
Composite Ceramics Based on Pastes Including Tricalcium Phosphate and an Aqueous Solution of Sodium Silicate

 ,
,  , , ,
, , ,
Abstract
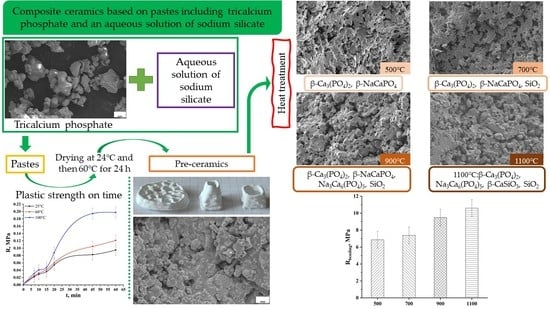
1. Introduction
- The paste should be homogeneous and free of air bubbles;
- It should possess a large volume fraction of the filler;
- It should possess sedimentation resistance;
- It should possess a fluidity suitable for extrusion;
- During extrusion, delamination should not be observed;
- It should maintain its shape after printing.
2. Materials and Methods
2.1. Samples Preparation
2.2. Characterization
2.2.1. Pastes Plastic Strength Determination
2.2.2. Determination of the Strength Properties
2.2.3. Structural Characterization
2.2.4. Thermogravimetric and Mass Spectrometric Analysis
3. Results and Discussion
3.1. Pastes Characterization
3.2. Characterization of the Preceramic Samples
3.3. Characterization of the Ceramic Samples
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bellucci, D.; Sola, A.; Cannillo, V. Hydroxyapatite and Tricalcium Phosphate Composites with Bioactive Glass as Second Phase: State of the Art and Current Applications. J. Biomed. Mater. Res. A 2016, 104, 1030–1056. [Google Scholar] [CrossRef] [PubMed]
- Arango-Ospina, M.; Boccaccini, A.R. Bioactive Glasses and Ceramics for Tissue Engineering. In Tissue Engineering Using Ceramics and Polymers; Woodhead Publishing: Sawston, UK, 2022; pp. 111–178. [Google Scholar] [CrossRef]
- Reza Rezaie, H.; Beigi Rizi, H.; Rezaei Khamseh, M.M.; Öchsner, A. A Review on Dental Materials. In Advanced Structured Materials; Springer International Publishing: Cham, Switzerland, 2020; Volume 123, ISBN 978-3-030-48930-4. [Google Scholar]
- Sventskaya, N.V.; Lukina, Y.S.; Larionov, D.S.; Andreev, D.V.; Sivkov, S.P. 3D-Matrix Based on Bioactive Glass and Calcium Phosphates with Controllable Resorption Rate for Bone Tissue Replacement. Glass Ceram. 2017, 73, 342–347. [Google Scholar] [CrossRef]
- Safronova, T.V.; Putlyaev, V.I.; Shekhirev, M.A.; Kuznetsov, A.V. Composite Ceramic Containing a Bioresorbable Phase. Glass Ceram. 2007, 64, 102–106. [Google Scholar] [CrossRef]
- Balamurugan, A.; El Mabrouk, K.; Pina, S.; Bousmina, M.M.; Ferreira, J.M.F. Melt-Derived Condensed Polymorphic Calcium Phosphate as Bone Substitute Material: An In Vitro Study. J. Am. Ceram. Soc. 2011, 94, 3023–3029. [Google Scholar] [CrossRef]
- Jalota, S.; Bhaduri, S.B.; Tas, A.C. A New Rhenanite (β-NaCaPO4) and Hydroxyapatite Biphasic Biomaterial for Skeletal Repair. J. Biomed. Mater. Res. B Appl. Biomater. 2007, 80B, 304–316. [Google Scholar] [CrossRef] [PubMed]
- Bose, S.; Ke, D.; Sahasrabudhe, H.; Bandyopadhyay, A. Additive Manufacturing of Biomaterials. Prog. Mater. Sci. 2018, 93, 45–111. [Google Scholar] [CrossRef] [PubMed]
