
Artificial Neural Network Analysis of Gene Expression Data Predicted Non-Hodgkin Lymphoma Subtypes with High Accuracy



Abstract
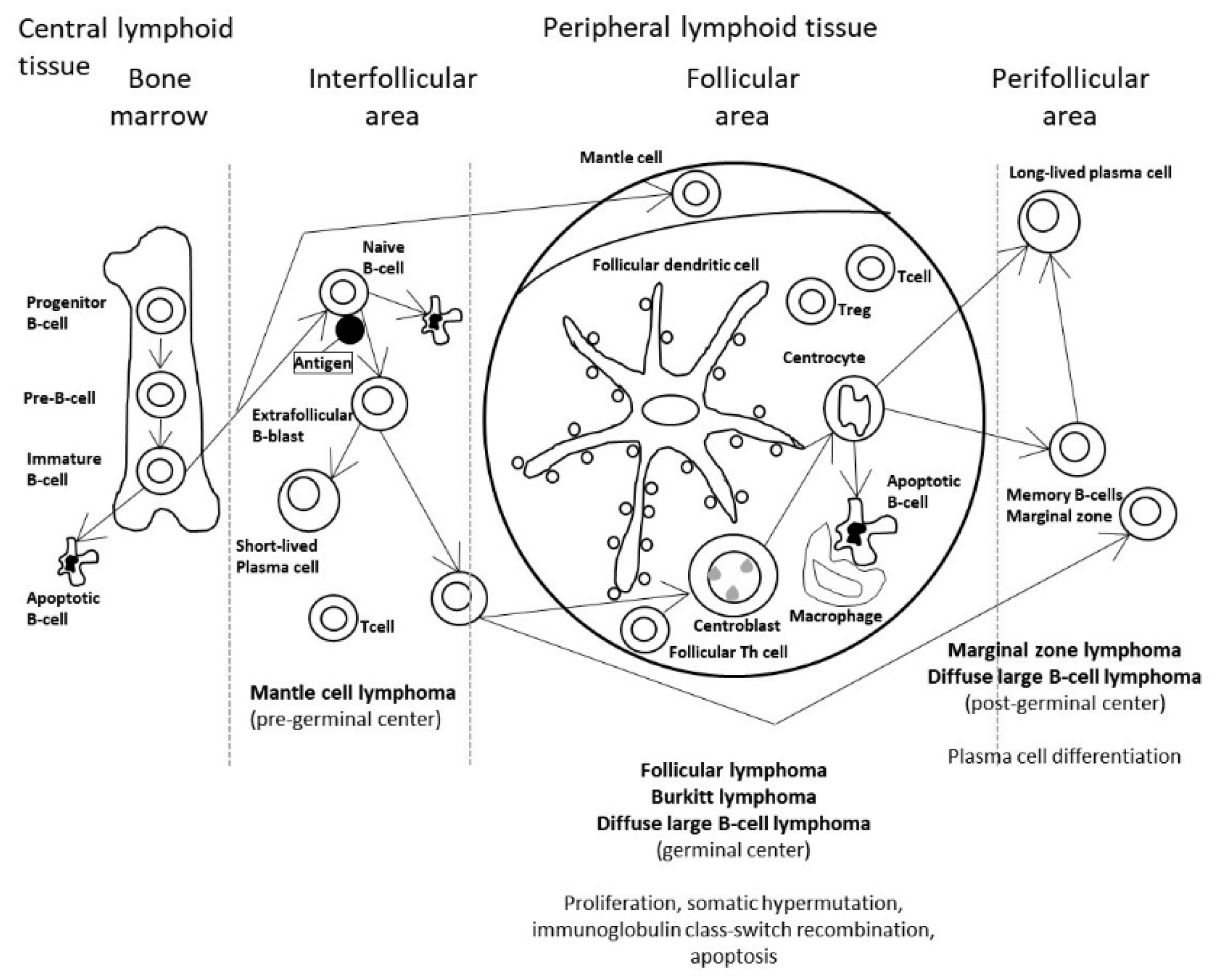
:1. Introduction
2. Materials and Methods
2.1. Gene Expression Data Set
2.2. Software
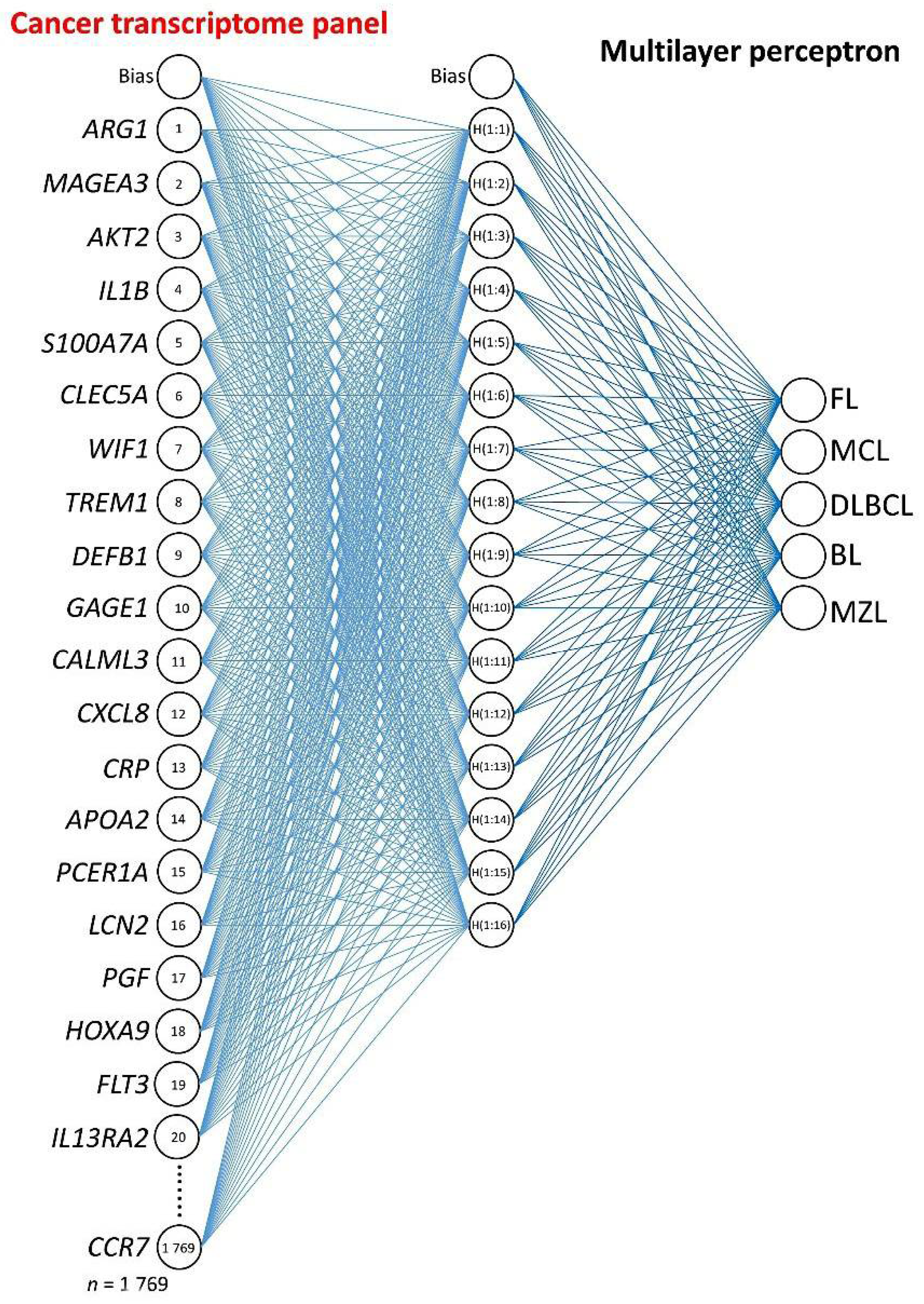
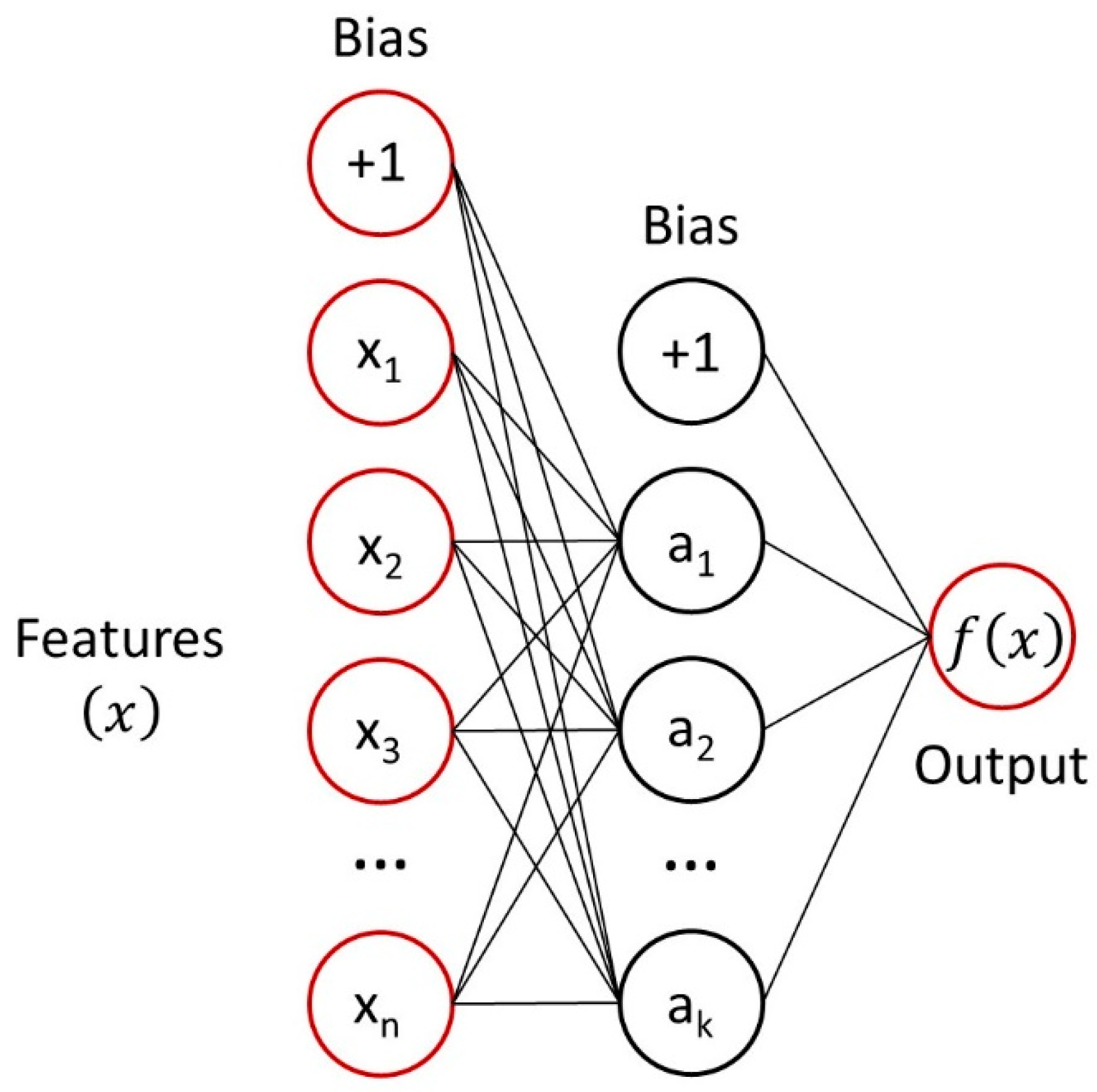
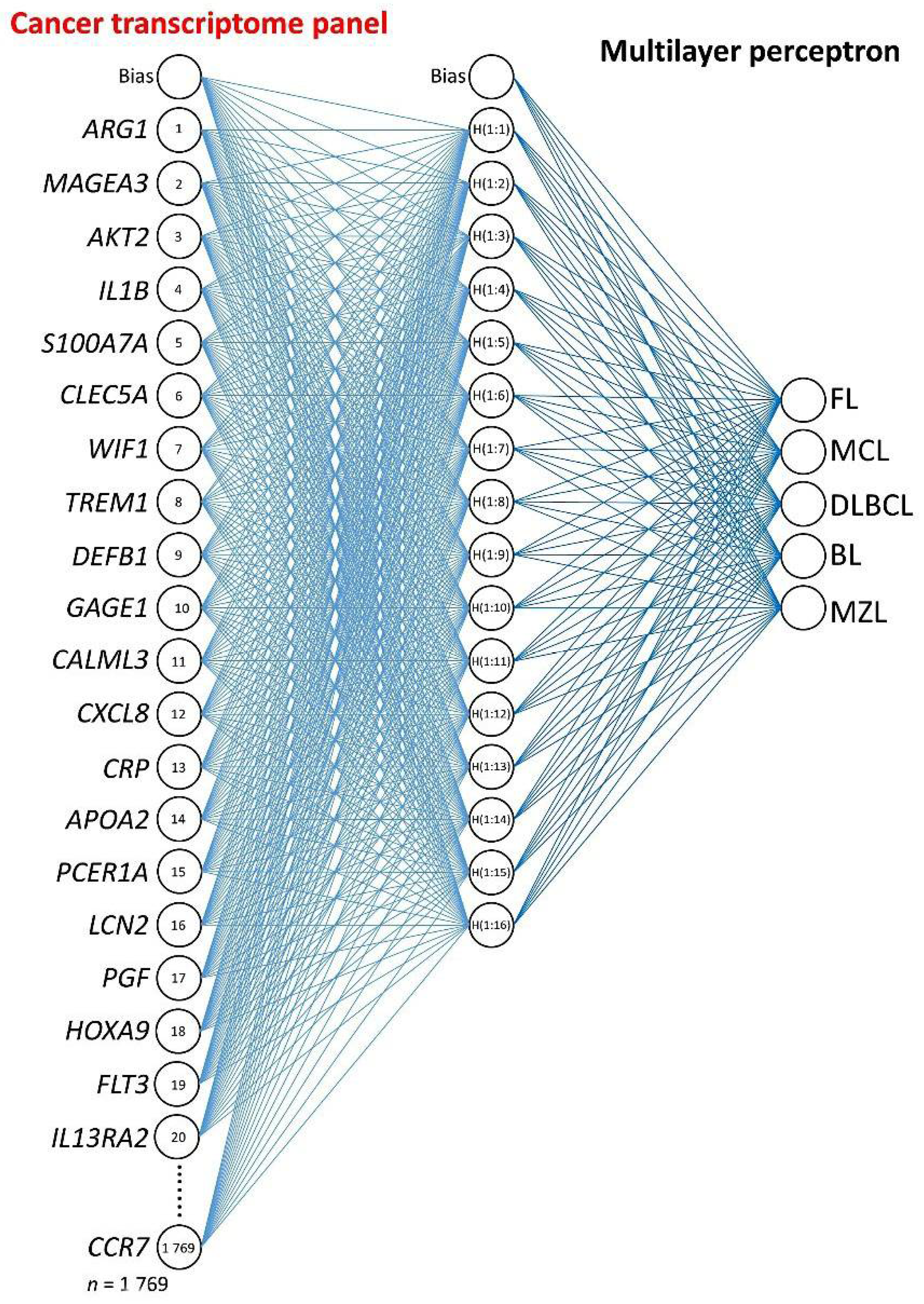
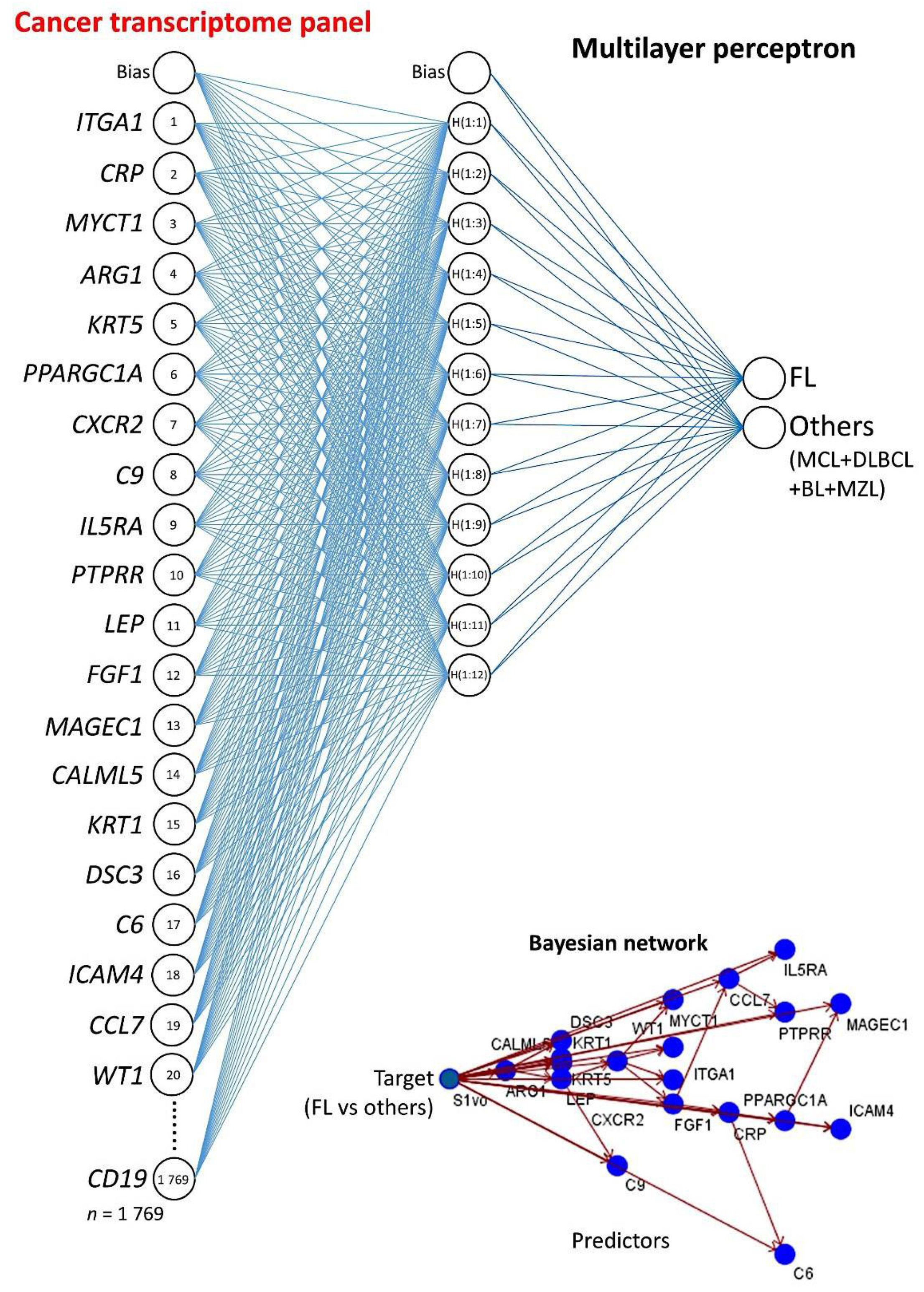
2.3. Multilayer Perceptron Analysis
2.4. Hardware
