
Fabrication and Characterization of Hollow Polysiloxane Microsphere Polymer Matrix Composites with Improved Energy Absorption

 , ,
, ,  ,
,  and
and 
Abstract
:1. Introduction
2. Materials and Methods
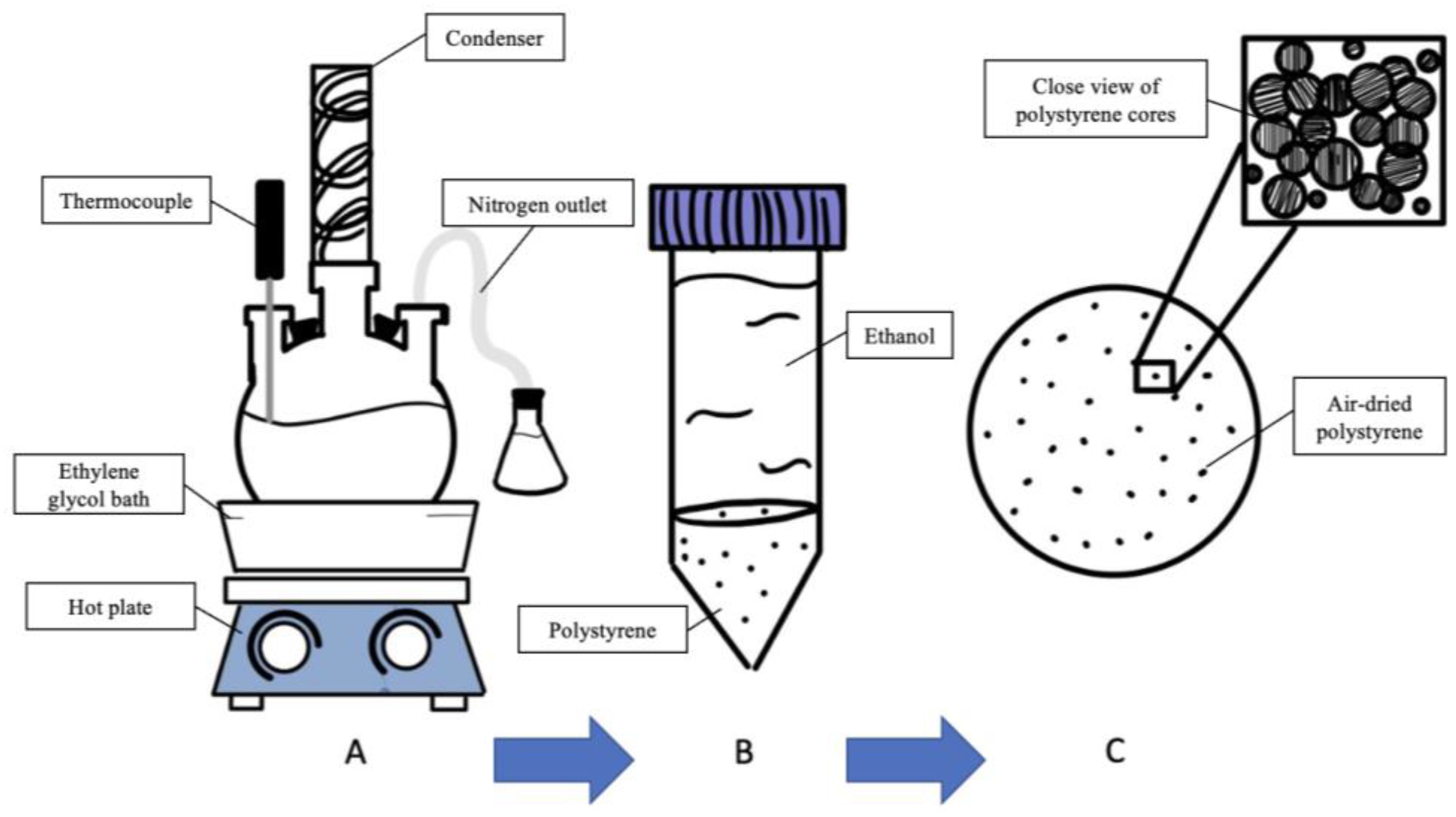
2.1. Polystyrene Core Synthesis
2.2. Polystyrene–Polysiloxane Core–Shell Composite Fabrication
2.3. Hollow Polysiloxane Microsphere Generation
2.4. Polymer Syntactic Foam Formation
3. Characterization
4. Results and Discussion
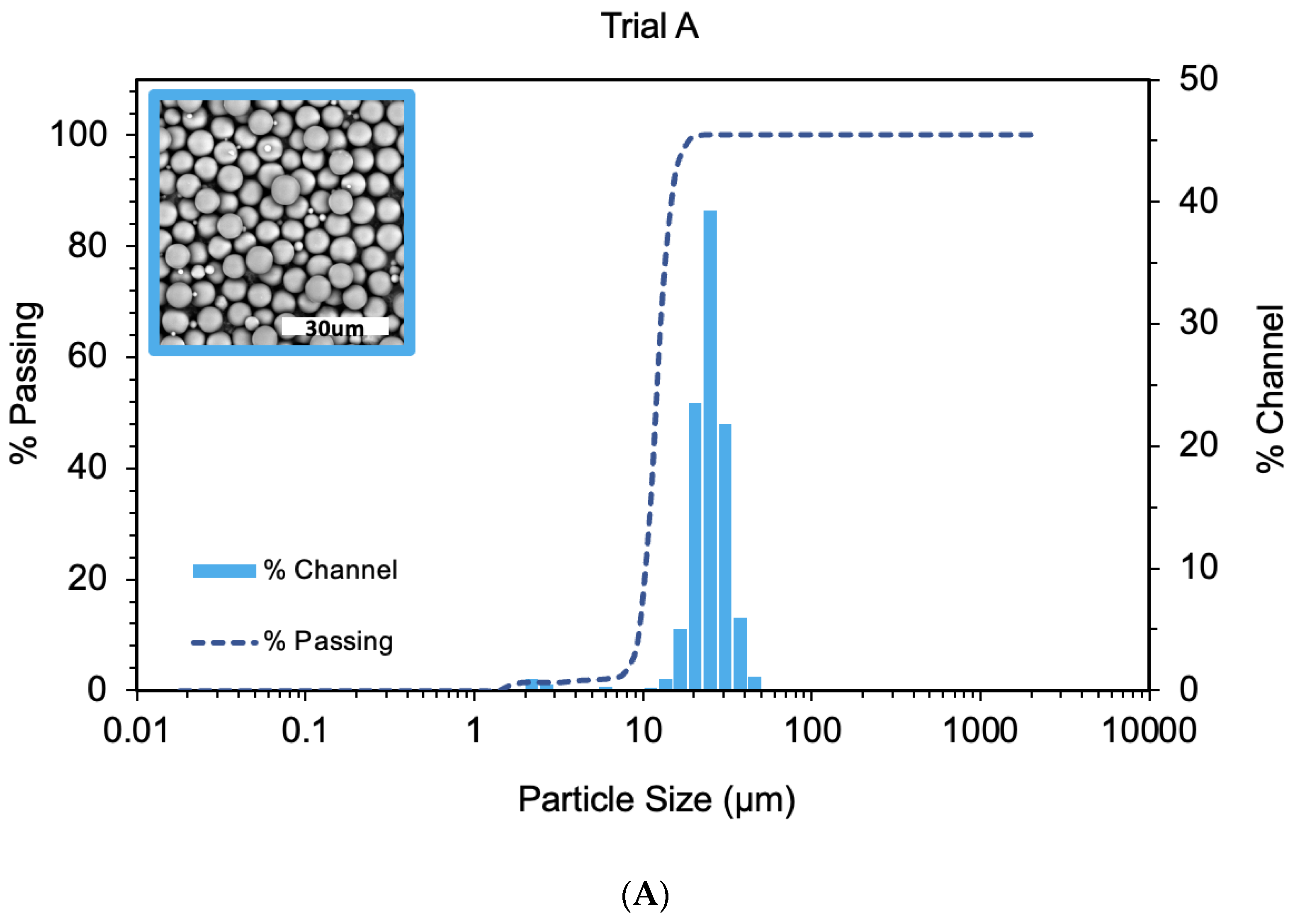
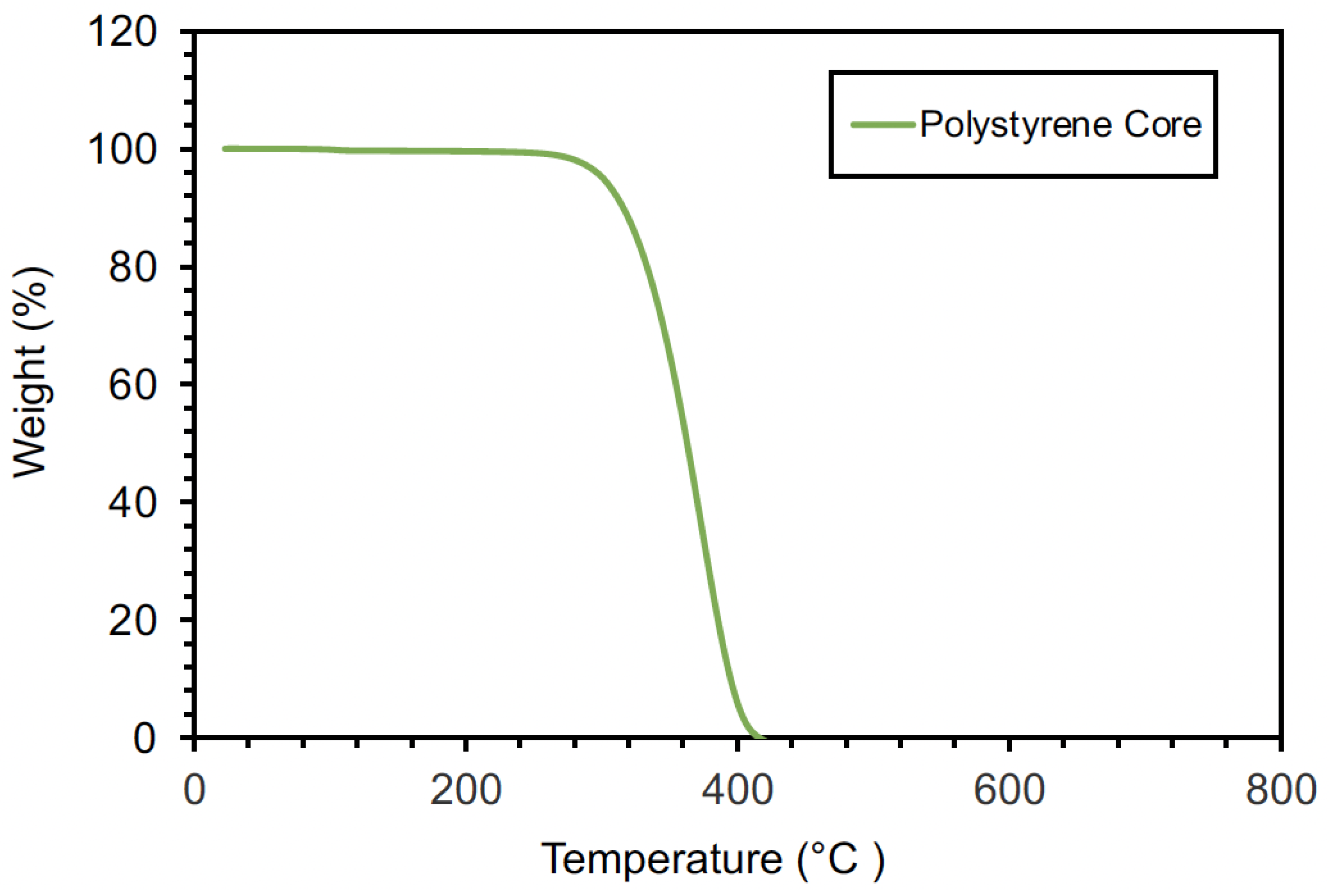
4.1. Polystyrene Cores
4.2. Core–Shell Composites
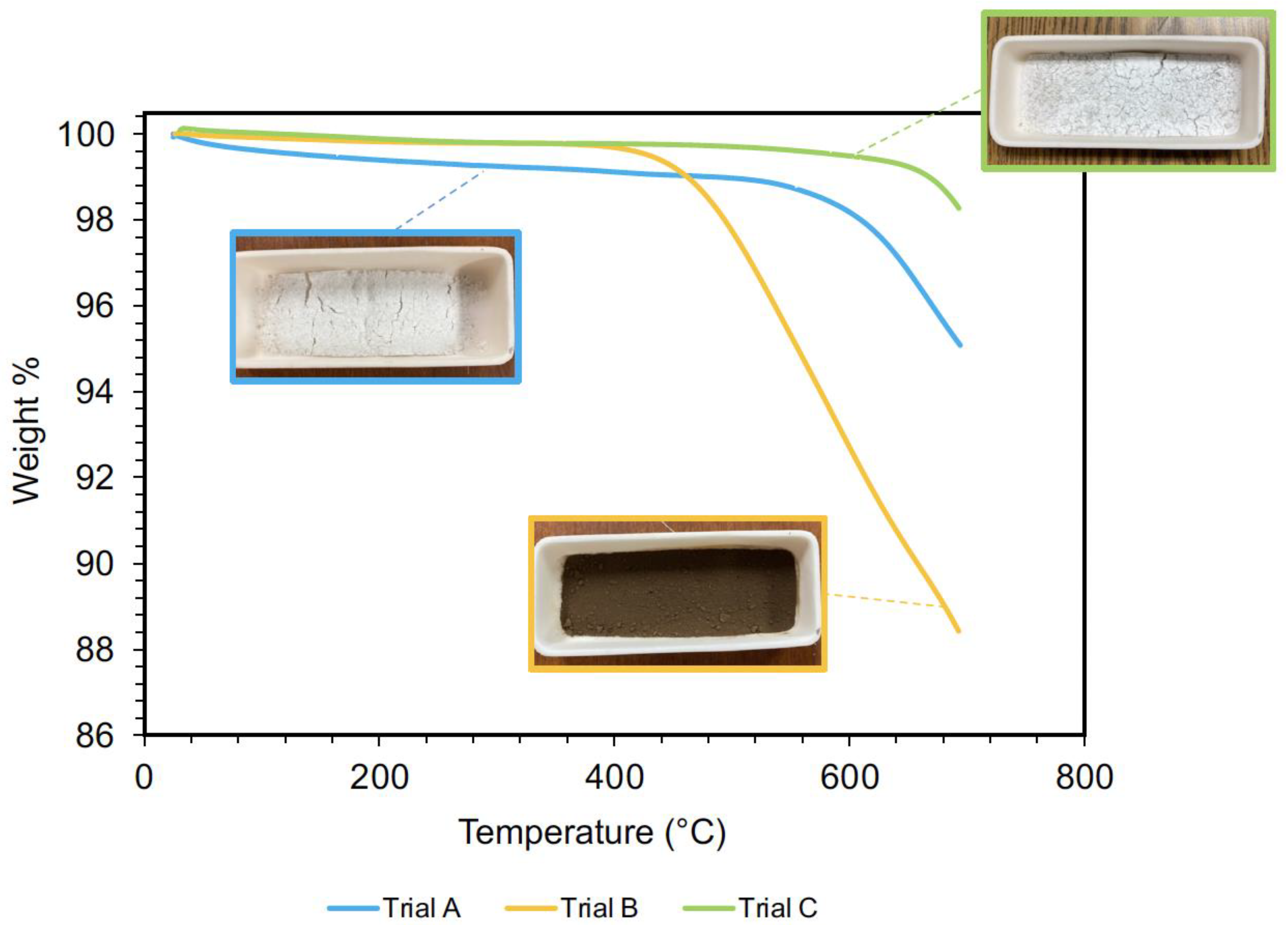
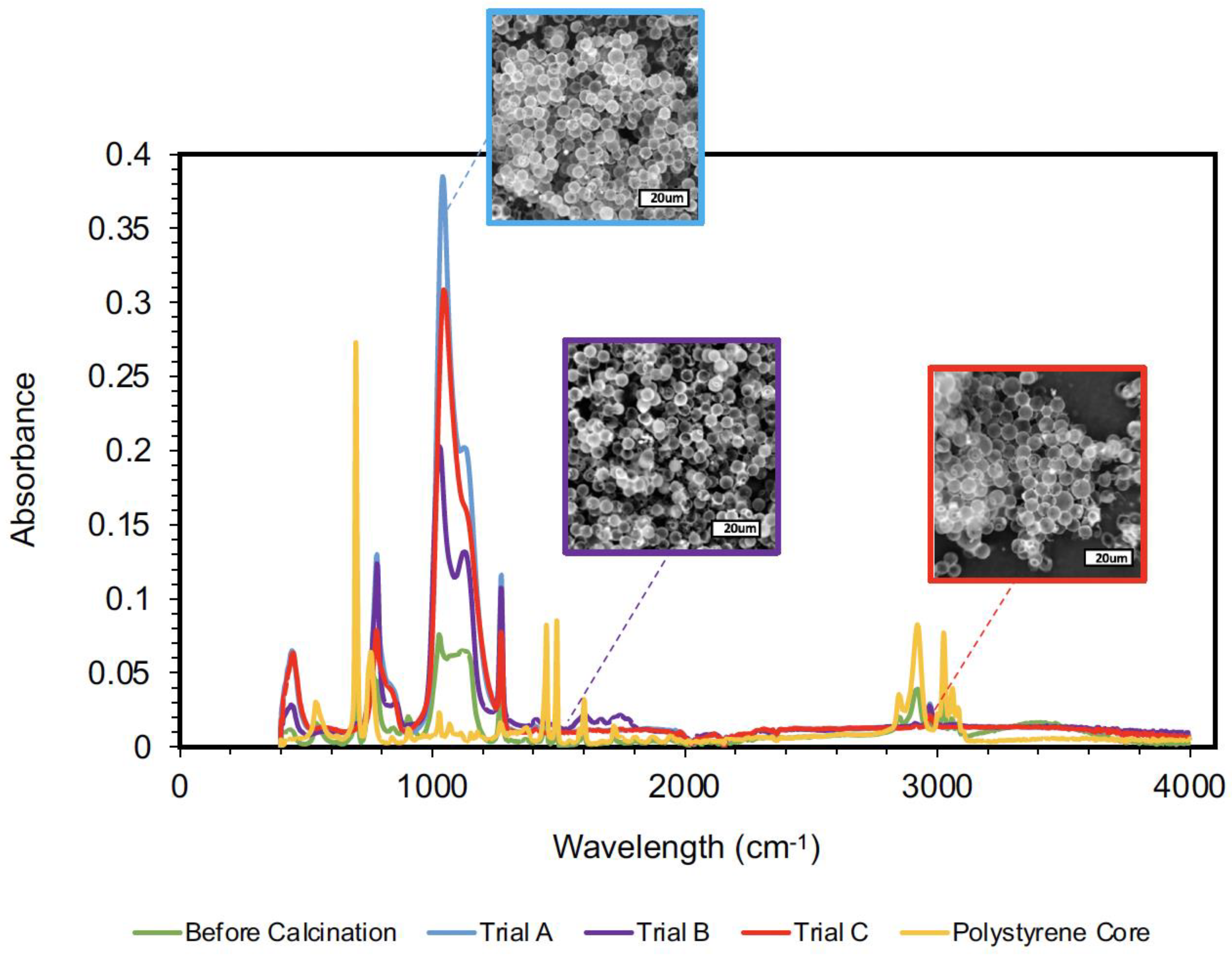
4.3. Polysiloxane Hollow Microspheres
4.4. PDMS–Polysiloxane Syntactic Foams
4.5. Energy Absorption and Elastic Recovery of Syntactic Foams
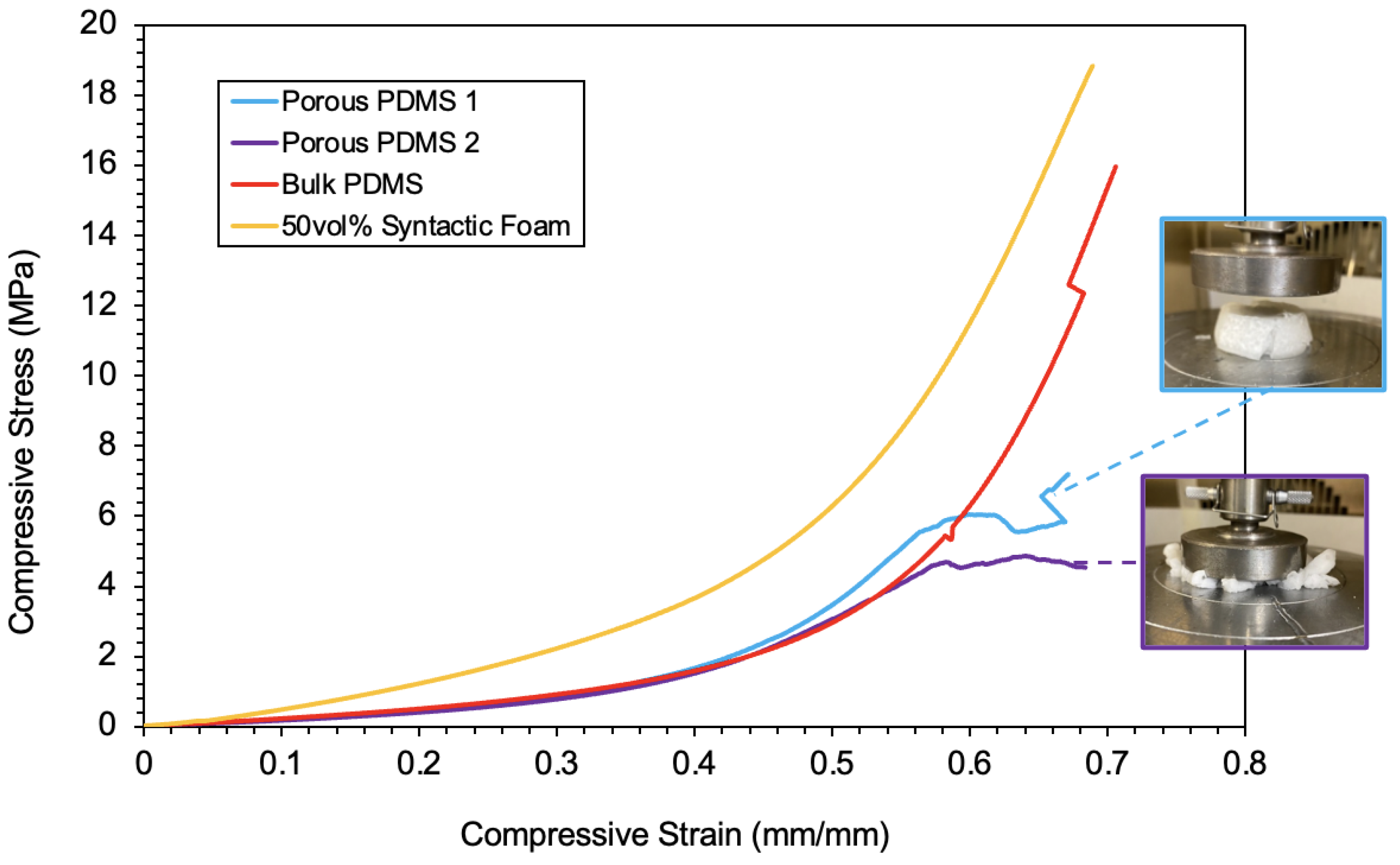
4.5.1. Macroscopic Compression Comparison
4.5.2. Uniaxial Cyclic Compression of Syntactic Foams at 70% Compression
4.5.3. Uniaxial Cyclic Compression Comparison
4.5.4. Time Recovery following Cyclic Compression Tests
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Wang, H.; Chen, P.; Zheng, X. Hollow permeable polysiloxane capsules: A novel approach for fabrication, guest encapsulation and morphology studies. J. Mater. Chem. 2004, 14, 1648–1651. [Google Scholar] [CrossRef]