- Safronova, T.V.; Putlyaev, V.I.; Filippov, Y.Y.; Shatalova, T.B.; Naberezhnyi, D.O.; Nasriddinov, A.F.; Larionov, D.S. Ceramics Based on Powder Mixtures Containing Calcium Hydrogen Phosphates and Sodium Salts (Na2CO3, Na4P2O7, and NaPO3). Inorg. Mater. 2018, 54, 724–735. [Google Scholar] [CrossRef]
- Safronova, T.V.; Putlyayev, V.I.; Knotko, A.V.; Filippov, Y.Y.; Klimashina, E.S.; Ryzhov, A.P.; Saidzhonov, B.M. Powder Mixtures Based on Calcium Hydroxyapatite and Sodium Salts. Inorg. Mater. Appl. Res. 2018, 9, 726–731. [Google Scholar] [CrossRef]
- Safronova, T.V. Inorganic Materials for Regenerative Medicine. Inorg. Mater. 2021, 57, 443–474. [Google Scholar] [CrossRef]
- Demirkiran, H.; Mohandas, A.; Dohi, M.; Fuentes, A.; Nguyen, K.; Aswath, P. Bioactivity and Mineralization of Hydroxyapatite with Bioglass as Sintering Aid and Bioceramics with Na3Ca6(PO4)5 and Ca5(PO4)2SiO4 in a Silicate Matrix. Mater. Sci. Eng. C 2010, 30, 263–272. [Google Scholar] [CrossRef]
- Afonko, A.A.; Kirillova, S.A.; Almjashev, V.I. Ceramic and Composite Nanomaterials Based on Calcium Orthophosphates. Nanosyst. Phys. Chem. Math. 2012, 3, 84–102. [Google Scholar]
- Bergmann, C.; Lindner, M.; Zhang, W.; Koczur, K.; Kirsten, A.; Telle, R.; Fischer, H. 3D Printing of Bone Substitute Implants Using Calcium Phosphate and Bioactive Glasses. J. Eur. Ceram. Soc. 2010, 30, 2563–2567. [Google Scholar] [CrossRef]
- Su, J.; Hua, S.; Chen, A.; Chen, P.; Yang, L.; Yuan, X.; Qi, D.; Zhu, H.; Yan, C.; Xiao, J.; et al. Three-Dimensional Printing of Gyroid-Structured Composite Bioceramic Scaffolds with Tuneable Degradability. Biomater. Adv. 2022, 133, 112595. [Google Scholar] [CrossRef]
- Cozza, N.; Monte, F.; Bonani, W.; Aswath, P.; Motta, A.; Migliaresi, C. Bioactivity and Mineralization of Natural Hydroxyapatite from Cuttlefish Bone and Bioglass® Co-Sintered Bioceramics. J. Tissue. Eng. Regen. Med. 2018, 12, e1131–e1142. [Google Scholar] [CrossRef]
- Ando, J. Phase Diagrams of Ca3(PO4)2-Mg3(PO4)2 and Ca3(PO4)2-CaNaPO4 Systems. Bull. Chem. Soc. Jpn. 1958, 31, 201–205. [Google Scholar] [CrossRef]
- Han, Y.C.; Wang, X.Y.; Li, S.P. Change of Phase Composition and Morphology of Sonochemically Synthesised Hydroxyapatite Nanoparticles with Glycosaminoglycans during Thermal Treatment. Adv. Appl. Ceram. 2009, 108, 400–405. [Google Scholar] [CrossRef]
- Doi, Y.; Shimizu, Y.; Moriwaki, Y.; Aga, M.; Iwanaga, H.; Shibutani, T.; Yamamoto, K.; Iwayama, Y. Development of a New Calcium Phosphate Cement That Contains Sodium Calcium Phosphate. Biomaterials 2001, 22, 847–854. [Google Scholar] [CrossRef]
- Cousin, O.; Rivenet, M.; Boivin, J.C.; Abraham, F. A Refractory Bonding Phase Fundamental Study In The Na2O-CaO-P2O5-SiO2 System. Phosphorus Res. Bull. 1999, 10, 183–188. [Google Scholar] [CrossRef][Green Version]