2.5. Ethical Compliance
3. Results
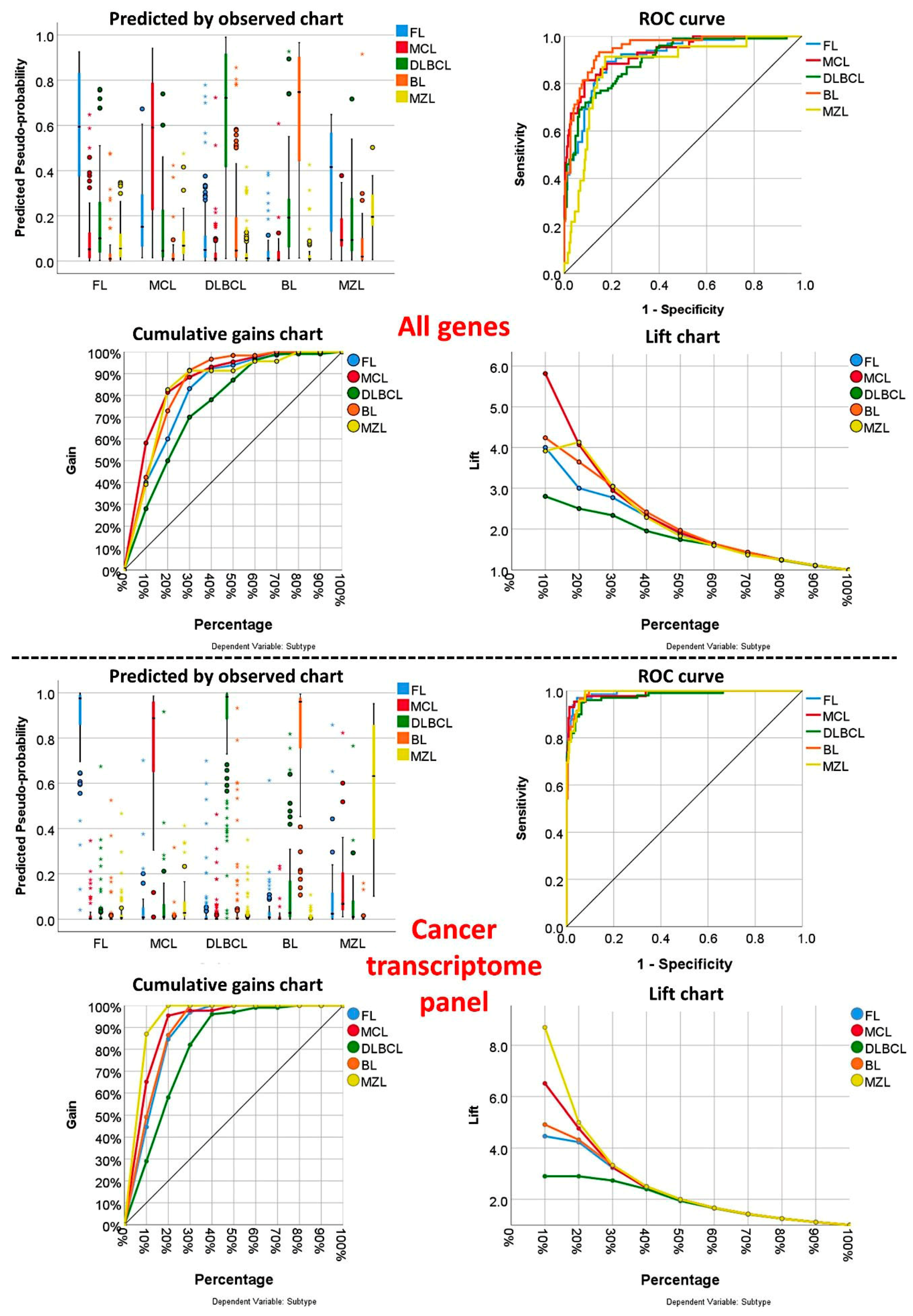
3.1. Multilayer Perceptron Analysis (MLP) for Predicting All NHL Subtypes
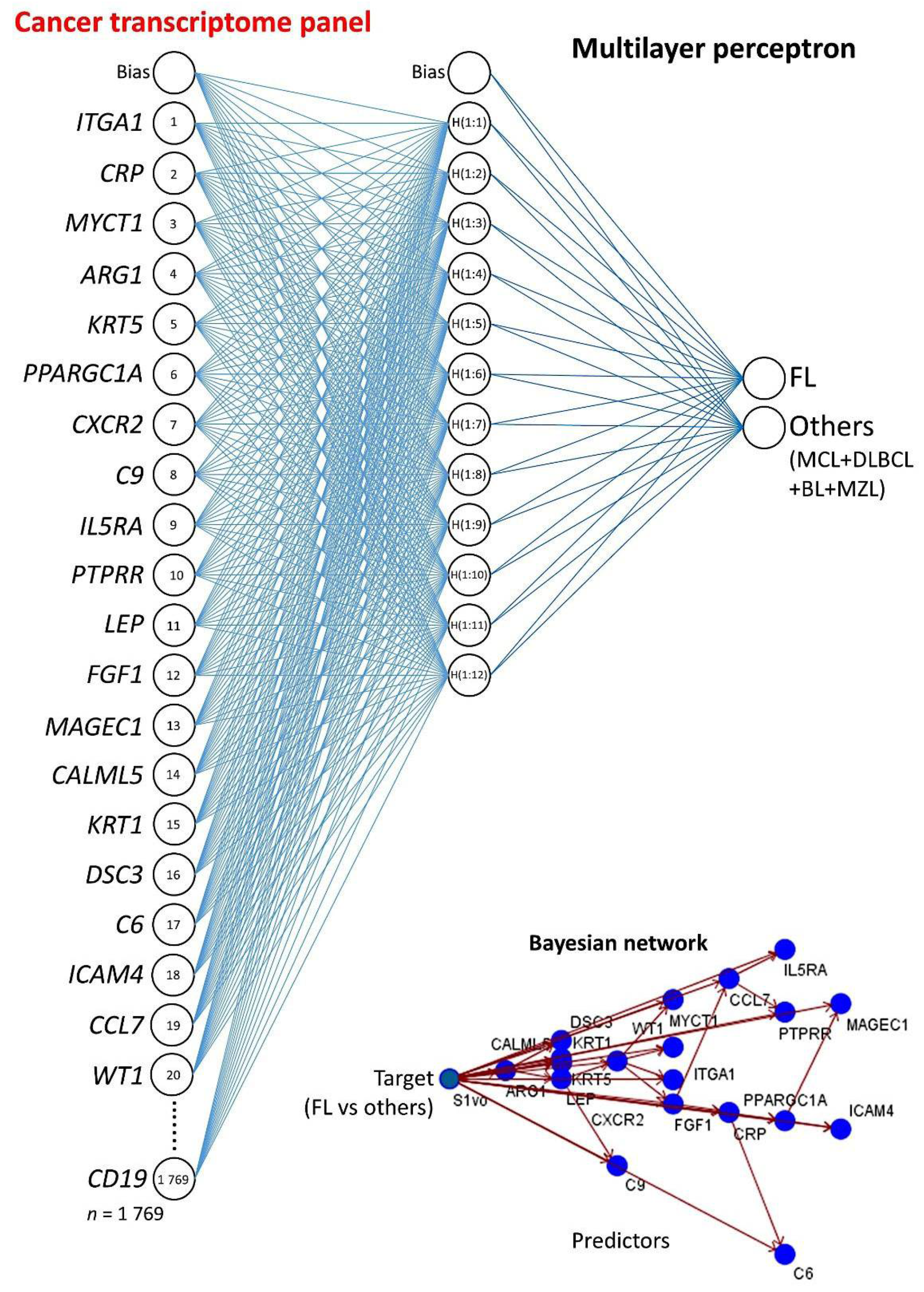
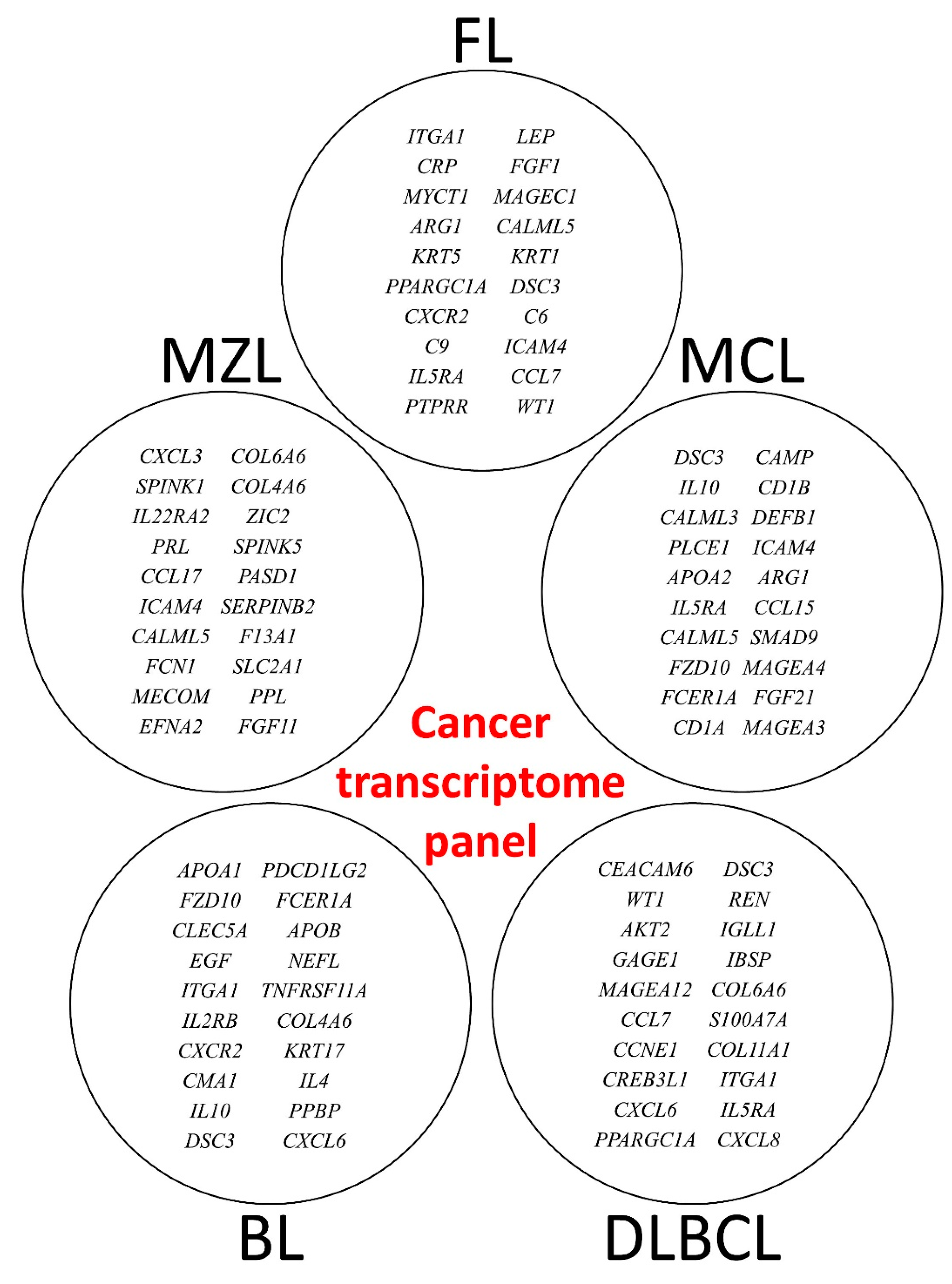
3.2. Multilayer Perceptron Analysis (MLP) for Predicting Each NHL Subtype against the Other Subtypes
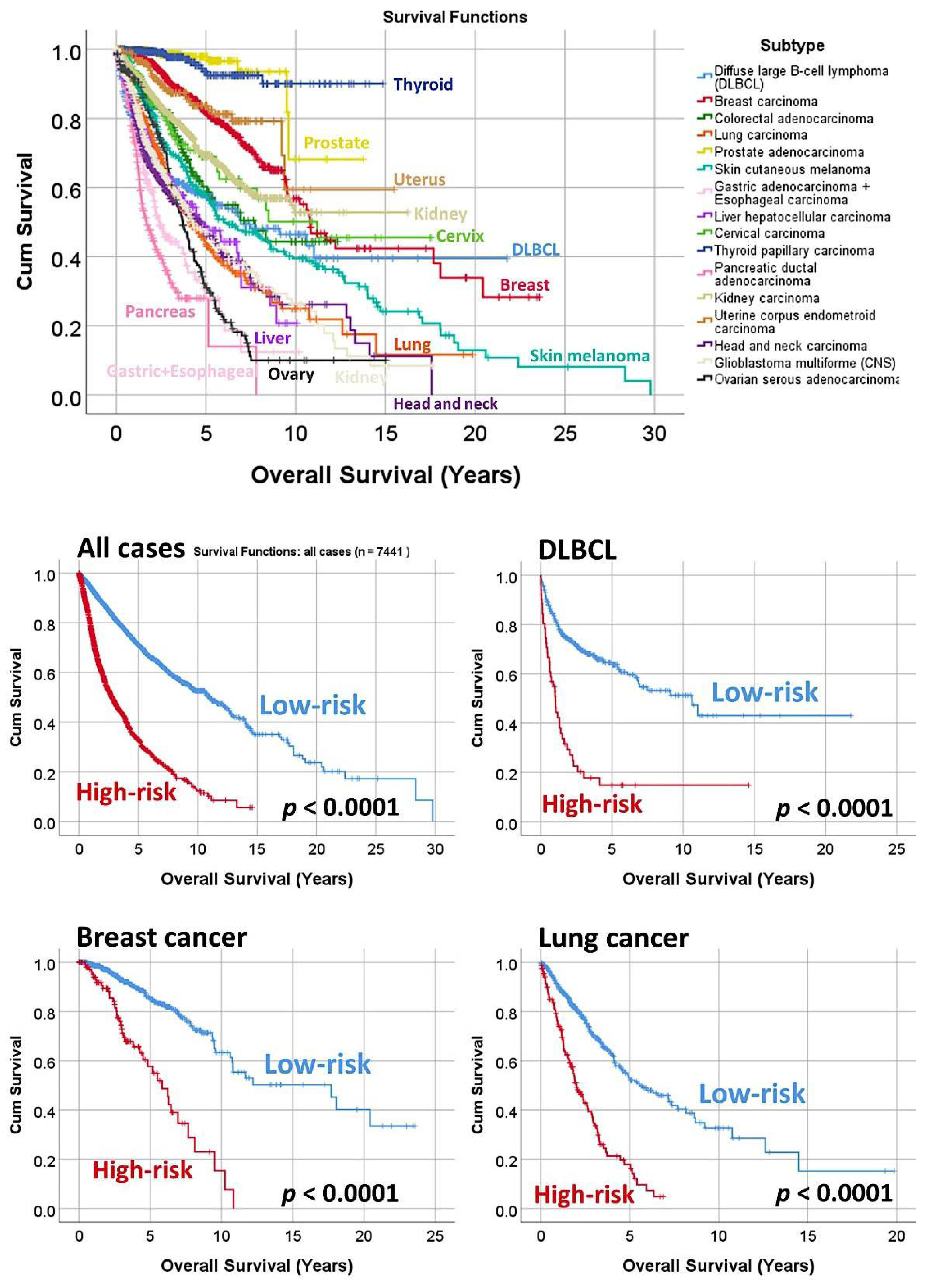
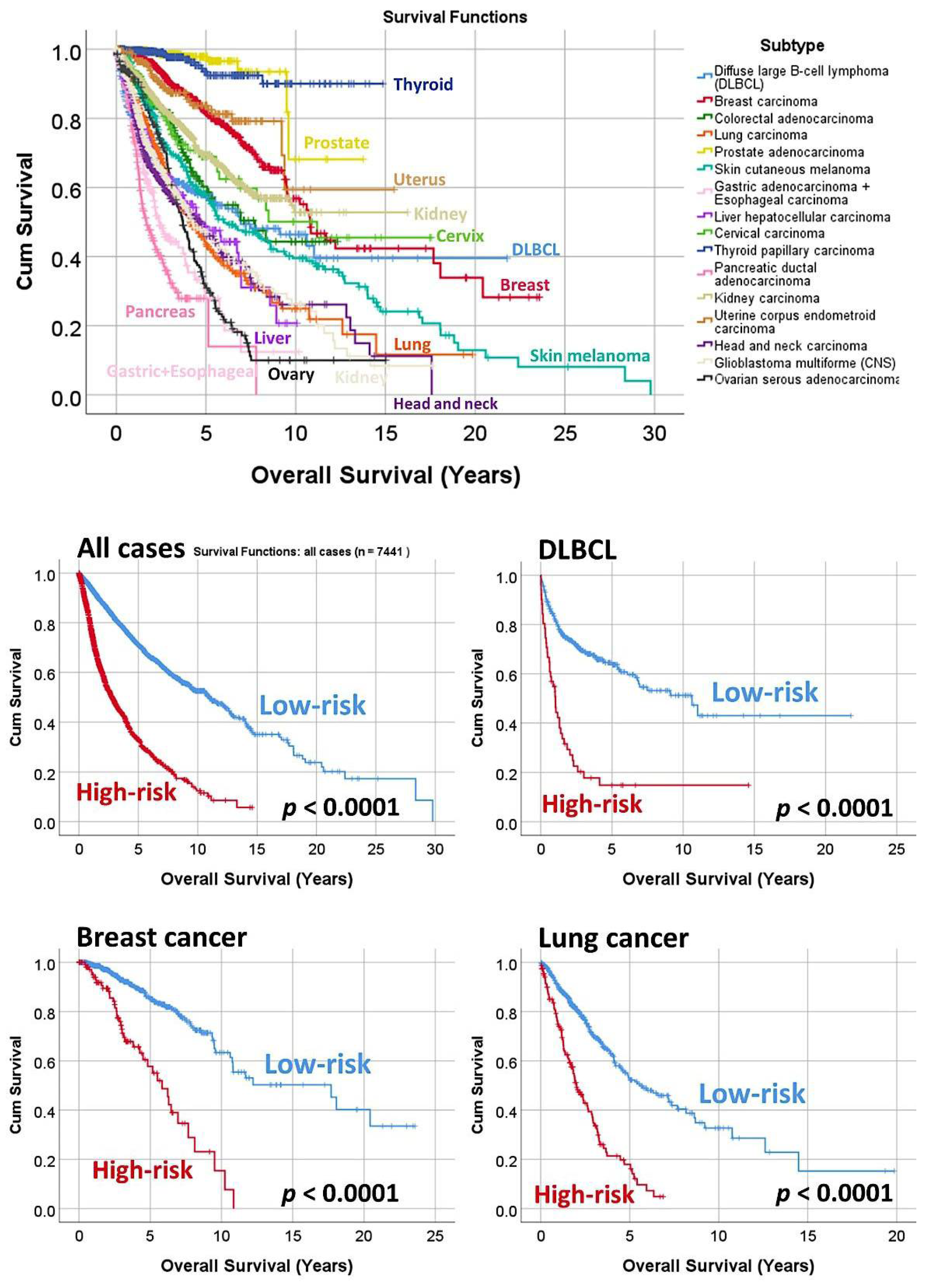
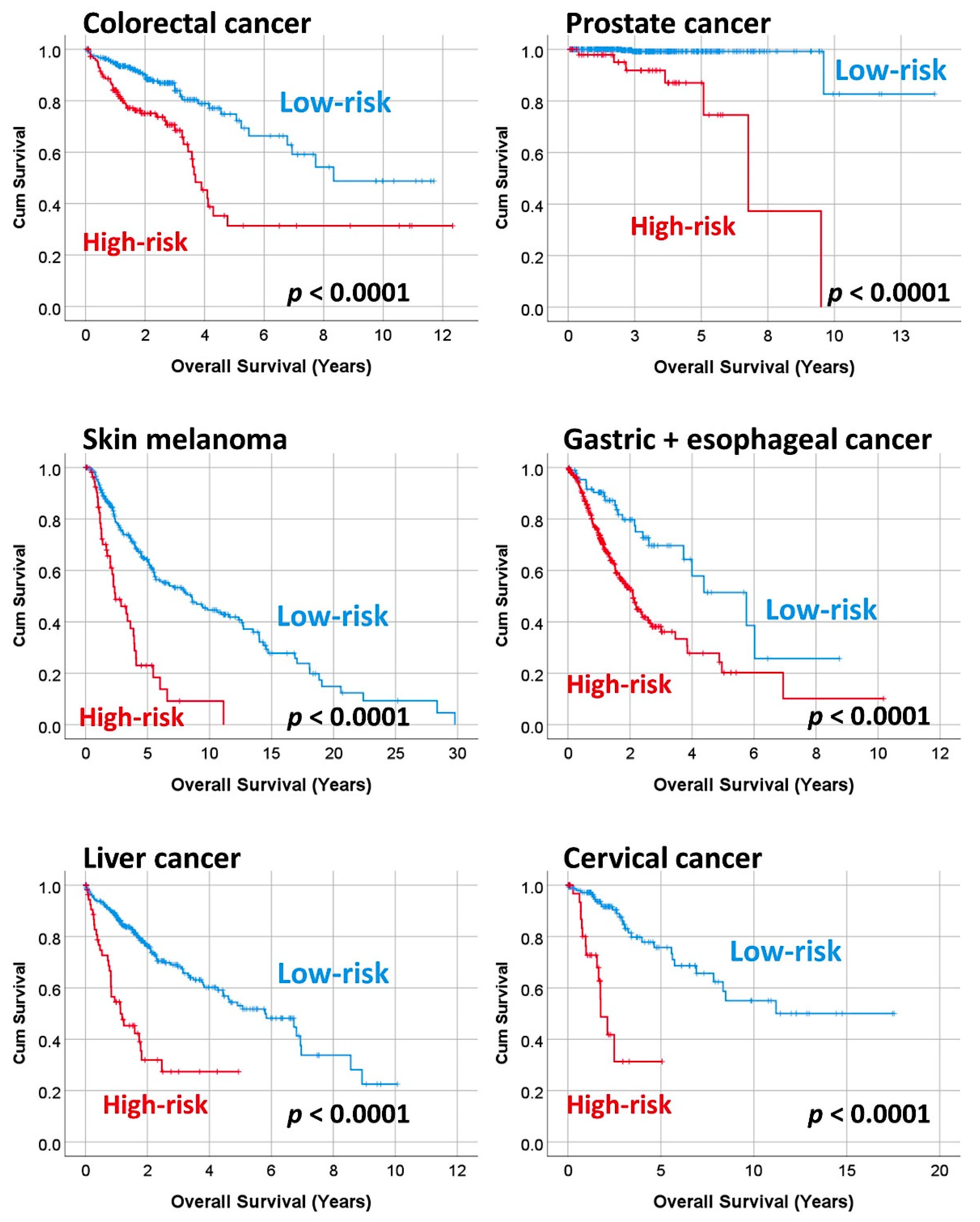
