
Central and Peripheral Immunity Responses in Parkinson’s Disease: An Overview and Update

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
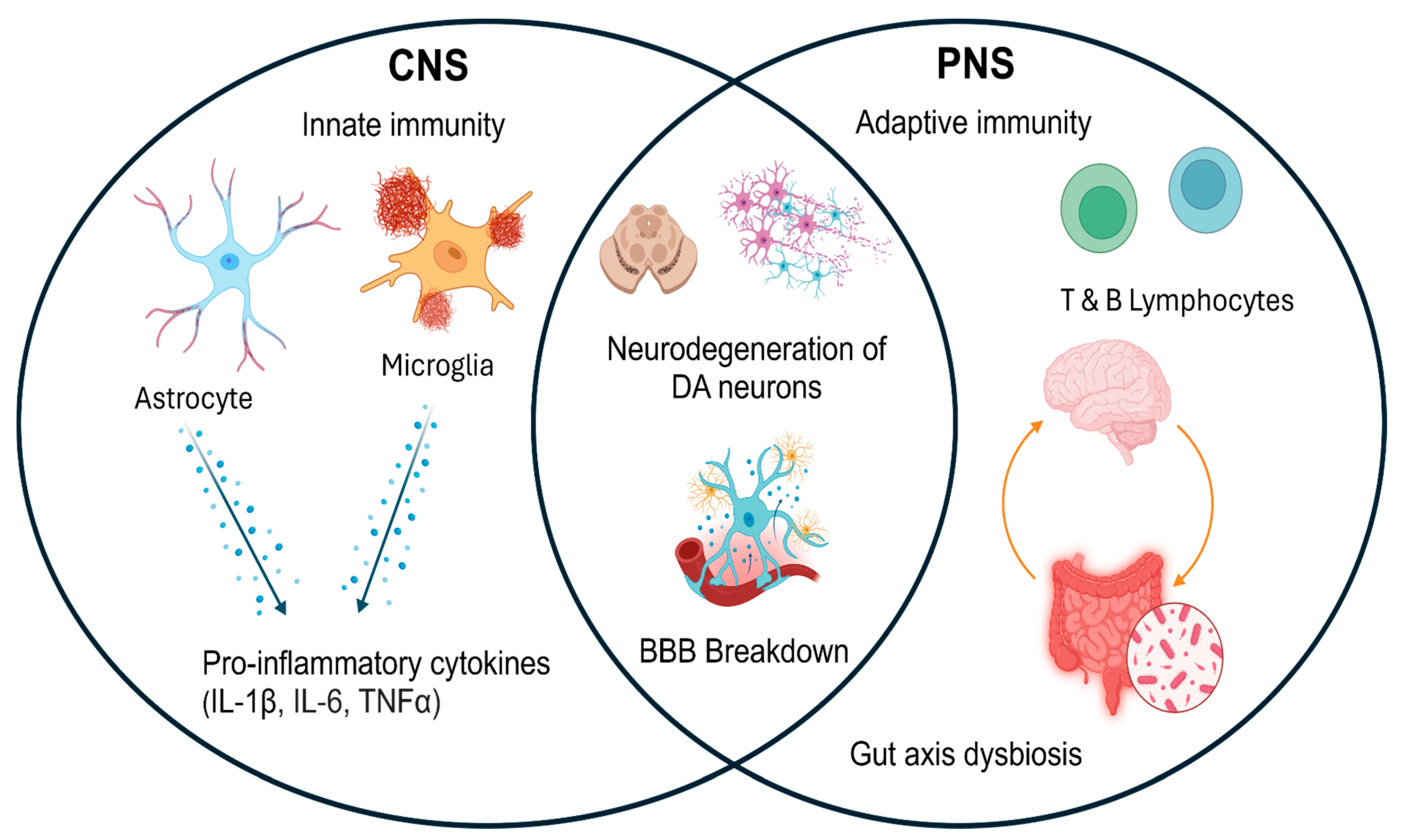
2. Bidirectional Central–Peripheral Immune Interplay
3. Peripheral Immune Dysregulation and Its Impact on PD Pathogenesis
4. Chronic Inflammation, Immune Senescence, and Central–Peripheral Immune Communication
5. Dopaminergic and Immunomodulatory Drug Therapies
6. Immune-Based Therapies in Clinical Trials
7. DBS and Immunomodulatory Effects
8. Integrating Immune Phenotyping in PD Management
9. Future Directions in Advancing Combination Therapies
10. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Chia, S.J.; Tan, E.K.; Chao, Y.X. Historical Perspective: Models of Parkinson’s Disease. Int. J. Mol. Sci. 2020, 21, 2464. [Google Scholar] [CrossRef] [PubMed]
- De Virgilio, A.; Greco, A.; Fabbrini, G.; Inghilleri, M.; Rizzo, M.I.; Gallo, A.; Conte, M.; Rosato, C.; Appiani, M.C.; de Vincentiis, M. Parkinson’s disease: Autoimmunity and neuroinflammation. Autoimmun. Rev. 2016, 15, 1005–1011. [Google Scholar] [CrossRef] [PubMed]
- Marogianni, C.; Sokratous, M.; Dardiotis, E.; Hadjigeorgiou, G.M.; Bogdanos, D.; Xiromerisiou, G. Neurodegeneration and Inflammation-An Interesting Interplay in Parkinson’s Disease. Int. J. Mol. Sci. 2020, 21, 8421. [Google Scholar] [CrossRef]
- Isik, S.; Yeman Kiyak, B.; Akbayir, R.; Seyhali, R.; Arpaci, T. Microglia Mediated Neuroinflammation in Parkinson’s Disease. Cells 2023, 12, 1012. [Google Scholar] [CrossRef] [PubMed]
- Araújo, B.; Caridade-Silva, R.; Soares-Guedes, C.; Martins-Macedo, J.; Gomes, E.D.; Monteiro, S.; Teixeira, F.G. Neuroinflammation and Parkinson’s Disease-From Neurodegeneration to Therapeutic Opportunities. Cells 2022, 11, 2908. [Google Scholar] [CrossRef]
- Zhu, B.; Yin, D.; Zhao, H.; Zhang, L. The immunology of Parkinson’s disease. Semin. Immunopathol. 2022, 44, 659–672. [Google Scholar] [CrossRef]
- Bartl, M.; Xylaki, M.; Bähr, M.; Weber, S.; Trenkwalder, C.; Mollenhauer, B. Evidence for immune system alterations in peripheral biological fluids in Parkinson’s disease. Neurobiol. Dis. 2022, 170, 105744. [Google Scholar] [CrossRef]
- Karpenko, M.N.; Vasilishina, A.A.; Gromova, E.A.; Muruzheva, Z.M.; Miliukhina, I.V.; Bernadotte, A. Interleukin-1β, interleukin-1 receptor antagonist, interleukin-6, interleukin-10, and tumor necrosis factor-α levels in CSF and serum in relation to the clinical diversity of Parkinson’s disease. Cell. Immunol. 2018, 327, 77–82. [Google Scholar] [CrossRef]
- Heidari, A.; Yazdanpanah, N.; Rezaei, N. The role of Toll-like receptors and neuroinflammation in Parkinson’s disease. J. Neuroinflamm. 2022, 19, 135. [Google Scholar] [CrossRef]
- Liu, T.W.; Chen, C.M.; Chang, K.H. Biomarker of Neuroinflammation in Parkinson’s Disease. Int. J. Mol. Sci. 2022, 23, 4148. [Google Scholar] [CrossRef]
- Mogi, M.; Harada, M.; Narabayashi, H.; Inagaki, H.; Minami, M.; Nagatsu, T. Interleukin (IL)-1 beta, IL-2, IL-4, IL-6 and transforming growth factor-alpha levels are elevated in ventricular cerebrospinal fluid in juvenile parkinsonism and Parkinson’s disease. Neurosci. Lett. 1996, 211, 13–16. [Google Scholar] [CrossRef] [PubMed]
- Harms, A.S.; Ferreira, S.A.; Romero-Ramos, M. Periphery and brain, innate and adaptive immunity in Parkinson’s disease. Acta Neuropathol. 2021, 141, 527–545. [Google Scholar] [CrossRef]