- El-Ghannam, A.R. Advanced Bioceramic Composite for Bone Tissue Engineering: Design Principles and Structure–Bioactivity Relationship. J. Biomed. Mater. Res. A 2004, 69A, 490–501. [Google Scholar] [CrossRef]
- Gupta, G.; El-Ghannam, A.; Kirakodu, S.; Khraisheh, M.; Zbib, H. Enhancement of Osteoblast Gene Expression by Mechanically Compatible Porous Si-Rich Nanocomposite. J. Biomed. Mater. Res. B Appl. Biomater. 2007, 81B, 387–396. [Google Scholar] [CrossRef]
- Jones, J.R. Review of Bioactive Glass: From Hench to Hybrids. Acta Biomater. 2013, 9, 4457–4486. [Google Scholar] [CrossRef] [PubMed]
- No, Y.; Li, J.; Zreiqat, H. Doped Calcium Silicate Ceramics: A New Class of Candidates for Synthetic Bone Substitutes. Materials 2017, 10, 153. [Google Scholar] [CrossRef]
- Zuev, D.M.; Klimashina, E.S.; Evdokimov, P.v.; Filippov, Y.Y.; Putlyaev, V.I. Preparation of β-Ca3(PO4)2/Poly(D,L-Lactide) and β-Ca3(PO4)2/Poly(ε-Caprolactone) Biocomposite Implants for Bone Substitution. Inorg. Mater. 2018, 54, 87–95. [Google Scholar] [CrossRef]
- Roohani-Esfahani, S.I.; Newman, P.; Zreiqat, H. Design and Fabrication of 3D Printed Scaffolds with a Mechanical Strength Comparable to Cortical Bone to Repair Large Bone Defects. Sci. Rep. 2016, 6, srep19468. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Lee, Y.; Yang, S.; Hao, Y.; Evans, J.R.G.; Parini, C.G. Solvent-Based Paste Extrusion Solid Freeforming. J. Eur. Ceram. Soc. 2010, 30, 1–10. [Google Scholar] [CrossRef]
- Fu, Q.; Saiz, E.; Tomsia, A.P. Direct Ink Writing of Highly Porous and Strong Glass Scaffolds for Load-Bearing Bone Defects Repair and Regeneration. Acta Biomater. 2011, 7, 3547–3554. [Google Scholar] [CrossRef]
- Al-Qutaifi, S.; Nazari, A.; Bagheri, A. Mechanical Properties of Layered Geopolymer Structures Applicable in Concrete 3D-Printing. Constr. Build. Mater. 2018, 176, 690–699. [Google Scholar] [CrossRef]
- Takadama, H.; Kim, H.M.; Kokubo, T.; Nakamura, T. Mechanism of Biomineralization of Apatite on a Sodium Silicate Glass: TEM−EDX Study In Vitro. Chem. Mater. 2001, 13, 1108–1113. [Google Scholar] [CrossRef]
- Golovanova, O.A.; Zaits, A.V. Biomimetic Coating of a Titanium Substrate with Silicon-Substituted Hydroxyapatite. Inorg. Mater. 2018, 54, 1124–1130. [Google Scholar] [CrossRef]
- Kaimonov, M.; Safronova, T.; Shatalova, T.; Filippov, Y.; Tikhomirova, I.; Sergeev, N. Composite Ceramics in the Na2O-CaO-SiO2-P2O5 System Obtained from Pastes Including Hydroxyapatite and an Aqueous Solution of Sodium Silicate. Ceramics 2022, 5, 550–561. [Google Scholar] [CrossRef]
- ICDD. PDF-4+ 2010 (Database); Kabekkodu, S., Ed.; International Centre for Diffraction Data: Newtown Square, PA, USA, 2010; Available online: https://www.icdd.com/pdf-2/ (accessed on 15 July 2022).