- de Figueiredo, A.M.; Glória, J.C.; Chaves, Y.O.; Neves, W.L.L.; Mariúba, L.A.M. Diagnostic applications of microsphere-based flow cytometry: A review. Exp. Biol. Med. (Maywood) 2022, 247, 1852–1861. [Google Scholar] [CrossRef] [PubMed]
- Díez, N.; Sevilla, M.; Fuertes, A.B. Highly Packed Monodisperse Porous Carbon Microspheres for Energy Storage in Supercapacitors and Li−S Batteries. ChemElectroChem 2020, 7, 3798–3810. [Google Scholar] [CrossRef]
- Li, X.P.; Deng, Z.; Li, Y.; Zhang, H.B.; Zhao, S.; Zhang, Y.; Wu, X.Y.; Yu, Z.Z. Controllable synthesis of hollow microspheres with Fe@Carbon dual-shells for broad bandwidth microwave absorption. Carbon 2019, 147, 172–181. [Google Scholar] [CrossRef]
- Leong, H.J.; Oh, S.-G. Preparation of antibacterial TiO2 particles by hybridization with azelaic acid for applications in cosmetics. J. Ind. Eng. Chem. 2018, 66, 242–247. [Google Scholar] [CrossRef]
- Ji, X.; Zhang, H.; Bai, Z.; Qiu, G.; Guo, M.; Cheng, F.; Zhang, M. Self-assembled multifunctional bulk hollow microspheres: Thermal insulation, sound absorption and fire resistance. Energy Build. 2019, 205, 109533. [Google Scholar] [CrossRef]