- Tan, E.K.; Chao, Y.X.; West, A.; Chan, L.L.; Poewe, W.; Jankovic, J. Parkinson disease and the immune system—Associations, mechanisms and therapeutics. Nat. Rev. Neurol. 2020, 16, 303–318. [Google Scholar] [CrossRef]
- Xiao, Y.; Wei, Q.; Ou, R.; Yang, T.; Jiang, Q.; Hou, Y.; Zhang, L.; Liu, K.; Wang, S.; Lin, J.; et al. Association between peripheral adaptive immune markers and disease progression in Parkinson’s disease. J. Neurol. 2023, 270, 4444–4450. [Google Scholar] [CrossRef]
- Yanamandra, K.; Gruden, M.A.; Casaite, V.; Meskys, R.; Forsgren, L.; Morozova-Roche, L.A. α-synuclein reactive antibodies as diagnostic biomarkers in blood sera of Parkinson’s disease patients. PLoS ONE 2011, 6, e18513. [Google Scholar] [CrossRef] [PubMed]
- Papachroni, K.K.; Ninkina, N.; Papapanagiotou, A.; Hadjigeorgiou, G.M.; Xiromerisiou, G.; Papadimitriou, A.; Kalofoutis, A.; Buchman, V.L. Autoantibodies to alpha-synuclein in inherited Parkinson’s disease. J. Neurochem. 2007, 101, 749–756. [Google Scholar] [CrossRef] [PubMed]
- McFarthing, K.; Buff, S.; Rafaloff, G.; Pitzer, K.; Fiske, B.; Navangul, A.; Beissert, K.; Pilcicka, A.; Fuest, R.; Wyse, R.K.; et al. Parkinson’s Disease Drug Therapies in the Clinical Trial Pipeline: 2024 Update. J. Parkinsons Dis. 2024, 14, 899–912. [Google Scholar] [CrossRef]
- Channer, B.; Matt, S.M.; Nickoloff-Bybel, E.A.; Pappa, V.; Agarwal, Y.; Wickman, J.; Gaskill, P.J. Dopamine, Immunity, and Disease. Pharmacol. Rev. 2023, 75, 62–158. [Google Scholar] [CrossRef]
- Matt, S.M.; Gaskill, P.J. Where Is Dopamine and how do Immune Cells See it?: Dopamine-Mediated Immune Cell Function in Health and Disease. J. Neuroimmune Pharmacol. 2020, 15, 114–164. [Google Scholar] [CrossRef]
- Levite, M. Dopamine and T cells: Dopamine receptors and potent effects on T cells, dopamine production in T cells, and abnormalities in the dopaminergic system in T cells in autoimmune, neurological and psychiatric diseases. Acta Physiol. 2016, 216, 42–89. [Google Scholar] [CrossRef]
- Sarkar, C.; Basu, B.; Chakroborty, D.; Dasgupta, P.S.; Basu, S. The immunoregulatory role of dopamine: An update. Brain Behav. Immun. 2010, 24, 525–528. [Google Scholar] [CrossRef]
- McFleder, R.L.; Musacchio, T.; Keller, J.; Knorr, S.; Petschner, T.; Chen, J.; Muthuraman, M.; Badr, M.; Harder-Rauschenberger, L.; Kremer, F.; et al. Deep brain stimulation halts Parkinson’s disease-related immune dysregulation in the brain and peripheral blood. Brain Behav. Immun. 2025, 123, 851–862. [Google Scholar] [CrossRef] [PubMed]
- Chang, L.; Dong, W.W.; Luo, B.; Qiu, C.; Lu, Y.; Lin, X.J.; Zhang, W.B. Deep brain stimulation improves central nervous system inflammation in Parkinson’s disease: Evidence and perspectives. CNS Neurosci. Ther. 2023, 29, 2177–2185. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Qi, B.; Xu, W.; Ma, B.; Li, L.; Chen, Q.; Qian, Q.; Liu, X.; Qu, H. Clinical correlation of peripheral CD4+-cell sub-sets, their imbalance and Parkinson’s disease. Mol. Med. Rep. 2015, 12, 6105–6111. [Google Scholar] [CrossRef]
- Saleh, M.; Markovic, M.; Olson, K.E.; Gendelman, H.E.; Mosley, R.L. Therapeutic Strategies for Immune Transformation in Parkinson’s Disease. J. Parkinsons Dis. 2022, 12, S201–S222. [Google Scholar] [CrossRef]
