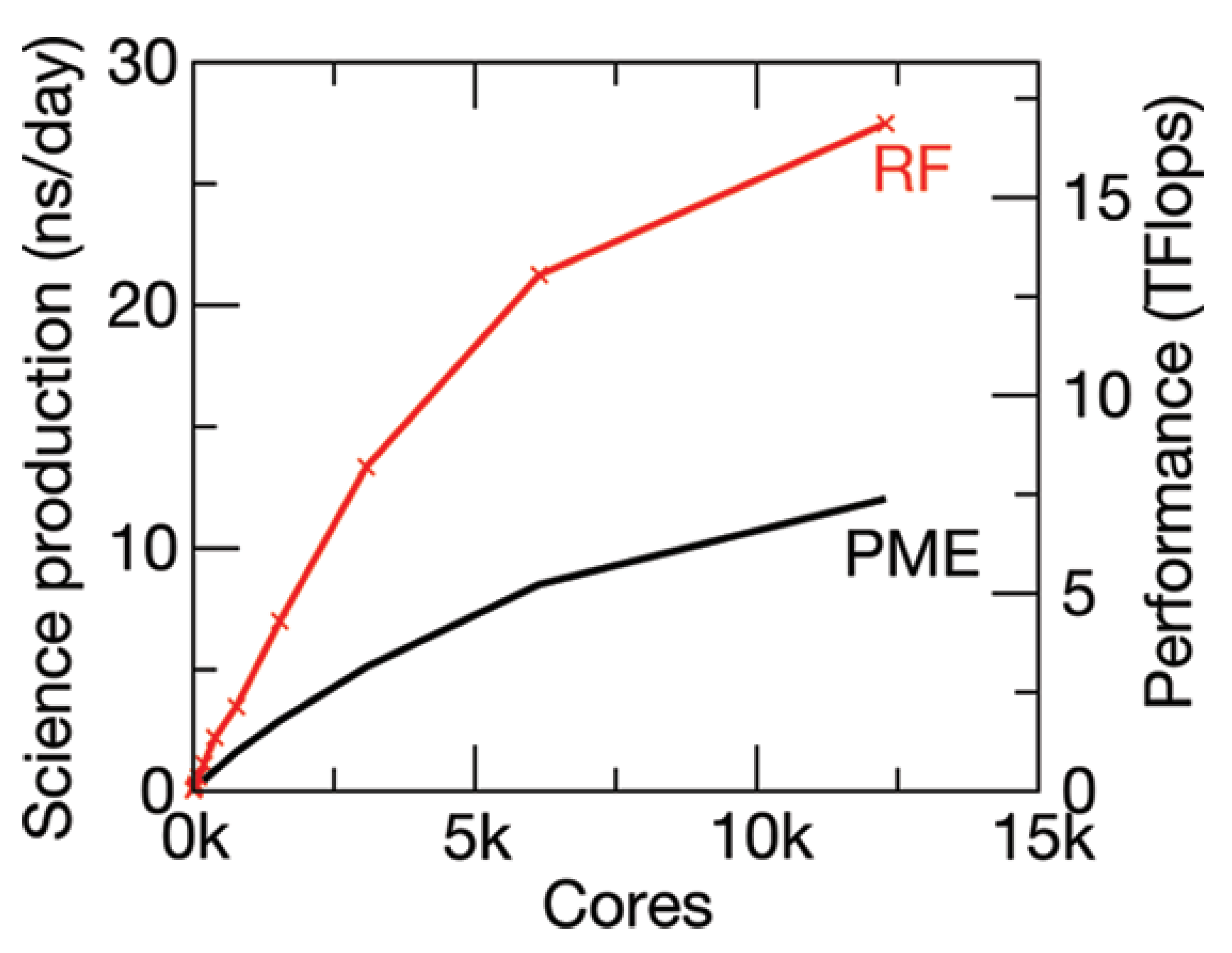
Using Equation (19), Schulz et al. [
37] were able to demonstrate the scalability and efficiency of their reaction field method over the conventional particle mesh Ewald summation method. Up to three million- and five million atom simulated systems were studied and ∼30 ns/day was achieved. This represents a massive improvement on existing MD methodologies. The scalability differences between the PME and the RF method [
38] can be clearly seen in
Figure 14.