3.3. Prediction of the Overall Survival of DLBCL and Other Types of Cancer
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Freedman, A.S.; Friedberg, J.W.; Aster, J.C. Classification of the hematopoietic neoplasms. In UpToDate; Lister, A., Rosmarin, A.G., Eds.; UpToDate: Waltham, MA, USA, 2020. [Google Scholar]
- Freedman, A.S.; Friedberg, J.W.; Aster, J.C. Clinical presentation and initial evaluation of non-Hodgkin lymphoma. In UpToDate; Lister, A., Rosmarin, A.G., Eds.; UpToDate: Waltham, MA, USA, 2021. [Google Scholar]
- Swerdlow, S.H.; Campo, E.; Pileri, S.A.; Harris, N.L.; Stein, H.; Siebert, R.; Advani, R.; Ghielmini, M.; Salles, G.A.; Zelenetz, A.D.; et al. The 2016 revision of the World Health Organization classification of lymphoid neoplasms. Blood 2016, 127, 2375–2390. [Google Scholar] [CrossRef] [Green Version]
- Freedman, A.S.; Aster, J.C. Prognosis of diffuse large B cell lymphoma. In UpToDate; Lister, A., Rosmarin, A.G., Eds.; UpToDate: Waltham, MA, USA, 2021. [Google Scholar]
- IBM Corporation. IBM SPSS Neural Networks. New Tools for Building Predictive Models; IBM Corporation: Armonk, NY, USA; New York, NY, USA, 2011. [Google Scholar]
- IBM Corporation. IBM SPSS Neural Networks 26. IBM SPSS Statistics 26 Documentation. Document Number 874712. Modified Date: 26 May 2021. Available online: https://www.ibm.com/support/pages/node/874712 (accessed on 9 September 2021).