- Bachiller, S.; Jiménez-Ferrer, I.; Paulus, A.; Yang, Y.; Swanberg, M.; Deierborg, T.; Boza-Serrano, A. Microglia in Neurological Diseases: A Road Map to Brain-Disease Dependent-Inflammatory Response. Front. Cell. Neurosci. 2018, 12, 488. [Google Scholar] [CrossRef]
- Li, Q.; Barres, B.A. Microglia and macrophages in brain homeostasis and disease. Nat. Rev. Immunol. 2018, 18, 225–242. [Google Scholar] [CrossRef] [PubMed]
- Harms, A.S.; Cao, S.; Rowse, A.L.; Thome, A.D.; Li, X.; Mangieri, L.R.; Cron, R.Q.; Shacka, J.J.; Raman, C.; Standeart, D.G. MHCII is required for α-synuclein-induced activation of microglia, CD4 T cell proliferation, and dopaminergic neurodegeneration. J. Neurosci. 2013, 33, 9592–9600. [Google Scholar] [CrossRef]
- Almolda, B.; González, B.; Castellano, B. Are Microglial Cells the Regulators of Lymphocyte Responses in the CNS? Front. Cell. Neurosci. 2015, 9, 440. [Google Scholar] [CrossRef]
- McGeer, P.L.; Itagaki, S.; Boyes, B.E.; McGeer, E.G. Reactive microglia are positive for HLA-DR in the substantia nigra of Parkinson’s and Alzheimer’s disease brains. Neurology 1988, 38, 1285–1291. [Google Scholar] [CrossRef]
- Sawada, M.; Imamura, K.; Nagatsu, T. Role of cytokines in inflammatory process in Parkinson’s disease. J. Neural Transm. Suppl. 2006, 70, 373–381. [Google Scholar] [CrossRef]
- Gray, M.T.; Woulfe, J.M. Striatal blood-brain barrier permeability in Parkinson’s disease. J. Cereb. Blood Flow. Metab. 2015, 35, 747–750. [Google Scholar] [CrossRef] [PubMed]
- Al-Bachari, S.; Vidyasagar, R.; Emsley, H.C.; Parkes, L.M. Structural and physiological neurovascular changes in idiopathic Parkinson’s disease and its clinical phenotypes. J. Cereb. Blood Flow. Metab. 2017, 37, 3409–3421. [Google Scholar] [CrossRef]
- Brochard, V.; Combadière, B.; Prigent, A.; Laouar, Y.; Perrin, A.; Beray-Berthat, V.; Bonduelle, O.; Alvarez-Fischer, D.; Callebert, J.; Launay, J.-M.; et al. Infiltration of CD4+ lymphocytes into the brain contributes to neurodegeneration in a mouse model of Parkinson disease. J. Clin. Investig. 2009, 119, 182–192. [Google Scholar] [CrossRef] [PubMed]
- Ho, M.S. Microglia in Parkinson’s Disease. Adv. Exp. Med. Biol. 2019, 1175, 335–353. [Google Scholar] [CrossRef]
- Houser, M.C.; Chang, J.; Factor, S.A.; Molho, E.S.; Zabetian, C.P.; Hill-Burns, E.M.; Payami, H.; Hertzberg, V.S.; Tansey, M.G. Stool Immune Profiles Evince Gastrointestinal Inflammation in Parkinson’s Disease. Mov. Disord. 2018, 33, 793–804. [Google Scholar] [CrossRef]
- Gopinath, A.; Mackie, P.; Hashimi, B.; Buchanan, M.A.; Smith, A.R.; Saadatpour, L.; Gittis, A.; Ramirez-Zamora, A.; Okun, M.S.; Streit, W.J.; et al. DAT and TH expression marks human Parkinson’s disease in peripheral immune cells. NPJ Parkinsons Dis. 2022, 8, 72. [Google Scholar] [CrossRef]
- Mackie, P.; Lebowitz, J.; Saadatpour, L.; Nickoloff, E.; Gaskill, P.; Khoshbouei, H. The dopamine transporter: An unrecognized nexus for dysfunctional peripheral immunity and signaling in Parkinson’s Disease. Brain Behav. Immun. 2018, 70, 21–35. [Google Scholar] [CrossRef]
- Butler, B.; Saha, K.; Rana, T.; Becker, J.P.; Sambo, D.; Davari, P.; Goodwin, J.S.; Khoshbouei, H. Dopamine Transporter Activity Is Modulated by α-Synuclein. J. Biol. Chem. 2015, 290, 29542–29554. [Google Scholar] [CrossRef]