The proof of concept established by Schulz et al. in [
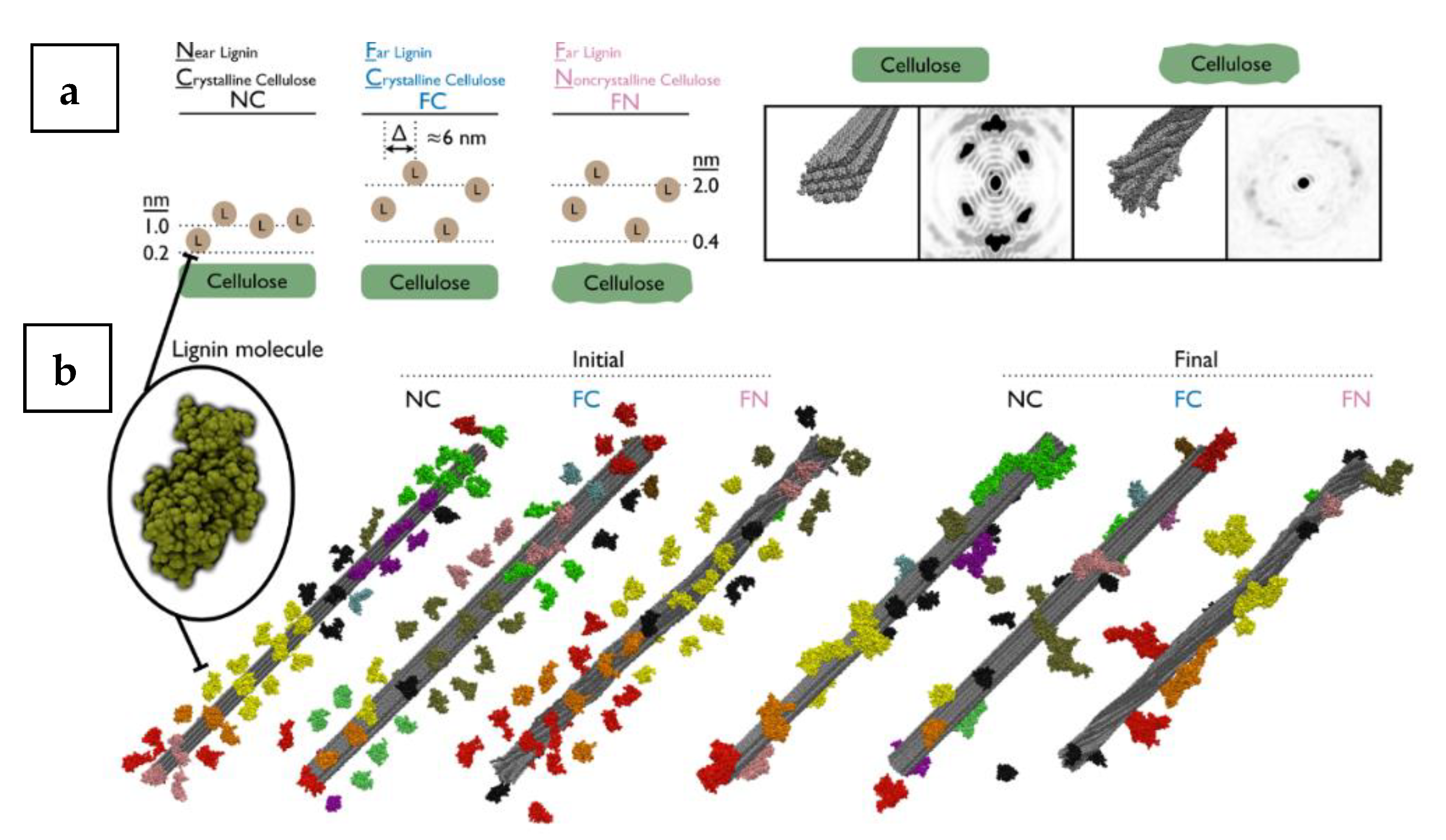
37] showed the viability of simulation of multi-million atom lignocellulosic systems by using the reaction field method. Building on that Lindner et al. [
40], built 3.31, 3.43, and 3.80 million atom lignocellulosic systems and ran the simulations on the Jaguar XT5 Petaflop Supercomputer using 40,000 cores at a peak performance of 27 ns/day similar to the work of Schulz et al. [
37]. The work of Lindner et al. [
40] is one of the largest-ever atomistic biomolecular MD simulations conducted to date. The lignin molecule modelled consists of 61 monomers, corresponding to a molecular weight of 13 kDa while the cellulose fibre model consisted of 36 chains with a chain length of 160 monomers (
Figure 15). The lignins were simulated with crystalline (FC, FN) and non-crystalline cellulose (NC) models. For each model, 52 lignin molecules were placed around the central cellulose fibre (at distances randomly chosen within the interval of 0.2 to 1.0 nm and 0.4 to 2.0 nm) for the NC model, the FC (crystalline cellulose with initial lignin position far from the central fibre) and FN (crystalline cellulose with initial lignin position far from the central fibre) models, respectively. The simulations were performed with the GROMACS [