- Ward, J.M.; Chormaic, S.N. Thermo-optical tuning of whispering gallery modes in Er:Yb co-doped phosphate glass microspheres. Appl. Phys. B 2010, 100, 847–850. [Google Scholar] [CrossRef] [Green Version]
- Zhao, P.-P.; Deng, C.; Zhao, Z.-Y.; Huang, S.-C.; Lu, P.; Wang, Y.-Z. Nanoflake-Constructed Supramolecular Hierarchical Porous Microspheres for Fire-Safety and Highly Efficient Thermal Energy Storage. ACS Appl. Mater. Interfaces 2020, 12, 28700–28710. [Google Scholar] [CrossRef] [PubMed]
- Feng, H.M.; Zheng, J.C.; Lei, N.Y.; Yu, L.; Kong, K.H.K.; Yu, H.Q.; Lau, T.C.; Lam, M.H. Photoassisted Fenton Degradation of Polystyrene. Environ. Sci. Technol. 2011, 45, 744–750. [Google Scholar] [CrossRef]
- Yang, W.; Gao, F.; Qiu, Y.; Liu, W.; Xu, H.; Yang, L.; Liu, Y. CsPbBr3-Quantum-Dots/Polystyrene@Silica Hybrid Microsphere Structures with Significantly Improved Stability for White LEDs. Adv. Opt. Mater. 2019, 7, 1900546. [Google Scholar] [CrossRef]
- Ding, D.; Wang, Y.; Li, X.; Qiang, R.; Xu, P.; Chu, W.; Han, X.; Du, Y. Rational design of core-shell Co@C microspheres for high-performance microwave absorption. Carbon 2017, 111, 722–732. [Google Scholar] [CrossRef]
- Li, G.; Zhu, J.; Tian, W.; Ma, C. Preparation and optical property of anatase hollow microsphere with mesoporosity. Mater. Res. Bull. 2009, 44, 271–275. [Google Scholar] [CrossRef]