- Korneev, V.I.; Danilov, V.V. Soluble and Liquid Glass; Stroy-izdat: St. Petersburg, Russia, 1996. [Google Scholar]
- Greaves, G.N.; Sen, S. Inorganic Glasses, Glass-Forming Liquids and Amorphizing Solids. Adv. Phys. 2007, 56, 1–166. [Google Scholar] [CrossRef]
- Yashima, M.; Sakai, A.; Kamiyama, T.; Hoshikawa, A. Crystal Structure Analysis of β-Tricalcium Phosphate Ca3(PO4)2 by Neutron Powder Diffraction. J. Solid State Chem. 2003, 175, 272–277. [Google Scholar] [CrossRef]
- Morozov, V.A.; Belik, A.A.; Kotov, R.N.; Presnyakov, I.A.; Khasanov, S.S.; Lazoryak, B.I. Crystal Structures of Double Calcium and Alkali Metal Phosphates Ca10 M(PO4)7(M = Li, Na, K). Crystallogr. Rep. 2000, 45, 13–20. [Google Scholar] [CrossRef]
- Catauro, M.; Bollino, F.; Papale, F. Synthesis of SiO2 System via Sol–Gel Process: Biocompatibility Tests with a Fibroblast Strain and Release Kinetics. J. Biomed. Mater. Res. A 2014, 102, 1677–1680. [Google Scholar] [CrossRef]
- Catauro, M.; Bollino, F.; Papale, F.; Gallicchio, M.; Pacifico, S. Influence of the Polymer Amount on Bioactivity and Biocompatibility of SiO2/PEG Hybrid Materials Synthesized by Sol–Gel Technique. Mater. Sci. Eng. C 2015, 48, 548–555. [Google Scholar] [CrossRef]
- Vapirov, V.V.; Feoktistov, V.M.; Venskovich, A.A.; Vapirova, N.V. On Silicon’s Behavior and Its Biological Role In Nature. Sci. Notes Petrozavodsk. State Univ. 2017, 2, 95–102. [Google Scholar]
- Ahmad, H. Biocompatible SiO2 in the Fabrication of Stimuli-Responsive Hybrid Composites and Their Application Potential. J. Chem. 2015, 2015, 846328. [Google Scholar] [CrossRef]
- Zhang, N.; Molenda, J.A.; Fournelle, J.H.; Murphy, W.L.; Sahai, N. Effects of Pseudowollastonite (CaSiO3) Bioceramic on in Vitro Activity of Human Mesenchymal Stem Cells. Biomaterials 2010, 31, 7653–7665. [Google Scholar] [CrossRef]
- Sarmento, C.; Luklinska, Z.B.; Brown, L.; Anseau, M.; de Aza, P.N.; de Aza, S.; Hughes, F.J.; McKay, I.J. In Vitro Behavior of Osteoblastic Cells Cultured in the Presence of Pseudowollastonite Ceramic. J. Biomed. Mater. Res. A 2004, 69A, 351–358. [Google Scholar] [CrossRef]
- Sahai, N.; Anseau, M. Cyclic Silicate Active Site and Stereochemical Match for Apatite Nucleation on Pseudowollastonite Bioceramic–Bone Interfaces. Biomaterials 2005, 26, 5763–5770. [Google Scholar] [CrossRef]
- Ni, S.; Chang, J.; Chou, L.; Zhai, W. Comparison of Osteoblast-like Cell Responses to Calcium Silicate and Tricalcium Phosphate Ceramics in Vitro. Wiley Online Libr. 2006, 80, 174–183. [Google Scholar] [CrossRef]
- de Aza, P.N.; Luklinska, Z.B.; Martinez, A.; Anseau, M.R.; Guitian, F.; de Aza, S.; de Aza, S. Morphological and Structural Study of Pseudowollastonite Implants in Bone. J. Microsc. 2000, 197, 60–67. [Google Scholar] [CrossRef]