- Swant, J.; Goodwin, J.S.; North, A.; Ali, A.A.; Gamble-George, J.; Chirwa, S.; Khoshbouei, H. α-Synuclein stimulates a dopamine transporter-dependent chloride current and modulates the activity of the transporter. J. Biol. Chem. 2011, 286, 43933–43943. [Google Scholar] [CrossRef]
- Yoo, H.S.; Chung, S.J.; Chung, S.J.; Moon, H.; Oh, J.S.; Kim, J.S.; Hong, J.Y.; Ye, B.S.; Sohn, Y.H.; Lee, P.H. Presynaptic dopamine depletion determines the timing of levodopa-induced dyskinesia onset in Parkinson’s disease. Eur. J. Nucl. Med. Mol. Imaging. 2018, 45, 423–431. [Google Scholar] [CrossRef]
- Marek, K.; Innis, R.; van Dyck, C.; Fussell, B.; Early, M.; Eberly, S.; Oakes, D.; Seibyl, J. [123I]beta-CIT SPECT imaging assessment of the rate of Parkinson’s disease progression. Neurology. 2001, 57, 2089–2094. [Google Scholar] [CrossRef] [PubMed]
- Savitt, D.; Jankovic, J. Targeting α-Synuclein in Parkinson’s Disease: Progress Towards the Development of Disease-Modifying Therapeutics. Drugs 2019, 79, 797–810. [Google Scholar] [CrossRef]
- Liu, Y.; Xie, X.; Xia, L.-P.; Lv, H.; Lou, F.; Ren, Y.; He, Z.-Y.; Luo, X.-G. Peripheral immune tolerance alleviates the intracranial lipopolysaccharide injection-induced neuroinflammation and protects the dopaminergic neurons from neuroinflammation-related neurotoxicity. J. Neuroinflamm. 2017, 14, 223. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Liu, Z.; Cao, B.B.; Qiu, Y.H.; Peng, Y.P. Treg Cells Protect Dopaminergic Neurons against MPP+ Neurotoxicity via CD47-SIRPA Interaction. Cell Physiol. Biochem. 2017, 41, 1240–1254. [Google Scholar] [CrossRef]
- Perry, V.H.; Holmes, C. Microglial priming in neurodegenerative disease. Nat. Rev. Neurol. 2014, 10, 217–224. [Google Scholar] [CrossRef] [PubMed]
- Xie, X.; Luo, X.; Liu, N.; Li, X.; Lou, F.; Zheng, Y.; Ren, Y. Monocytes, microglia, and CD200-CD200R1 signaling are essential in the transmission of inflammation from the periphery to the central nervous system. J. Neurochem. 2017, 141, 222–235. [Google Scholar] [CrossRef]
- Takata, F.; Nakagawa, S.; Matsumoto, J.; Dohgu, S. Blood-Brain Barrier Dysfunction Amplifies the Development of Neuroinflammation: Understanding of Cellular Events in Brain Microvascular Endothelial Cells for Prevention and Treatment of BBB Dysfunction. Front. Cell. Neurosci. 2021, 15, 661838. [Google Scholar] [CrossRef]
- Rochfort, K.D.; Collins, L.E.; Murphy, R.P.; Cummins, P.M. Downregulation of blood-brain barrier phenotype by proinflammatory cytokines involves NADPH oxidase-dependent ROS generation: Consequences for interendothelial adherens and tight junctions. PLoS ONE 2014, 9, e101815. [Google Scholar] [CrossRef]
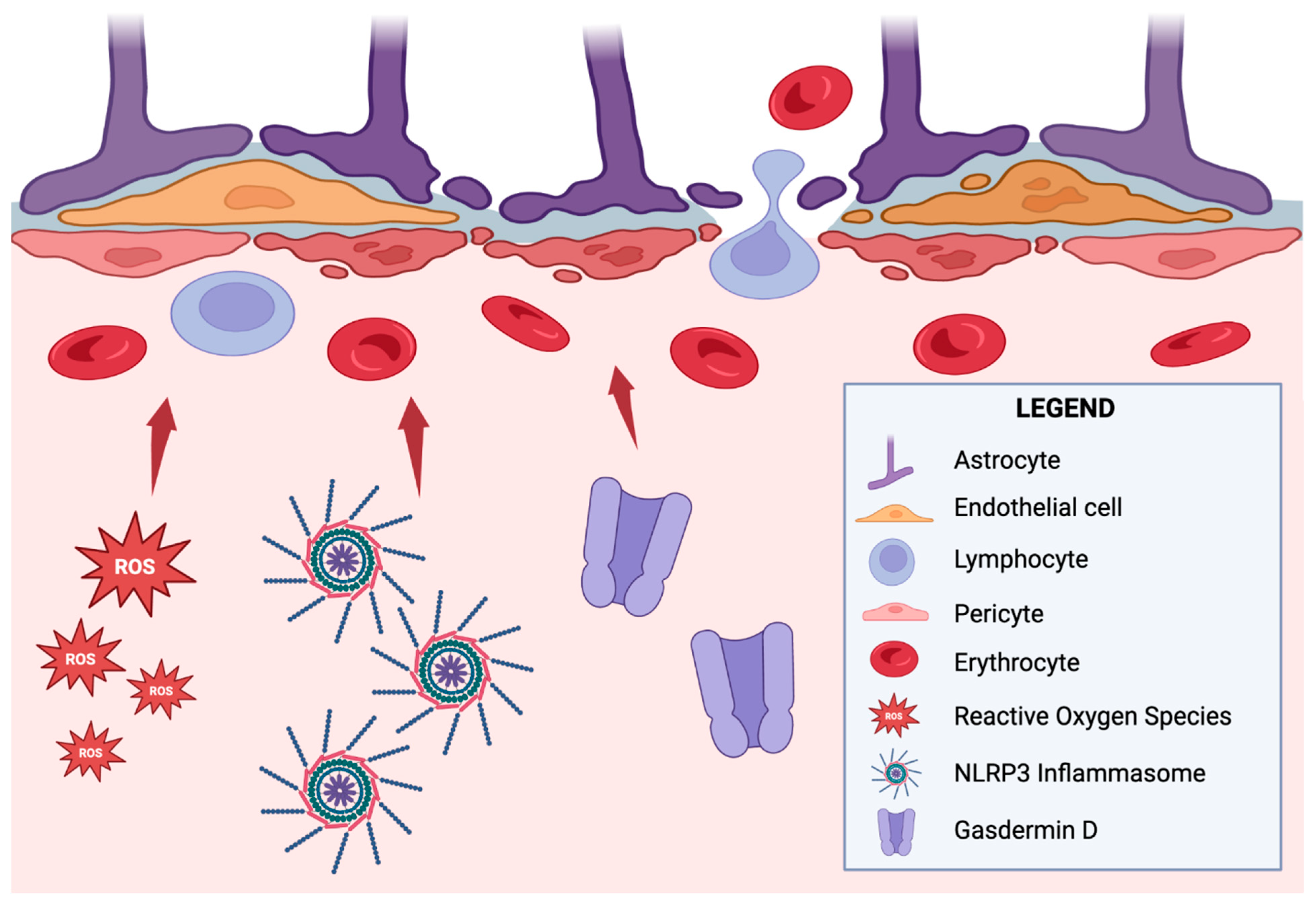
- Wei, C.; Jiang, W.; Wang, R.; Zhong, H.; He, H.; Gao, X.; Zhong, S.; Yu, F.; Guo, Q.; Zhang, L.; et al. Brain endothelial GSDMD activation mediates inflammatory BBB breakdown. Nature 2024, 629, 893–900. [Google Scholar] [CrossRef]
- Yoon, S.-H.; Kim, C.Y.; Lee, E.; Lee, C.; Lee, K.-S.; Lee, J.; Park, H.; Choi, B.; Hwang, I.; Kim, J.; et al. Microglial NLRP3-gasdermin D activation impairs blood-brain barrier integrity through interleukin-1β-independent neutrophil chemotaxis upon peripheral inflammation in mice. Nat. Commun. 2025, 16, 699. [Google Scholar] [CrossRef]
- Al-Bachari, S.; Naish, J.H.; Parker, G.J.M.; Emsley, H.C.A.; Parkes, L.M. Blood-Brain Barrier Leakage Is Increased in Parkinson’s Disease. Front. Physiol. 2020, 11, 593026. [Google Scholar] [CrossRef]