- Wu, A.S.; Small, W., IV; Bryson, T.M.; Cheng, E.; Metz, T.R.; Schulze, S.E.; Duoss, E.B.; Wilson, T.S. 3D Printed Silicones with Shape Memory. Sci. Rep. 2017, 7, 4664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tian, Q.; Yu, D.; Zhu, K.; Hu, G.; Zhang, L.; Liu, Y. Multi-hollow polymer microspheres with enclosed surfaces and compartmentalized voids prepared by seeded swelling polymerization method. J. Colloid Interface Sci. 2016, 473, 44–51. [Google Scholar] [CrossRef] [PubMed]
- Wei, B.; Wang, S.; Song, H.; Liu, H.; Li, J.; Liu, N. A review of recent progress in preparation of hollow polymer microspheres. Pet. Sci. 2009, 6, 306–312. [Google Scholar] [CrossRef] [Green Version]
- Lu, X.; Xin, Z. Preparation and characterization of micron-sized hollow polysiloxane spheres. Chem. Eng. Sci. 2007, 62, 4880–4884. [Google Scholar] [CrossRef]
- Gupta, N.; Zeltmann, S.E.; Shunmugasamy, V.C.; Pinisetty, D. Applications of Polymer Matrix Syntactic Foams. JOM 2014, 66, 245–254. [Google Scholar] [CrossRef]
- Li, G.; Jones, N. Development of rubberized syntactic foam. Compos. Part A Appl. Sci. Manuf. 2007, 38, 1483–1492. [Google Scholar] [CrossRef]
- Xu, T.; Li, G. A shape memory polymer based syntactic foam with negative Poisson’s ratio. Mater. Sci. Eng. A 2011, 528, 6804–6811. [Google Scholar] [CrossRef]