- Xue, W.; Liu, X.; Zheng, X.; Ding, C. In Vivo Evaluation of Plasma-Sprayed Wollastonite Coating. Biomaterials 2005, 26, 3455–3460. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Lin, K.; Wang, Z.; Chang, J.; Wang, L.; Lu, J.; Ning, C. Reconstruction of Calvarial Defect of Rabbits Using Porous Calcium Silicate Bioactive Ceramics. Biomaterials 2008, 29, 2588–2596. [Google Scholar] [CrossRef]
- Srinath, P.; Abdul Azeem, P.; Venugopal Reddy, K. Review on Calcium Silicate-Based Bioceramics in Bone Tissue Engineering. Int. J. Appl. Ceram. Technol. 2020, 17, 2450–2464. [Google Scholar] [CrossRef]
- Dickens, B.; Schroeder, L.W.; Brown, W.E. Crystallographic Studies of the Role of Mg as a Stabilizing Impurity in β-Ca3(PO4)2. The Crystal Structure of Pure β-Ca3(PO4)2. J. Solid State Chem. 1974, 10, 232–248. [Google Scholar] [CrossRef]
- ben Amara, M.; Vlasse, M.; le Flem, G.; Hagenmuller, P. Structure of the Low-Temperature Variety of Calcium Sodium Orthophosphate, NaCaPO4. Acta Cryst. C 1983, 39, 1483–1485. [Google Scholar] [CrossRef]
- Liu, X.; Ei-Ghannam, A. Effect of Processing Parameters on the Microstructure and Mechanical Behavior of Silica-Calcium Phosphate Nanocomposite. J. Mater. Sci. Mater. Med. 2010, 21, 2087–2094. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Preceramic | 500 °C | 700 °C | 900 °C | 1100 °C |
|---|---|---|---|---|
| β-Ca3(PO4)2 Nonidentified phase of hydrated Na2O · 2,87SiO2 | β-Ca3(PO4)2 β-NaCaPO4 | β-Ca3(PO4)2 β-NaCaPO4 SiO2 | β-Ca3(PO4)2 β-NaCaPO4 Na3Ca6(PO4)5 SiO2 | β-Ca3(PO4)2 Na3Ca6(PO4)5 β-CaSiO3 SiO2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kaimonov, M.; Safronova, T.; Shatalova, T.; Filippov, Y.; Tikhomirova, I.; Lukina, Y. Composite Ceramics Based on Pastes Including Tricalcium Phosphate and an Aqueous Solution of Sodium Silicate. J. Compos. Sci. 2022, 6, 267. https://doi.org/10.3390/jcs6090267
Kaimonov M, Safronova T, Shatalova T, Filippov Y, Tikhomirova I, Lukina Y. Composite Ceramics Based on Pastes Including Tricalcium Phosphate and an Aqueous Solution of Sodium Silicate. Journal of Composites Science. 2022; 6(9):267. https://doi.org/10.3390/jcs6090267
Chicago/Turabian StyleKaimonov, Maksim, Tatiana Safronova, Tatiana Shatalova, Yaroslav Filippov, Irina Tikhomirova, and Yulia Lukina. 2022. "Composite Ceramics Based on Pastes Including Tricalcium Phosphate and an Aqueous Solution of Sodium Silicate" Journal of Composites Science 6, no. 9: 267. https://doi.org/10.3390/jcs6090267
APA StyleKaimonov, M., Safronova, T., Shatalova, T., Filippov, Y., Tikhomirova, I., & Lukina, Y. (2022). Composite Ceramics Based on Pastes Including Tricalcium Phosphate and an Aqueous Solution of Sodium Silicate. Journal of Composites Science, 6(9), 267. https://doi.org/10.3390/jcs6090267