11] suite using the TIP3P water model and the CHARMM Carbohydrate Solution Force Field (CSFF) for cellulose and the CHARMM Lignin Force Field similar to previous works on lignocellulosics [
21,
34,
37]. The MD simulation was able to produce simulated directional X-ray scattering peaks diagram along the fibre axis. This is shown for both the crystalline and the non-crystalline cellulose, (
Figure 15a). It was found that the models with the crystalline cellulose exhibited strong Bragg peaks, which were absent in the non-crystalline model.
The results of Lindner et al. [
40] shone a light on the intrinsic processes that determine the nature of lignin precipitation onto cellulose and the dependence on cellulose crystallinity thereof. Their results showed that lignin has a strong tendency toward aggregation with lignin and cellulose. The simulations also revealed that the average direct interaction energy between lignin and cellulose is independent of cellulose crystallinity. However, lignin aggregation onto non-crystalline cellulose was found to be less favourable than for the crystalline form, owing to the significantly different solvation properties of crystalline and amorphous cellulose.
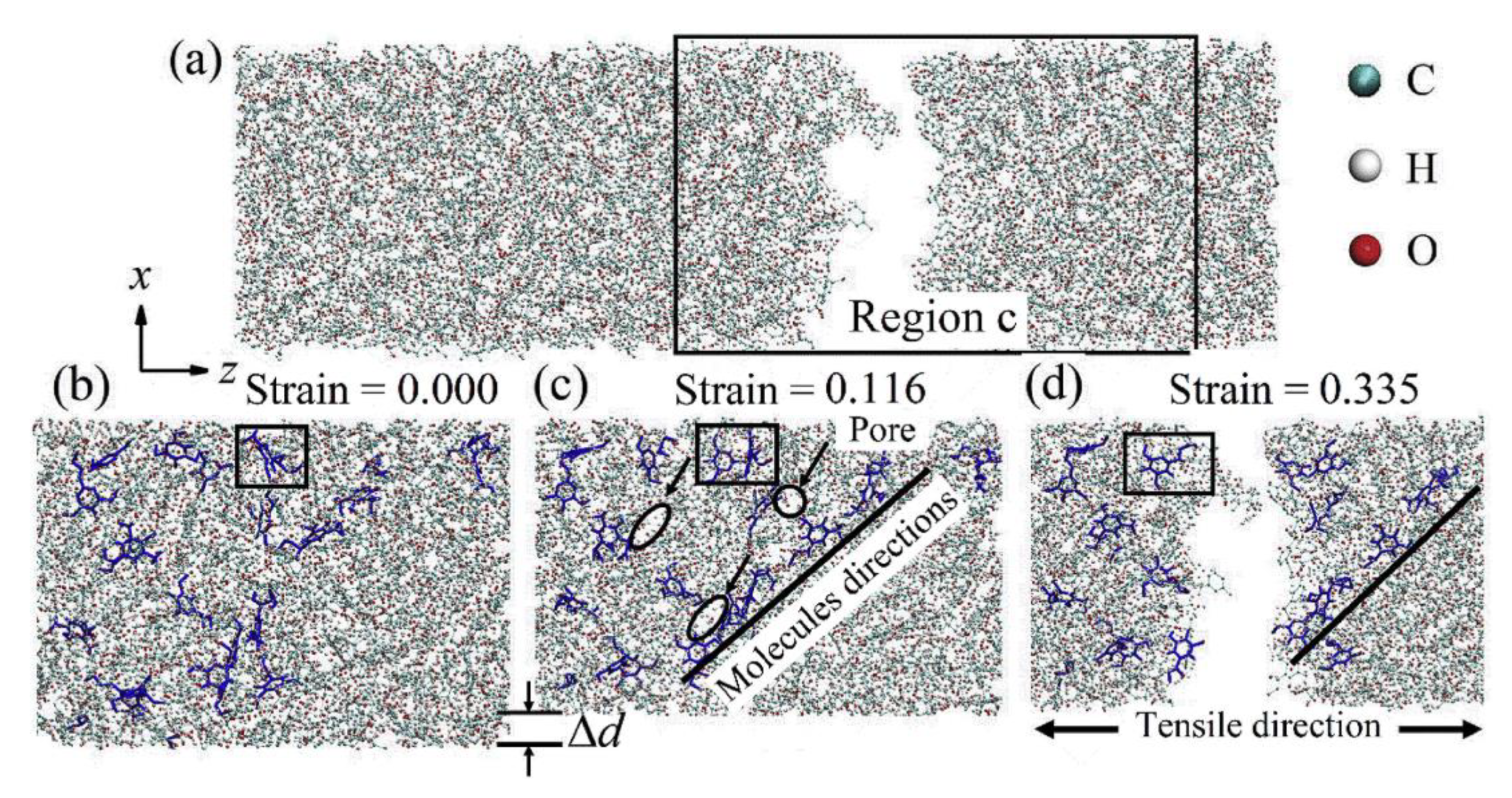
Biomass is also converted into functional products using enzymatic methods, specifically hydrolysis. This process is also inhibited by the presence of lignin similar to [
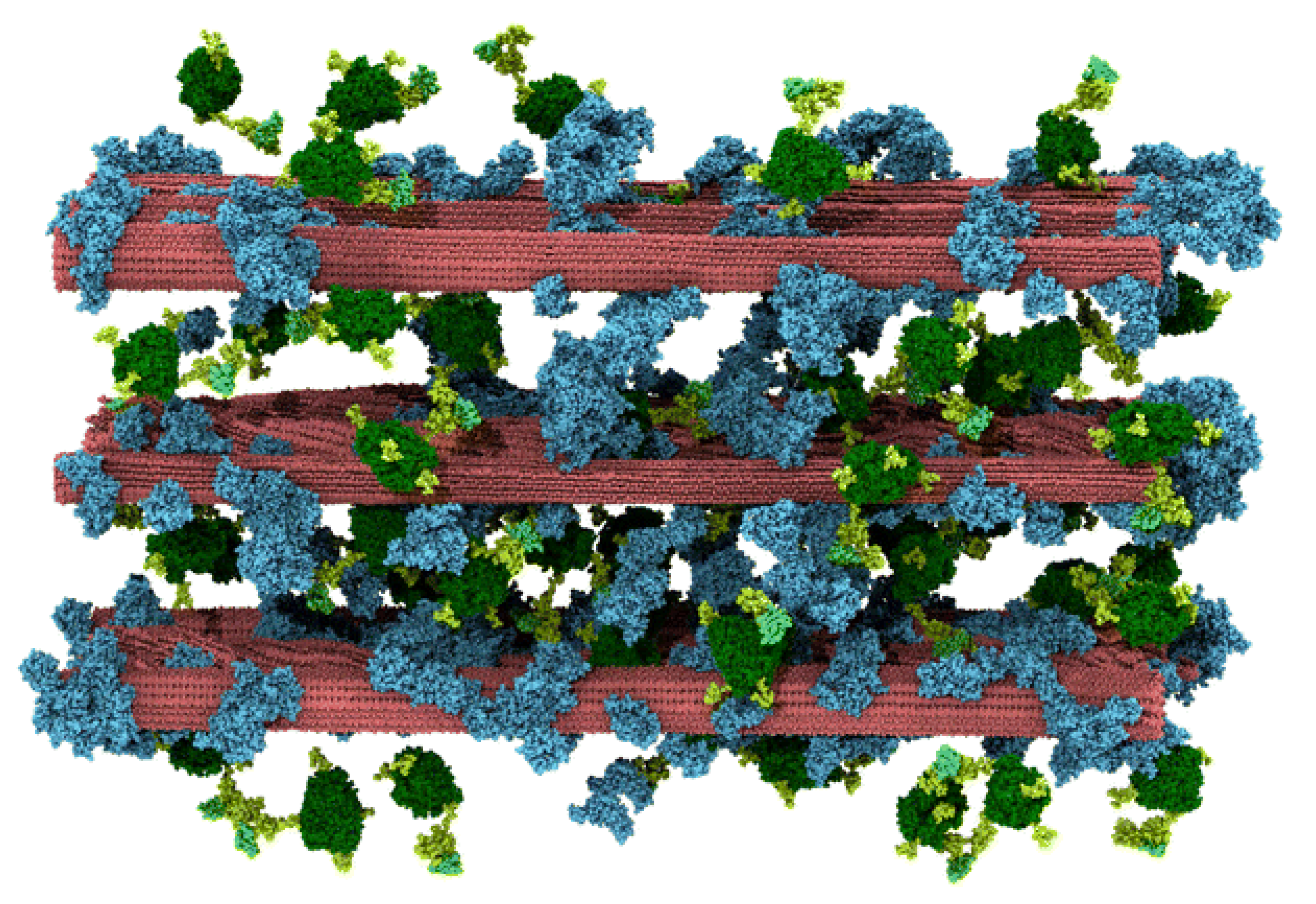
40]. To this end, Vermass et al. [
41] used MD for elucidating the atomistic/molecular aspects of lignin inhibition of enzymatic hydrolysis. The RFZ equation was used for electrostatic interactions similar to Schulz et al. [
37]. The constructed model shown in
Figure 16 represents the largest biomass MD simulation at 23.7-million atom. The total simulation time was 1312 ns. The simulations were carried out on the TITAN XC6 Supercomputer at Oak Ridge National Laboratory, using 60,000 cores at a peak performance of 45 ns/day higher than the 27 ns/day achieved in [
37] and [
40]. The multi-component simulation model was built to represent a pre-treated biomass system of cellulose and lignin at room temperature upon the addition of cellulolytic enzyme and is shown in