- Laurent, C.; Dorothée, G.; Hunot, S.; Martin, E.; Monnet, Y.; Duchamp, M.; Dong, Y.; Légeron, F.-P.; Leboucher, A.; Burnouf, S.; et al. Hippocampal T cell infiltration promotes neuroinflammation and cognitive decline in a mouse model of tauopathy. Brain 2017, 140, 184–200. [Google Scholar] [CrossRef]
- Mou, Y.; Du, Y.; Zhou, L.; Yue, J.; Hu, X.; Liu, Y.; Chen, S.; Lin, X.; Zhang, G.; Xiao, H.; et al. Gut Microbiota Interact With the Brain Through Systemic Chronic Inflammation: Implications on Neuroinflammation, Neurodegeneration, and Aging. Front. Immunol. 2022, 13, 796288. [Google Scholar] [CrossRef]
- Kearns, R. Gut-Brain Axis and Neuroinflammation: The Role of Gut Permeability and the Kynurenine Pathway in Neurological Disorders. Cell. Mol. Neurobiol. 2024, 44, 64. [Google Scholar] [CrossRef]
- Padhi, P.; Worth, C.; Zenitsky, G.; Jin, H.; Sambamurti, K.; Anantharam, V.; Kanthasamy, A.; Kanthasamy, A.G. Mechanistic Insights Into Gut Microbiome Dysbiosis-Mediated Neuroimmune Dysregulation and Protein Misfolding and Clearance in the Pathogenesis of Chronic Neurodegenerative Disorders. Front. Neurosci. 2022, 16, 836605. [Google Scholar] [CrossRef] [PubMed]
- Lukiw, W.J.; Arceneaux, L.; Li, W.; Bond, T.; Zhao, Y. Gastrointestinal (GI)-Tract Microbiome Derived Neurotoxins and their Potential Contribution to Inflammatory Neurodegeneration in Alzheimer’s Disease (AD). J. Alzheimers Dis. Parkinsonism. 2021, 11, 525. [Google Scholar]
- Hestad, K.; Alexander, J.; Rootwelt, H.; Aaseth, J.O. The Role of Tryptophan Dysmetabolism and Quinolinic Acid in Depressive and Neurodegenerative Diseases. Biomolecules 2022, 12, 998. [Google Scholar] [CrossRef]
- Sanjari Moghaddam, H.; Ghazi Sherbaf, F.; Mojtahed Zadeh, M.; Ashraf-Ganjouei, A.; Aarabi, M.H. Association Between Peripheral Inflammation and DATSCAN Data of the Striatal Nuclei in Different Motor Subtypes of Parkinson Disease. Front. Neurol. 2018, 9, 234. [Google Scholar] [CrossRef]
- Kim, R.; Kang, N.; Byun, K.; Park, K.; Jun, J.S. Prognostic significance of peripheral neutrophils and lymphocytes in early untreated Parkinson’s disease: An 8-year follow-up study. J. Neurol. Neurosurg. Psychiatry 2023, 94, 1040–1046. [Google Scholar] [CrossRef]
- Zhang, F.; Chen, B.; Ren, W.; Yan, Y.; Zheng, X.; Jin, S.; Chang, Y. Association analysis of dopaminergic degeneration and the neutrophil-to-lymphocyte ratio in Parkinson’s disease. Front. Aging Neurosci. 2024, 16, 1377994. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Delgado, L.; Labrador-Espinosa, M.Á.; Macías-García, D.; Jesús, S.; Zamora, B.B.; Fernández-Rodríguez, P.; Adarmes-Gómez, A.D.; Castillo, M.I.R.; Castro-Labrador, S.; Silva-Rodríguez, J.; et al. Peripheral Inflammation Is Associated with Dopaminergic Degeneration in Parkinson’s Disease. Mov. Disord. 2023, 38, 755–763. [Google Scholar] [CrossRef] [PubMed]
- Calabrese, V.; Santoro, A.; Monti, D.; Crupi, R.; di Paola, R.; Latteri, S.; Cuzzocrea, S.; Zappia, M.; Giordano, J.; Calabrese, E.J.; et al. Aging and Parkinson’s Disease: Inflammaging, neuroinflammation and biological remodeling as key factors in pathogenesis. Free Radic. Biol. Med. 2018, 115, 80–91. [Google Scholar] [CrossRef]