- Ullah, I.; Manzo, M.; Shah, M.; Madden, M. Graph Convolutional Networks: Analysis, improvements and results. arXiv 2019, arXiv:1912.09592. [Google Scholar]
- Breen, K.H.; James, S.C.; White, J.D.; Allen, P.M.; Arnold, J.G. A Hybrid Artificial Neural Network to Estimate Soil Moisture Using SWAT+ and SMAP Data. Mach. Learn. Knowl. Extr. 2020, 2, 283–306. [Google Scholar] [CrossRef]
- Lin, H.; Zheng, W.; Peng, X. Orientation-Encoding CNN for Point Cloud Classification and Segmentation. Mach. Learn. Knowl. Extr. 2021, 3, 601–614. [Google Scholar] [CrossRef]
- Mayr, F.; Yovine, S.; Visca, R. Property Checking with Interpretable Error Characterization for Recurrent Neural Networks. Mach. Learn. Knowl. Extr. 2021, 3, 205–227. [Google Scholar] [CrossRef]
- Pickens, A.; Sengupta, S. Benchmarking Studies Aimed at Clustering and Classification Tasks Using K-Means, Fuzzy C-Means and Evolutionary Neural Networks. Mach. Learn. Knowl. Extr. 2021, 3, 695–719. [Google Scholar] [CrossRef]
- Shah, S.A.A.; Manzoor, M.A.; Bais, A. Canopy Height Estimation at Landsat Resolution Using Convolutional Neural Networks. Mach. Learn. Knowl. Extr. 2020, 2, 23–36. [Google Scholar] [CrossRef] [Green Version]
- Silva Araújo, V.J.; Guimarães, A.J.; de Campos Souza, P.V.; Rezende, T.S.; Araújo, V.S. Using Resistin, Glucose, Age and BMI and Pruning Fuzzy Neural Network for the Construction of Expert Systems in the Prediction of Breast Cancer. Mach. Learn. Knowl. Extr. 2019, 1, 466–482. [Google Scholar] [CrossRef] [Green Version]
- Škrlj, B.; Kralj, J.; Lavrač, N.; Pollak, S. Towards Robust Text Classification with Semantics-Aware Recurrent Neural Architecture. Mach. Learn. Knowl. Extr. 2019, 1, 575–589. [Google Scholar] [CrossRef] [Green Version]
- Ma, M.C.J.; Tadros, S.; Bouska, A.; Heavican, T.; Yang, H.; Deng, Q.; Moore, D.; Akhter, A.; Hartert, K.; Jain, N.; et al. Subtype-specific and co-occurring genetic alterations in B-cell non-Hodgkin lymphoma. Haematologica 2021. [Google Scholar] [CrossRef]
- Aguirre-Gamboa, R.; Gomez-Rueda, H.; Martinez-Ledesma, E.; Martinez-Torteya, A.; Chacolla-Huaringa, R.; Rodriguez-Barrientos, A.; Tamez-Pena, J.G.; Trevino, V. SurvExpress: An online biomarker validation tool and database for cancer gene expression data using survival analysis. PLoS ONE 2013, 8, e74250. [Google Scholar] [CrossRef] [Green Version]
- Carreras, J.; Kikuti, Y.Y.; Miyaoka, M.; Hiraiwa, S.; Tomita, S.; Ikoma, H.; Kondo, Y.; Ito, A.; Shiraiwa, S.; Hamoudi, R.; et al. A Single Gene Expression Set Derived from Artificial Intelligence Predicted the Prognosis of Several Lymphoma Subtypes; and High Immunohistochemical Expression of TNFAIP8 Associated with Poor Prognosis in Diffuse Large B-Cell Lymphoma. AI 2020, 1, 342–360. [Google Scholar] [CrossRef]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carreras, J.; Hamoudi, R.; Nakamura, N. Artificial Intelligence Analysis of Gene Expression Data Predicted the Prognosis of Patients with Diffuse Large B-Cell Lymphoma. Tokai J. Exp. Clin. Med. 2020, 45, 37–48. Available online: http://mj-med-u-tokai.com/pdf/450107.pdf (accessed on 9 September 2021). [PubMed]
- Carreras, J.; Kikuti, Y.Y.; Miyaoka, M.; Hiraiwa, S.; Tomita, S.; Ikoma, H.; Kondo, Y.; Ito, A.; Nakamura, N.; Hamoudi, R. Artificial Intelligence Analysis of the Gene Expression of Follicular Lymphoma Predicted the Overall Survival and Correlated with the Immune Microenvironment Response Signatures. Mach. Learn. Knowl. Extr. 2020, 2, 647–671. [Google Scholar] [CrossRef]
- Carreras, J.; Kikuti, Y.Y.; Miyaoka, M.; Hiraiwa, S.; Tomita, S.; Ikoma, H.; Kondo, Y.; Ito, A.; Nakamura, N.; Hamoudi, R. A Combination of Multilayer Perceptron, Radial Basis Function Artificial Neural Networks and Machine Learning Image Segmentation for the Dimension Reduction and the Prognosis Assessment of Diffuse Large B-Cell Lymphoma. AI 2021, 2, 106–134. [Google Scholar] [CrossRef]
- Carreras, J.; Kikuti, Y.Y.; Miyaoka, M.; Roncador, G.; Garcia, J.F.; Hiraiwa, S.; Tomita, S.; Ikoma, H.; Kondo, Y.; Ito, A.; et al. Integrative Statistics, Machine Learning and Artificial Intelligence Neural Network Analysis Correlated CSF1R with the Prognosis of Diffuse Large B-Cell Lymphoma. Hemato 2021, 2, 182–206. [Google Scholar] [CrossRef]
- Carreras, J.; Kikuti, Y.Y.; Roncador, G.; Miyaoka, M.; Hiraiwa, S.; Tomita, S.; Ikoma, H.; Kondo, Y.; Ito, A.; Shiraiwa, S.; et al. High Expression of Caspase-8 Associated with Improved Survival in Diffuse Large B-Cell Lymphoma: Machine Learning and Artificial Neural Networks Analyses. BioMedInformatics 2021, 1, 18–46. [Google Scholar] [CrossRef]
- UniProt, C. UniProt: The universal protein knowledgebase in 2021. Nucleic Acids Res. 2021, 49, D480–D489. [Google Scholar] [CrossRef]
- Stelzer, G.; Rosen, N.; Plaschkes, I.; Zimmerman, S.; Twik, M.; Fishilevich, S.; Stein, T.I.; Nudel, R.; Lieder, I.; Mazor, Y.; et al. The GeneCards Suite: From Gene Data Mining to Disease Genome Sequence Analyses. Curr. Protoc. Bioinform. 2016, 54, 1.30.1–1.30.33. [Google Scholar] [CrossRef]
- Matas-Cespedes, A.; Rodriguez, V.; Kalko, S.G.; Vidal-Crespo, A.; Rosich, L.; Casserras, T.; Balsas, P.; Villamor, N.; Gine, E.; Campo, E.; et al. Disruption of follicular dendritic cells-follicular lymphoma cross-talk by the pan-PI3K inhibitor BKM120 (Buparlisib). Clin. Cancer Res. 2014, 20, 3458–3471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawaguchi, Y.; Saito, B.; Nakata, A.; Matsui, T.; Sasaki, Y.; Shimada, S.; Abe, M.; Watanuki, M.; Baba, Y.; Murai, S.; et al. Elevated C-reactive protein level is associated with poor prognosis in follicular lymphoma patients undergoing rituximab-containing chemotherapy. Int. J. Hematol. 2020, 112, 341–348. [Google Scholar] [CrossRef] [PubMed]
- Arlauckas, S.P.; Garren, S.B.; Garris, C.S.; Kohler, R.H.; Oh, J.; Pittet, M.J.; Weissleder, R. Arg1 expression defines immunosuppressive subsets of tumor-associated macrophages. Theranostics 2018, 8, 5842–5854. [Google Scholar] [CrossRef] [PubMed]
- Valero, J.G.; Matas-Cespedes, A.; Arenas, F.; Rodriguez, V.; Carreras, J.; Serrat, N.; Guerrero-Hernandez, M.; Yahiaoui, A.; Balague, O.; Martin, S.; et al. The receptor of the colony-stimulating factor-1 (CSF-1R) is a novel prognostic factor and therapeutic target in follicular lymphoma. Leukemia 2021. [Google Scholar] [CrossRef] [PubMed]
- Metzger, K.; Moratin, J.; Freier, K.; Hoffmann, J.; Zaoui, K.; Plath, M.; Stogbauer, F.; Freudlsperger, C.; Hess, J.; Horn, D. A six-gene expression signature related to angiolymphatic invasion is associated with poor survival in laryngeal squamous cell carcinoma. Eur. Arch. Otorhinolaryngol. 2021, 278, 1199–1207. [Google Scholar] [CrossRef]