Figure 16. The model consists of cellulose fibres, lignin molecules, and Cel7A cellulases. Other components of biomass, such as pectins and hemicellulose, were assumed to have been removed by dilute acid pre-treatment.
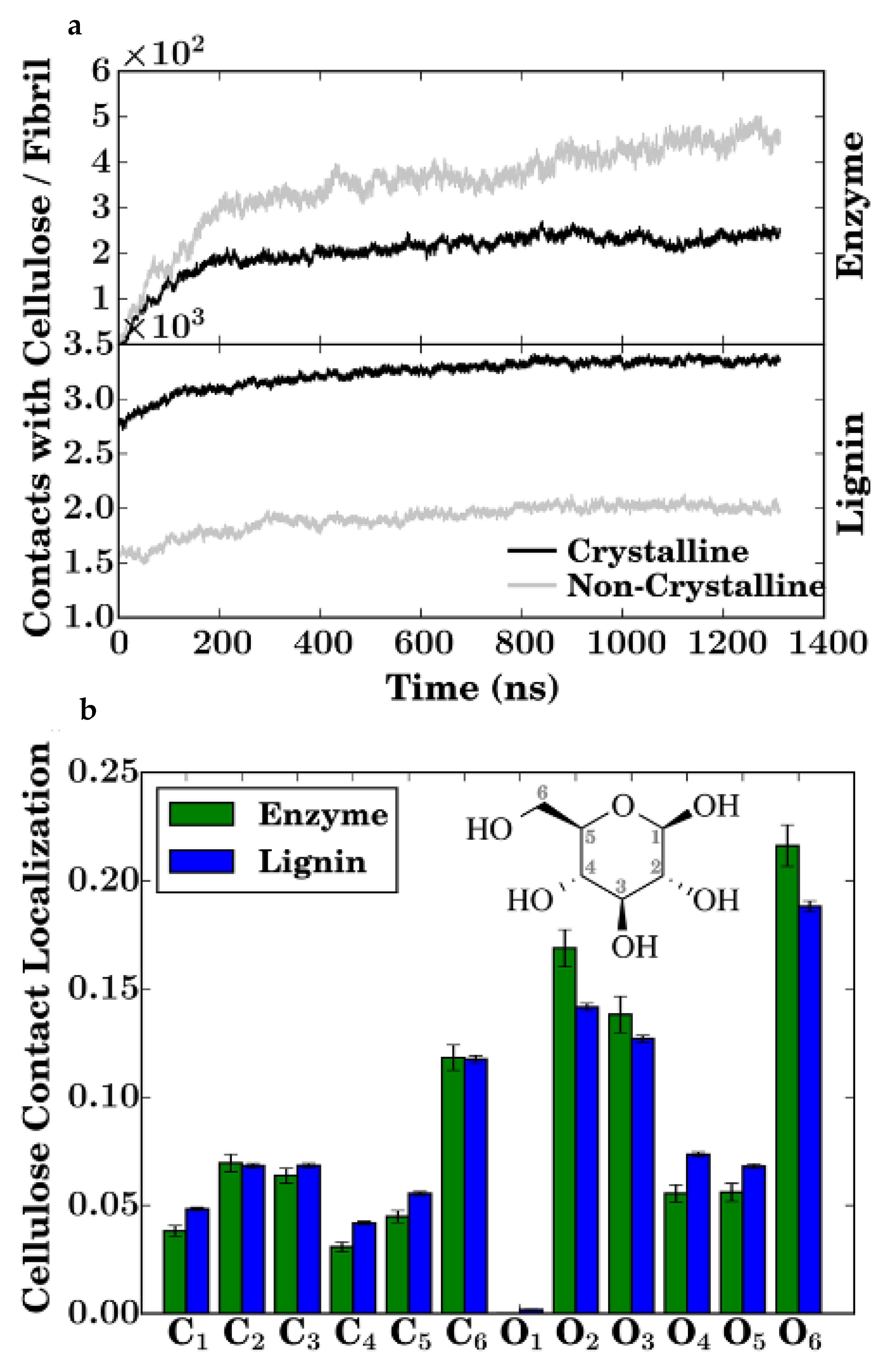
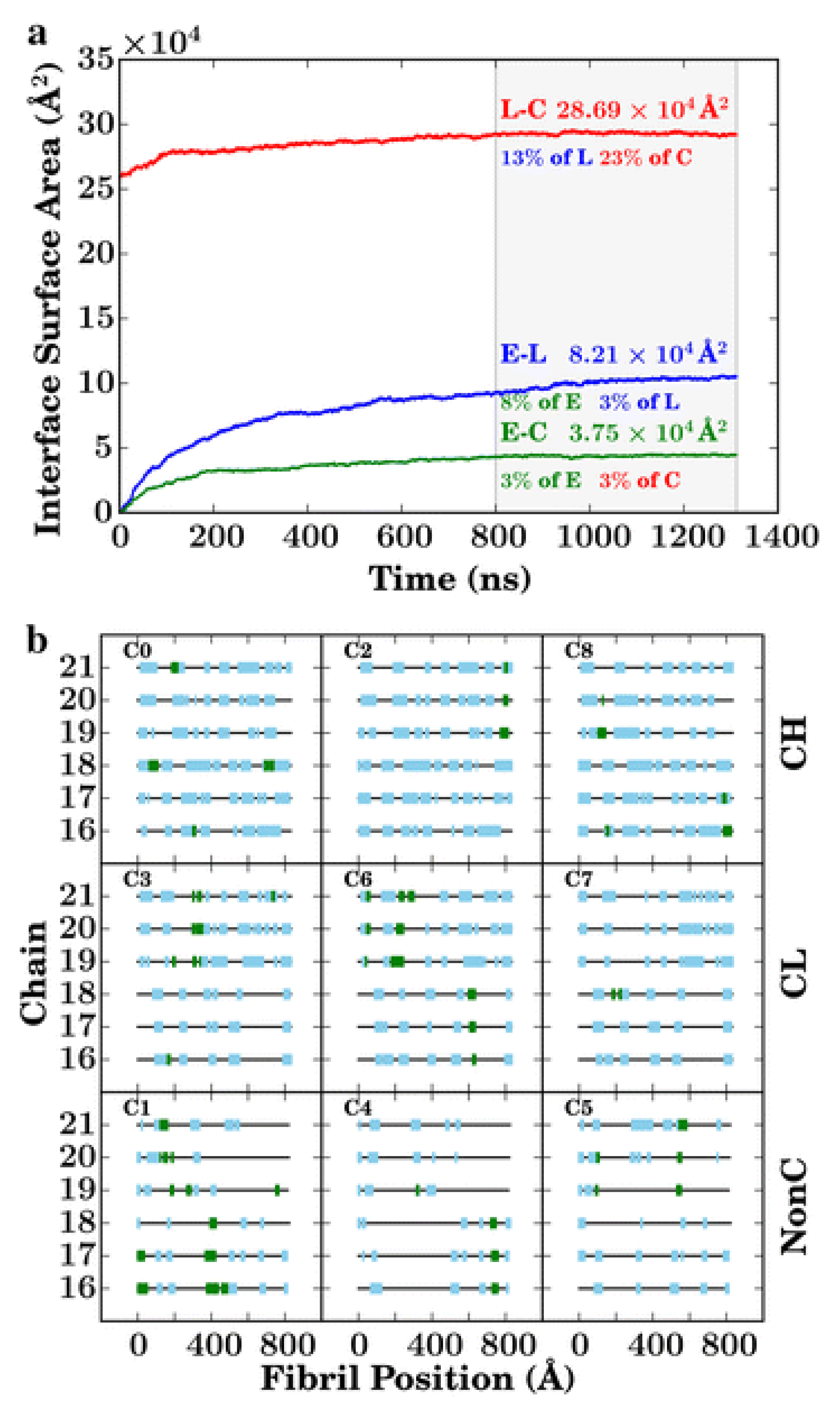
In the initial parts of the simulation, the lignin-lignin and lignin-cellulose interactions did not change significantly. As the simulation progresses a gradual increase in the number of enzymatic contacts to the lignin and cellulose phases were observed as the enzymes diffused to the lignocellulose. It was observed that the lignin mediates the formation of a fully interconnected network of cellulose, lignin, and enzyme, with each molecule linked to all others directly or indirectly. The enzymatic diffusion was also mediated by the lignin. The binding of the enzyme to cellulose or lignin led to a three orders magnitude reduction in the translational diffusion coefficient of the enzyme (∼10
−6 cm
2/s initially to ∼10
−9 cm
2/s at the end of the simulation). After equilibration, the surfaces of the cellulose fibrils in the simulated model were analysed. Similar to the process of cellulose precipitation investigated by Linder et al. [