- Ziabska, K.; Ziemka-Nalecz, M.; Pawelec, P.; Sypecka, J.; Zalewska, T. Aberrant Complement System Activation in Neurological Disorders. Int. J. Mol. Sci. 2021, 22, 4675. [Google Scholar] [CrossRef] [PubMed]
- Weiss, F.; Labrador-Garrido, A.; Dzamko, N.; Halliday, G. Immune responses in the Parkinson’s disease brain. Neurobiol. Dis. 2022, 168, 105700. [Google Scholar] [CrossRef]
- Rauschenberger, L.; Behnke, J.; Grotemeyer, A.; Knorr, S.; Volkmann, J.; Ip, C.W. Age-dependent neurodegeneration and neuroinflammation in a genetic A30P/A53T double-mutated α-synuclein mouse model of Parkinson’s disease. Neurobiol. Dis. 2022, 171, 105798. [Google Scholar] [CrossRef]
- Zhou, Y.; Lu, M.; Du, R.-H.; Qiao, C.; Jiang, C.-Y.; Zhang, K.-Z.; Ding, J.-H.; Hu, G. MicroRNA-7 targets Nod-like receptor protein 3 inflammasome to modulate neuroinflammation in the pathogenesis of Parkinson’s disease. Mol. Neurodegener. 2016, 11, 28. [Google Scholar] [CrossRef]
- Chou, J.P.; Effros, R.B. T cell replicative senescence in human aging. Curr. Pharm. Des. 2013, 19, 1680–1698. [Google Scholar] [CrossRef]
- Williams-Gray, C.H.; Wijeyekoon, R.S.; Scott, K.M.; Hayat, S.; Barker, R.A.; Jones, J.L. Abnormalities of age-related T cell senescence in Parkinson’s disease. J. Neuroinflamm. 2018, 15, 166. [Google Scholar] [CrossRef]
- Rodriguez, M.; Rodriguez-Sabate, C.; Morales, I.; Sanchez, A.; Sabate, M. Parkinson’s disease as a result of aging. Aging Cell 2015, 14, 293–308. [Google Scholar] [CrossRef]
- Sakiyama, H.; Baba, K.; Kimura, Y.; Ogawa, K.; Nishiike, U.; Hayakawa, H.; Yoshida, M.; Aguirre, C.; Ikenaka, K.; Nagano, S.; et al. Accelerated senescence exacerbates α-synucleinopathy in senescence-accelerated prone 8 mice via persistent neuroinflammation. Neurochem. Int. 2025, 182, 105906. [Google Scholar] [CrossRef]
- Chinta, S.J.; Woods, G.; Demaria, M.; Rane, A.; Zou, Y.; McQuade, A.; Rajagopalan, S.; Limbad, C.; Madden, D.T.; Campisi, J.; et al. Cellular Senescence Is Induced by the Environmental Neurotoxin Paraquat and Contributes to Neuropathology Linked to Parkinson’s Disease. Cell Rep. 2018, 22, 930–940. [Google Scholar] [CrossRef]
- Pringsheim, T.; Day, G.S.; Smith, D.B.; Rae-Grant, A.; Licking, N.; Armstrong, M.J.; Roze, E.; Miyasaki, J.M.; Hauser, R.A.; Espay, A.J.; et al. Dopaminergic Therapy for Motor Symptoms in Early Parkinson Disease Practice Guideline Summary: A Report of the AAN Guideline Subcommittee. Neurology 2021, 97, 942–957. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, D.P.; Bortolanza, M.; Del Bel, E.A. Interferon-γ Involvement in the Neuroinflammation Associated with Parkinson’s Disease and L-DOPA-Induced Dyskinesia. Neurotox. Res. 2021, 39, 705–719. [Google Scholar] [CrossRef] [PubMed]
- Tran, T.N.; Vo, T.N.N.; Frei, K.; Truong, D.D. Levodopa-induced dyskinesia: Clinical features, incidence, and risk factors. J. Neural Transm. 2018, 125, 1109–1117. [Google Scholar] [CrossRef]
- Jones-Tabah, J.; Mohammad, H.; Hadj-Youssef, S.; Kim, L.E.H.; Martin, R.D.; Benaliouad, F.; Tanny, J.C.; Clarke, P.B.S.; Hébert, T.E. Dopamine D1 receptor signalling in dyskinetic Parkinsonian rats revealed by fiber photometry using FRET-based biosensors. Sci. Rep. 2020, 10, 14426. [Google Scholar] [CrossRef]
- Vega-Angeles, V.T.; Morales-Ruiz, V.; Adalid-Peralta, L.V. Immunomodulation as a treatment for parkinson’s disease in current trials: A systematic review and meta-analysis. Rev. Investig. Clin. 2024, 76, 159–169. [Google Scholar] [CrossRef]