- Romano, A.; Parrinello, N.L.; Chiarenza, A.; Motta, G.; Tibullo, D.; Giallongo, C.; La Cava, P.; Camiolo, G.; Puglisi, F.; Palumbo, G.A.; et al. Immune off-target effects of Brentuximab Vedotin in relapsed/refractory Hodgkin Lymphoma. Br. J. Haematol. 2019, 185, 468–479. [Google Scholar] [CrossRef]
- Visser, H.P.; Tewis, M.; Willemze, R.; Kluin-Nelemans, J.C. Mantle cell lymphoma proliferates upon IL-10 in the CD40 system. Leukemia 2000, 14, 1483–1489. [Google Scholar] [CrossRef] [Green Version]
- Takimoto-Shimomura, T.; Tsukamoto, T.; Maegawa, S.; Fujibayashi, Y.; Matsumura-Kimoto, Y.; Mizuno, Y.; Chinen, Y.; Shimura, Y.; Mizutani, S.; Horiike, S.; et al. Dual targeting of bromodomain-containing 4 by AZD5153 and BCL2 by AZD4320 against B-cell lymphomas concomitantly overexpressing c-MYC and BCL2. Investig. New Drugs 2019, 37, 210–222. [Google Scholar] [CrossRef]
- Shaw, K.T.; Greig, N.H. Chemokine receptor mRNA expression at the in vitro blood-brain barrier during HIV infection. Neuroreport 1999, 10, 53–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trentin, L.; Cabrelle, A.; Facco, M.; Carollo, D.; Miorin, M.; Tosoni, A.; Pizzo, P.; Binotto, G.; Nicolardi, L.; Zambello, R.; et al. Homeostatic chemokines drive migration of malignant B cells in patients with non-Hodgkin lymphomas. Blood 2004, 104, 502–508. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| MLP | All Genes Set (n = 20,863) | Cancer Transcriptome Panel 1 (n = 1769) |
|---|---|---|
| Case processing summary | ||
| Training | 199 (68.6%) | 199 (68.6%) |
| Testing | 91 (31.4%) | 91 (31.4%) |
| Valid | 290 (100%) | 290 (100%) |
| Network information | ||
| Input layer | ||
| Covariates | 20,863 | 1769 |
| Units | 20,863 | 1769 |
| Rescaling | Standardized | Standardized |
| Hidden layer | ||
| Number | 1 | 1 |
| Units | 12 | 16 |
| Activation function | Hyperbolic tangent | Hyperbolic tangent |
| Output layer | ||
| Dependent variable | 1, Subtype | 1, Subtype |
| Units | 5 | 5 |
| Activation function | Softmax | Softmax |
| Error function | Cross-entropy | Cross-entropy |
| Model summary | ||
| Training | ||
| Cross-entropy error | 147.201 | 34.967 |
| Incorrect predictions | 28.1% | 5.5% |
| Stopping rule | 1 consecutive step with no decrease in error | 1 consecutive step with no decrease in error |
| Time | 0:02:04.01 | 0:00:08.77 |
| Testing | ||
| Cross-entropy error | 78.305 | 56.043 |
| Incorrect predictions | 35.2% | 24.2% |
| Classification 2 | ||
| Training | 71.9% | 94.5% |
| Testing | 64.8% | 75.8% |
| Area under the curve | ||
| FL | 0.911 | 0.991 |
| MCL | 0.927 | 0.987 |
| DLBCL | 0.899 | 0.977 |
| BL | 0.947 | 0.990 |
| MZL | 0.872 | 0.989 |
| Gene | NI | Keyword | Function 1 |
|---|---|---|---|
| LCE2B | 1 | 1q21 | Late cornified envelope protein 2B, belongs to the LCE cluster on 1q21 |
| KNG1 | 0.909 | Apoptosis | Kininogen-1, negative regulation of cell adhesion, positive regulation of apoptotic process |
| IGHV7_81 | 0.863 | B-cell receptor | Probable nonfunctional immunoglobulin heavy variable 7-81, B-cell receptor signaling pathway, phagocytosis |
| TG | 0.842 | Hormone | Thyroglobulin, hormone activity |
| C6 | 0.837 | Membrane attack complex | Complement component C6, constituent of membrane attack complex (MAC), adaptive immune response by forming process |
| FGB | 0.834 | Apoptosis | Fibrinogen beta chain, blood coagulation, adaptive immune response, positive regulation ERK1/ERK2 cascade, negative regulation of extrinsic apoptotic signaling pathway |
| ZNF750 | 0.828 | RNA pol. | Zinc finger protein 750, regulation of RNA polymerase II |
| CTSV | 0.819 | MHC II | Cathepsin L2, cysteine protease, antigen processing and presentation of exogenous peptide antigen via MHC class II |
| INGX | 0.818 | Tumor suppressor gene | Inhibitor of growth family, X-linked (Pseudogene), ING1-like tumor suppressor protein |
| COL4A6 | 0.816 | Extracellular matrix | Collagen alpha-6 (IV) chain, extracellular structural constituent |
| ZG16B | 0.816 | Carbohydrate | Zymogen granule protein 16 homolog B, carbohydrate binding |
| SERPINB13 | 0.811 | Apoptosis | Serpin B13, negative regulation of endopeptidase activity and apoptotic process |
| TKTL1 | 0.809 | Metabolism | Transketolase-like protein 1, glucose metabolism |
| TPPP3 | 0.808 | Microtubule | Tubulin polymerization-promoting protein family member 3, microtubule-binding activity |
| PRL | 0.797 | Apoptosis | Prolactin, growth regulator, suppression of apoptosis |
| MYOM2 | 0.795 | Actin | Myomesin-2, actin filament binding, muscle contraction |
| EGF | 0.795 | Cell growth | Epidermal growth factor, plays an important role in the growth, proliferation, and differentiation of numerous cell types |
| VAT1L | 0.782 | Zinc | Synaptic vesicle membrane protein VAT-1 homolog-like, oxidoreductase activity, zinc ion binding |
| HTN1 | 0.775 | Humoral response | Histatin-1, antimicrobial humoral response |
| RBM20 | 0.770 | RNA splicing | RNA-binding protein 20, positive regulation of RNA splicing |
| Gene | NI | Function 1 |
|---|---|---|
| ARG1 | 1 | Arginase-1, critical regulator of innate and adaptive immune responses, T-cell and NK-cells suppression |
| MAGEA3 | 0.996 | Melanoma-associated antigen 3, tumor progression, negative regulation of endoplasmic reticulum stress-induced intrinsic apoptosis |
| AKT2 | 0.956 | RAC-beta serine/threonine-protein kinase, ATP binding, cell cycle, cell migration, apoptosis, B-cell signaling, glucose metabolism |
| IL1B | 0.935 | Interleukin-1 beta, potent proinflammatory cytokine |
| S100A7A | 0.925 | Protein S100A7A, calcium-dependent protein binding |
| CLEC5A | 0.898 | C-type lectin domain family 5 member A, recruitment of macrophages and neutrophils, proinflammatory cytokine release |
| WIF1 | 0.894 | Wnt inhibitory factor 1, negative regulation of Wnt signaling pathway |
| TREM1 | 0.884 | Triggering receptor expressed on myeloid cells 1, regulation of innate and humoral immune responses, amplification of immune response |
| DEFB1 | 0.874 | Beta-defensin 1, innate immune response |
| GAGE1 | 0.865 | G antigen 1, antigen recognized by autologous cytolytic T-lymphocytes (melanoma) |
| CALML3 | 0.862 | Calmodulin-like protein 3, calcium ion binding |
| CXCL8 | 0.856 | Interleukin-8, chemotaxis (neutrophils, basophils, T-cells) |
| CRP | 0.849 | C-reactive protein, host defense, acute-phase response, inflammatory response |
| APOA2 | 0.848 | Apolipoprotein A-II, cholesterol metabolic process, the negative regulation of cytokine production involved in immune response |
| FCER1A | 0.845 | High-affinity immunoglobulin epsilon receptor subunit alpha, binding to the FC region of IG epsilon, initiation of allergic responses |
| LCN2 | 0.843 | Neutrophil gelatinase-associated lipocalin, apoptosis, and innate immunity |
| PGF | 0.834 | Prostaglandin F2-alpha receptor, response to estradiol, inflammatory response, positive regulation of apoptotic process, positive regulation of gene expression |