- Yousaf, Z.; Smith, M.; Potluri, P.; Parnell, W. Compression properties of polymeric syntactic foam composites under cyclic loading. Compos. Part B Eng. 2020, 186, 107764. [Google Scholar] [CrossRef]
- Gupta, N.; Pinisetty, D. A Review of Thermal Conductivity of Polymer Matrix Syntactic Foams—Effect of Hollow Particle Wall Thickness and Volume Fraction. JOM 2013, 65, 234–245. [Google Scholar] [CrossRef]
- Madenci, E.; Özkılıç, Y.O.; Aksoylu, C.; Safonov, A. The Effects of Eccentric Web Openings on the Compressive Performance of Pultruded GFRP Boxes Wrapped with GFRP and CFRP Sheets. Polymers 2022, 14, 4567. [Google Scholar] [CrossRef] [PubMed]
- John, M.; Li, G. Self-healing of sandwich structures with a grid stiffened shape memory polymer syntactic foam core. Smart Mater. Struct. 2010, 19, 075013. [Google Scholar] [CrossRef]
- Sarrafan, S.; Feng, X.; Li, G. A soft syntactic foam actuator with high recovery stress, actuation strain, and energy output. Mater. Today Commun. 2022, 31, 103303. [Google Scholar] [CrossRef]
- Liu, Y.; Li, L.; Liu, H.; Zhang, M.; Liu, A.; Liu, L.; Tang, L.; Wang, G.; Zhou, S. Hollow polymeric microsphere-filled silicone-modified epoxy as an internally insulated material for composite cross-arm applications. Compos. Sci. Technol. 2020, 200, 108418. [Google Scholar] [CrossRef]