- Investigators, N.N.-P. A randomized clinical trial of coenzyme Q10 and GPI-1485 in early Parkinson disease. Neurology 2007, 68, 20–28. [Google Scholar] [CrossRef]
- Writing Group for the NINDS Exploratory Trials in Parkinson Disease (NET-PD) Investigators; Kieburtz, K.; Tilley, B.C.; Elm, J.J.; Babcock, D.; Hauser, R.; Ross, G.W.; Augustine, A.H.; Augustine, E.U.; Aminoff, M.J.; et al. Effect of creatine monohydrate on clinical progression in patients with Parkinson disease: A randomized clinical trial. JAMA 2015, 313, 584–593. [Google Scholar] [CrossRef]
- Neurol, L. Pioglitazone in early Parkinson’s disease: A phase 2, multicentre, double-blind, randomised trial. Lancet Neurol. 2015, 14, 795–803. [Google Scholar] [CrossRef]
- Cankaya, S.; Cankaya, B.; Kilic, U.; Kilic, E.; Yulug, B. The therapeutic role of minocycline in Parkinson’s disease. Drugs Context. 2019, 8, 212553. [Google Scholar] [CrossRef] [PubMed]
- Richard, E.; Bloem, B.R. Monoclonal Antibodies in Neurodegenerative Disease May Work, But They Don’t Help: A Perspective from Physicians. J. Parkinsons Dis. 2022, 12, 2289–2291. [Google Scholar] [CrossRef] [PubMed]
- Games, D.; Valera, E.; Spencer, B.; Rockenstein, E.; Mante, M.; Adame, A.; Patrick, C.; Ubhi, K.; Nuber, S.; Sacayon, P.; et al. Reducing C-terminal-truncated alpha-synuclein by immunotherapy attenuates neurodegeneration and propagation in Parkinson’s disease-like models. J. Neurosci. 2014, 34, 9441–9454. [Google Scholar] [CrossRef] [PubMed]
- Pagano, G.; Taylor, K.I.; Anzures-Cabrera, J.; Marchesi, M.; Simuni, T.; Marek, K.; Postuma, R.B.; Pavese, N.; Stocchi, F.; Azulay, J.-P.; et al. Trial of Prasinezumab in Early-Stage Parkinson’s Disease. N. Engl. J. Med. 2022, 387, 421–432. [Google Scholar] [CrossRef]
- Weihofen, A.; Liu, Y.; Arndt, J.W.; Huy, C.; Quan, C.; Smith, B.A.; Baeriswyl, J.-L.; Cavegn, N.; Senn, L.; Su, L.; et al. Development of an aggregate-selective, human-derived α-synuclein antibody BIIB054 that ameliorates disease phenotypes in Parkinson’s disease models. Neurobiol. Dis. 2019, 124, 276–288. [Google Scholar] [CrossRef]
- Lang, A.E.; Siderowf, A.D.; Macklin, E.A.; Poewe, W.; Brooks, D.J.; Fernandez, H.H.; Rascol, O.; Giladi, N.; Stocchi, F.; Tanner, C.M.; et al. Trial of Cinpanemab in Early Parkinson’s Disease. N. Engl. J. Med. 2022, 387, 408–420. [Google Scholar] [CrossRef]
- Kharel, S.; Ojha, R. Future of Monoclonal Antibody Therapy in Parkinson’s Disease. Ann. Neurosci. 2023, 30, 8–10. [Google Scholar] [CrossRef]
- Hickey, P.; Stacy, M. Deep Brain Stimulation: A Paradigm Shifting Approach to Treat Parkinson’s Disease. Front. Neurosci. 2016, 10, 173. [Google Scholar] [CrossRef]
- Chiken, S.; Nambu, A. Mechanism of Deep Brain Stimulation: Inhibition, Excitation, or Disruption? Neuroscientist 2016, 22, 313–322. [Google Scholar] [CrossRef]
- Chiken, S.; Nambu, A. High-frequency pallidal stimulation disrupts information flow through the pallidum by GABAergic inhibition. J. Neurosci. 2013, 33, 2268–2280. [Google Scholar] [CrossRef]
- Reese, R.; Leblois, A.; Steigerwald, F.; Pötter-Nerger, M.; Herzog, J.; Mehdorn, H.M.; Deuschl, G.; Meissner, W.G.; Volkmann, J. Subthalamic deep brain stimulation increases pallidal firing rate and regularity. Exp. Neurol. 2011, 229, 517–521. [Google Scholar] [CrossRef] [PubMed]