40], the enzymatic destruction of lignocellulosic biomass is also inherently dependent on the binding of the enzyme (in [
41], being TrCel7A cellulase) to the cellulose. There exists a close interaction between lignin and the enzyme. This, in addition with the mediation of the enzyme diffusion by the lignin results in the lignin outcompeting the cellulose in terms of binding to the cellulose-binding module (CBM) of the enzyme (
Figure 17a,b). From the simulation it could be observed that the flat hydrophobic surface on the CBM (formed by three tyrosine residues) which should promote binding to the hydrophobic surfaces of the cellulose fibres instead forms extensive contacts with the lignin. The cellulose surface, also, is crowded by the lignin with nearly 25% of the total cellulose surface area being consistently covered by lignin, significantly reducing the area accessible to the enzymes (
Figure 18a,b). Therefore, the presence of lignin molecules on the cellulose surface interferes with the mechanism of cellulose hydrolysis. The lignin molecules adhere to the surface of the cellulose fibres, slow the diffusion of the enzymes and out-compete the cellulose for contact with the enzyme CDM and thus, reduce the distance an enzyme bound to cellulose can travel before its path is blocked by a lignin molecule. Large scale simulations such as this one carried out by Vermass et al. [
41] and Lindner et al. [
40] provide a visual and graphical view of the fine molecular interactions that, in turn, affect industrial process such as enzymatic hydrolysis.
Therefore, over the past quarter of a century, the simulation and modelling of lignocellulosic systems has come a long way. Lignocellulosic systems ranging from a few hundred to several million molecules have been constructed, simulated, validated through molecular characterization. Fine structural characteristics of these systems that control the physical and biochemical properties of the lignocellulosic systems in the presence of water and other solvents, ions and enzymes have been unearthed through a combination of MD with mathematical and visual methods. The next step is to use these validated simulations to predict mechanical behaviour of the lignocellulosic phases namely, lignin and cellulose and the lignocellulosic composites which is covered in the next section.


















































{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}