- Simič, R.; Mandal, J.; Zhang, K.; Spencer, N.D. Oxygen inhibition of free-radical polymerization is the dominant mechanism behind the ‘mold effect’ on hydrogels. Soft Matter 2021, 17, 6394–6403. [Google Scholar] [CrossRef]
- Kim, J.-W.; Suh, K.-D. Highly monodisperse crosslinked polystyrene microparticles by dispersion polymerization. Colloid Polym. Sci. 1998, 276, 870–878. [Google Scholar] [CrossRef]
- Wilburn, B.R. “SynThesis and Advanced Manufacturing of Polymer Structures and Their Composites” (2020). Open Access Theses & Dissertations. 3060. Available online: https://scholarworks.utep.edu/open_etd/3060 (accessed on 1 March 2022).
- Reza, E.I. “SynThesis and Characterization of Functiona Materials for 3D Printed Composites” (2021). Open Access Theses & Dissertations. 3332. Available online: https://scholarworks.utep.edu/open_etd/3332 (accessed on 1 March 2022).
- Irigoyen, A. “Synthesis, Characterization, and Direct-Ink-Writing of Syntactic Foams” (2021). Open Access Theses & Dissertations. 3420. Available online: https://scholarworks.utep.edu/open_etd/3420 (accessed on 1 March 2022).