| HOXA9 | 0.827 | Homeobox protein Hox-A9, endothelial cell activation during inflammation |
| FLT3 | 0.817 | Receptor-type tyrosine-protein kinase FLT3, MAPK cascade, regulation of apoptosis, lymphocyte activation |
| IL13RA2 | 0.816 | Interleukin-13 receptor subunit alpha-2, cytokine-mediated signaling pathway, negative regulation of immunoglobulin production |
| MLP | FL vs. Others | MCL vs. Others | DLBCL vs. Others | BL vs. Others | MZL vs. Others |
|---|---|---|---|---|---|
| Case processing summary | |||||
| Training | 212 (73.1%) | 200 (69%) | 198 (68.3%) | 199 (68.6%) | 206 (71%) |
| Testing | 78 (26.9%) | 90 (31%) | 92 (31.7%) | 91 (31.4%) | 84 (29%) |
| Valid | 290 (100%) | 290 (100%) | 290 (100%) | 290 (100%) | 290 (100%) |
| Network information | |||||
| Input layer | |||||
| Covariates | 20,863 | 20,863 | 20,863 | 20,863 | 20,863 |
| Units | 20,863 | 20,863 | 20,863 | 20,863 | 20,863 |
| Rescaling | Standardized | Standardized | Standardized | Standardized | Standardized |
| Hidden layer | |||||
| Number | 1 | 1 | 1 | 1 | 1 |
| Units | 13 | 11 | 15 | 7 | 12 |
| Activation function | Hyperbolic tangent | Hyperbolic tangent | Hyperbolic tangent | Hyperbolic tangent | Hyperbolic tangent |
| Output layer | |||||
| Dependent variable | 1, Subtype | 1, Subtype | 1, Subtype | 1, Subtype | 1, Subtype |
| Units | 2 | 2 | 2 | 2 | 2 |
| Activation function | Softmax | Softmax | Softmax | Softmax | Softmax |
| Error function | Cross-entropy | Cross-entropy | Cross-entropy | Cross-entropy | Cross-entropy |
| Model summary | |||||
| Training | |||||
| Cross-entropy error | 49.720 | 38.996 | 59.031 | 29.720 | 10.144 |
| Incorrect predictions | 7.5% | 6.5% | 13.1% | 6.0% | 1.5% |
| Stopping rule | 1 consecutive step with no decrease in error | 1 consecutive step with no decrease in error | 1 consecutive step with no decrease in error | 1 consecutive step with no decrease in error | 1 consecutive step with no decrease in error |
| Time | 0:02:17.43 | 0:01:58.26 | 0:02:07.45 | 0:02:09.94 | 0:02:14.59 |
| Testing | |||||
| Cross-entropy error | 12.743 | 12.635 | 33.489 | 16.744 | 10.506 |
| Incorrect predictions | 3.8% | 5.6% | 17.4% | 4.4% | 6.0% |
| Classification 1 | |||||
| Training | 92.5% | 93.5% | 86.9% | 94.0% | 98.5% |
| Testing | 96.2% | 94.4% | 82.6% | 95.6% | 94% |
| Area under the curve | |||||
| FL | 0.955 | 0.941 | 0.927 | 0.976 | 0.990 |
| Others | 0.955 | 0.941 | 0.927 | 0.976 | 0.990 |
| MLP | FL vs. Others 1 | MCL vs. Others | DLBCL vs. Others | BL vs. Others | MZL vs. Others |
|---|---|---|---|---|---|
| Case processing summary | |||||
| Training | 212 (73.1%) | 200 (69.0%) | 198 (68.3%) | 199 (68.6%) | 206 (71.0%) |
| Testing | 78 (26.9%) | 90 (31.0%) | 92 (31.7%) | 91 (31.4%) | 84 (29.0%) |
| Valid | 290 (100%) | 290 (100%) | 290 (100%) | 290 (100%) | 290 (100%) |
| Network information | |||||
| Input layer | |||||
| Covariates | 1769 | 1769 | 1769 | 1769 | 1769 |
| Units | 1769 | 1769 | 1769 | 1769 | 1769 |
| Rescaling | Standardized | Standardized | Standardized | Standardized | Standardized |
| Hidden layer | |||||
| Number | 1 | 1 | 1 | 1 | 1 |
| Units | 12 | 12 | 13 | 10 | 14 |
| Activation function | Hyperbolic tangent | Hyperbolic tangent | Hyperbolic tangent | Hyperbolic tangent | Hyperbolic tangent |
| Output layer | |||||
| Dependent variable | 1, Subtype | 1, Subtype | 1, Subtype | 1, Subtype | 1, Subtype |
| Units | 2 | 2 | 2 | 2 | 2 |
| Activation function | Softmax | Softmax | Softmax | Softmax | Softmax |
| Error function | Cross-entropy | Cross-entropy | Cross-entropy | Cross-entropy | Cross-entropy |
| Model summary | |||||
| Training | |||||
| Cross-entropy error | 40.509 | 34.655 | 47.814 | 16.855 | 6.660 |
| Incorrect predictions | 7.1% | 5.0% | 9.1% | 3.5% | 1.0% |
| Stopping rule | 1 consecutive step with no decrease in error | 1 consecutive step with no decrease in error | 1 consecutive step with no decrease in error | 1 consecutive step with no decrease in error | 1 consecutive step with no decrease in error |
| Time | 0:00:08.02 | 0:00:09.00 | 0:00:07.92 | 0:00:08.69 | 0:00:08.56 |
| Testing | |||||
| Cross-entropy error | 16.923 | 6.950 | 21.492 | 15.320 | 11.794 |
| Incorrect predictions | 9.0% | 3.3% | 7.6% | 8.8% | 7.1% |
| Classification 1 | |||||
| Training | 92.9% | 95.0% | 90.0% | 96.5% | 99.0% |
| Testing | 91.0% | 96.7% | 92.4% | 91.2% | 92.9% |
| Area under the curve | |||||
| FL | 0.964 | 0.970 | 0.964 | 0.990 | 0.993 |
| Others | 0.964 | 0.970 | 0.964 | 0.990 | 0.993 |
| Subtype | Num. | p-Value | Hazard Risk | 95% CI | |
|---|---|---|---|---|---|
| Diffuse large B-cell lymphoma (DLBCL) | 414 | <0.0001 | 3.8 | 2.6 | 5.4 |
| Breast carcinoma | 962 | <0.0001 | 4.2 | 2.9 | 6.1 |
| Colorectal carcinoma | 466 | <0.0001 | 2.6 | 1.7 | 3.8 |
| Lung carcinoma | 650 | <0.0001 | 3.2 | 2.4 | 4.1 |
| Prostate adenocarcinoma | 497 | <0.0001 | 31.9 | 6.5 | 154.5 |
| Skin cutaneous melanoma | 335 | <0.0001 | 3.2 | 2.2 | 4.7 |
| Gastric adenocarcinoma + esophageal carcinoma | 440 | <0.0001 | 2.5 | 1.6 | 3.9 |
| Liver hepatocellular carcinoma | 361 | <0.0001 | 3.6 | 2.4 | 5.4 |
| Cervical carcinoma | 191 | <0.0001 | 6.7 | 3.3 | 13.8 |
| Thyroid papillary carcinoma | 489 | <0.0001 | 20.9 | 6.9 | 62.6 |
| Pancreatic ductal adenocarcinoma | 189 | <0.0001 | 3.4 | 2.2 | 5.2 |
| Kidney carcinoma | 792 | <0.0001 | 2.9 | 2.2 | 3.9 |
| Uterine corpus endometrioid carcinoma | 247 | <0.0001 | 8.7 | 3.9 | 18.9 |
| Head and neck carcinoma | 502 | <0.0001 | 2.3 | 1.7 | 3.0 |
| Central nervous system glioblastoma multiforme | 659 | <0.0001 | 3.8 | 2.7 | 5.3 |
| Ovarian serous carcinoma | 247 | <0.0001 | 3.6 | 2.3 | 5.7 |
| All cases | 7441 | <0.0001 | 3.6 | 3.3 | 3.9 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carreras, J.; Hamoudi, R. Artificial Neural Network Analysis of Gene Expression Data Predicted Non-Hodgkin Lymphoma Subtypes with High Accuracy. Mach. Learn. Knowl. Extr. 2021, 3, 720-739. https://doi.org/10.3390/make3030036
Carreras J, Hamoudi R. Artificial Neural Network Analysis of Gene Expression Data Predicted Non-Hodgkin Lymphoma Subtypes with High Accuracy. Machine Learning and Knowledge Extraction. 2021; 3(3):720-739. https://doi.org/10.3390/make3030036
Chicago/Turabian StyleCarreras, Joaquim, and Rifat Hamoudi. 2021. "Artificial Neural Network Analysis of Gene Expression Data Predicted Non-Hodgkin Lymphoma Subtypes with High Accuracy" Machine Learning and Knowledge Extraction 3, no. 3: 720-739. https://doi.org/10.3390/make3030036
APA StyleCarreras, J., & Hamoudi, R. (2021). Artificial Neural Network Analysis of Gene Expression Data Predicted Non-Hodgkin Lymphoma Subtypes with High Accuracy. Machine Learning and Knowledge Extraction, 3(3), 720-739. https://doi.org/10.3390/make3030036