- Gorsd, M.; Sathicq, G.; Romanelli, G.; Pizzio, L.; Blanco, M. Tungstophosphoric acid supported on core-shell polystyrene-silica microspheres or hollow silica spheres catalyzed trisubstituted imidazole synthesis by multicomponent reaction. J. Mol. Catal. A Chem. 2016, 420, 294–302. [Google Scholar] [CrossRef]
- Li, Y.; Liu, E.C.Y.; Pickett, N.; Skabara, P.J.; Cummins, S.S.; Ryley, S.; Sutherland, A.J.; O’Brien, P. Synthesis and characterization of CdS quantum dots in polystyrene microbeads. J. Mater. Chem. 2005, 15, 1238–1243. [Google Scholar] [CrossRef]
- Hopf, R.; Bernardi, L.; Menze, J.; Zündel, M.; Mazza, E.; Ehret, A.E. Experimental and theoretical analyses of the age-dependent large-strain behavior of Sylgard 184 (10:1) silicone elastomer. J. Mech. Behav. Biomed. Mater. 2016, 60, 425–437. [Google Scholar] [CrossRef]
- Zou, D.; Derlich, V.; Gandhi, K.; Park, M.; Sun, L.; Kriz, D.; Lee, Y.D.; Kim, G.; Aklonis, J.J.; Salovey, R. Model filled polymers. I. Synthesis of crosslinked monodisperse polystyrene beads. J. Polym. Sci. Part A Polym. Chem. 1990, 28, 1909–1921. [Google Scholar] [CrossRef]
- SYLGARDTM 184 Silicone Elastomer—Technical Data Sheet. The Dow Chemical Company. 2017. Available online: https://www.dow.com/content/dam/dcc/documents/en-us/productdatasheet/11/11-31/11-3184-sylgard-184-elastomer.pdf (accessed on 20 February 2023).
- Yun, D.S.; Lee, H.S.; Jang, H.G.; Yoo, J.W. Controlling Size and Distribution for Nano-sized Polystyrene Spheres. Bull. Korean Chem. Soc. 2010, 31, 1345–1348. [Google Scholar] [CrossRef] [Green Version]
- Sertchook, H.; Avnir, D. Submicron Silica/Polystyrene Composite Particles Prepared by a One-Step Sol−Gel Process. Chem. Mater. 2003, 15, 1690–1694. [Google Scholar] [CrossRef]
- Delgado, A.; González-Caballero, F.; Salcedo, J. On the zeta potential of spherical polystyrene particles from electrophoresis theories. Acta Polym. 1986, 37, 361–364. [Google Scholar] [CrossRef]
- Fang, J.; Xuan, Y.; Li, Q. Preparation of polystyrene spheres in different particle sizes and assembly of the PS colloidal crystals. Sci. China-Technol. Sci. 2010, 53, 3088–3093. [Google Scholar] [CrossRef]
- Meng, J.; Liu, T.-W.; Wang, H.-Y.; Dai, L.-H. Ultra-high energy absorption high-entropy alloy syntactic foam. Compos. Part B Eng. 2021, 207, 108563. [Google Scholar] [CrossRef]
- Chojnowski, J.; Slomkowski, S.; Fortuniak, W.; Mizerska, U.; Pospiech, P. Hydrophilic Polysiloxane Microspheres and Ceramic SiOC Microspheres Derived from Them. J. Inorg. Organomet. Polym. Mater. 2020, 30, 56–68. [Google Scholar] [CrossRef] [Green Version]
- Nam, T.H.; Petríková, I.; Marvalová, B. Experimental and numerical research of stress relaxation behavior of magnetorheological elastomer. Polym. Test. 2021, 93, 106886. [Google Scholar] [CrossRef]
- Tewani, H.; Hinaus, M.; Talukdar, M.; Sone, H.; Prabhakar, P. Architected syntactic foams: A tale of additive manufacturing and reinforcement parameters. arXiv 2022, arXiv:2211.00955. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Specimen Type | Young’s Modulus | Energy Absorption (Plateau Region) | Ultimate Compressive Strength |
|---|---|---|---|
| Porous PDMS 1 | 1.612 MPa | 0.748 J/mm3 | 7.21 MPa |
| Porous PDMS 2 | 1.315 MPa | 0.652 J/mm3 | 4.54 MPa |
| Bulk PDMS | 1.749 MPa | 0.684 J/mm3 | 15.9 MPa |
| 50 vol% Syntactic Foam | 3.575 MPa | 1.505 J/mm3 | 18.9 MPa |
| Compression # | Young’s Modulus | Energy Absorption (Plateau Region) | Ultimate Compressive Strength |
|---|---|---|---|
| 1 | 2.569 MPa | 1.461 J/mm3 | 22.374 MPa |
| 2 | 2.131 MPa | 0.844 J/mm3 | 25.308 MPa |
| 3 | 1.959 MPa | 0.642 J/mm3 | 24.841 MPa |
| Specimen Type | Energy Absorption at σ = 0.5 MPa | Energy Absorption at ε = 0.5 mm/mm |
|---|---|---|
| 50 vol% Syntactic Foam C1 | 0.024 J/mm3 | 1.279 J/mm3 |
| 50 vol% Syntactic Foam C2 | 0.026 J/mm3 | 1.193 J/mm3 |
| 50 vol% Syntactic Foam C3 | 0.027 J/mm3 | 1.162 J/mm3 |
| Bulk PDMS C1 | 0.077 J/mm3 | 0.207 J/mm3 |
| Bulk PDMS C2 | 0.076 J/mm3 | 0.211 J/mm3 |
| Bulk PDMS C3 | 0.076 J/mm3 | 0.220 J/mm3 |
| Porous PDMS C1 | 0.069 J/mm3 | 0.234 J/mm3 |
| Porous PDMS C2 | 0.070 J/mm3 | 0.224 J/mm3 |
| Porous PDMS C3 | 0.071 J/mm3 | 0.225 J/mm3 |
| Height | Specimen 1 | Specimen 2 | Specimen 3 |
|---|---|---|---|
| Original | 12.41 mm | 12.11 mm | 12.10 mm |
| After 1st compressions | 12.34 mm | 12.02 mm | 11.99 mm |
| After 1 week of recovery | 12.4 mm | 12.11 mm | 12.08 mm |
| After 2nd compressions | 12.31 mm | 11.97 mm | 11.95 mm |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gomez, S.G.; Irigoyen, A.; Gonzalez, S.; Estala-Rodriguez, K.; Shafirovich, E.; Hassan, M.S.; Zaman, S.; Lin, Y. Fabrication and Characterization of Hollow Polysiloxane Microsphere Polymer Matrix Composites with Improved Energy Absorption. J. Compos. Sci. 2023, 7, 98. https://doi.org/10.3390/jcs7030098
Gomez SG, Irigoyen A, Gonzalez S, Estala-Rodriguez K, Shafirovich E, Hassan MS, Zaman S, Lin Y. Fabrication and Characterization of Hollow Polysiloxane Microsphere Polymer Matrix Composites with Improved Energy Absorption. Journal of Composites Science. 2023; 7(3):98. https://doi.org/10.3390/jcs7030098
Chicago/Turabian StyleGomez, Sofia Gabriela, Andrea Irigoyen, Stephanie Gonzalez, Kevin Estala-Rodriguez, Evgeny Shafirovich, Md Sahid Hassan, Saqlain Zaman, and Yirong Lin. 2023. "Fabrication and Characterization of Hollow Polysiloxane Microsphere Polymer Matrix Composites with Improved Energy Absorption" Journal of Composites Science 7, no. 3: 98. https://doi.org/10.3390/jcs7030098
APA StyleGomez, S. G., Irigoyen, A., Gonzalez, S., Estala-Rodriguez, K., Shafirovich, E., Hassan, M. S., Zaman, S., & Lin, Y. (2023). Fabrication and Characterization of Hollow Polysiloxane Microsphere Polymer Matrix Composites with Improved Energy Absorption. Journal of Composites Science, 7(3), 98. https://doi.org/10.3390/jcs7030098