- Eser, P.; Kocabicak, E.; Bekar, A.; Temel, Y. Insights into neuroinflammatory mechanisms of deep brain stimulation in Parkinson’s disease. Exp. Neurol. 2024, 374, 114684. [Google Scholar] [CrossRef] [PubMed]
- Vedam-Mai, V.; Rodgers, C.; Gureck, A.; Vincent, M.; Ippolito, G.; Elkouzi, A.; Yachnis, A.T.; Foote, K.D.; Okun, M.S. Deep Brain Stimulation associated gliosis: A post-mortem study. Parkinsonism Relat. Disord. 2018, 54, 51–55. [Google Scholar] [CrossRef] [PubMed]
- Temel, Y.; Visser-Vandewalle, V.; Kaplan, S.; Kozan, R.; Daemen, M.A.; Blokland, A.; Schmitz, C.; Steinbusch, H.W. Protection of nigral cell death by bilateral subthalamic nucleus stimulation. Brain Res. 2006, 1120, 100–105. [Google Scholar] [CrossRef]
- Mazumder, S.; Bahar, A.Y.; Shepherd, C.E.; Prasad, A.A. Post-mortem brain histological examination in the substantia nigra and subthalamic nucleus in Parkinson’s disease following deep brain stimulation. Front. Neurosci. 2022, 16, 948523. [Google Scholar] [CrossRef]
- Pal, G.D.; Ouyang, B.; Serrano, G.; Shill, H.A.; Goetz, C.; Stebbins, G.; Metman, L.V.; Driver-Dunckley, E.; Mehta, S.H.; Caviness, J.N.; et al. Comparison of neuropathology in Parkinson’s disease subjects with and without deep brain stimulation. Mov. Disord. 2017, 32, 274–277. [Google Scholar] [CrossRef]
- Hilker, R.; Voges, J.; Ghaemi, M.; Lehrke, R.; Rudolf, J.; Koulousakis, A.; Herholz, K.; Wienhard, K.; Sturm, V.; Heiss, W. Deep brain stimulation of the subthalamic nucleus does not increase the striatal dopamine concentration in parkinsonian humans. Mov. Disord. 2003, 18, 41–48. [Google Scholar] [CrossRef]
- Kwiatek-Majkusiak, J.; Geremek, M.; Koziorowski, D.; Tomasiuk, R.; Szlufik, S.; Friedman, A. Higher serum levels of pro-hepcidin in patients with Parkinson’s disease treated with deep brain stimulation. Neurosci. Lett. 2018, 684, 205–209. [Google Scholar] [CrossRef]
- Ownby, R.L. Neuroinflammation and cognitive aging. Curr. Psychiatry Rep. 2010, 12, 39–45. [Google Scholar] [CrossRef]
- Zhao, Z.; Fu, Q.; Guo, X.; He, H.; Yang, G. Potential Biomarkers and Treatment of Neuroinflammation in Parkinson’s Disease. Actas Esp. Psiquiatr. 2025, 53, 181–188. [Google Scholar] [CrossRef]
- De Lella Ezcurra, A.L.; Chertoff, M.; Ferrari, C.; Graciarena, M.; Pitossi, F. Chronic expression of low levels of tumor necrosis factor-alpha in the substantia nigra elicits progressive neurodegeneration, delayed motor symptoms and microglia/macrophage activation. Neurobiol. Dis. 2010, 37, 630–640. [Google Scholar] [CrossRef]
- Tansey, M.G.; Wallings, R.L.; Houser, M.C.; Herrick, M.K.; Keating, C.E.; Joers, V. Inflammation and immune dysfunction in Parkinson disease. Nat. Rev. Immunol. 2022, 22, 657–673. [Google Scholar] [CrossRef] [PubMed]
- Ren, L.; Yi, J.; Yang, J.; Li, P.; Cheng, X.; Mao, P. Nonsteroidal anti-inflammatory drugs use and risk of Parkinson disease: A dose-response meta-analysis. Medicine 2018, 97, e12172. [Google Scholar] [CrossRef]
- Tansey, M.G.; Romero-Ramos, M. Immune system responses in Parkinson’s disease: Early and dynamic. Eur. J. Neurosci. 2019, 49, 364–383. [Google Scholar] [CrossRef] [PubMed]
- Fleury, V.; Zekeridou, A.; Lazarevic, V.; Gaïa, N.; Giannopoulou, C.; Genton, L.; Cancela, J.; Girard, M.; Goldstein, R.; Bally, J.F.; et al. Oral Dysbiosis and Inflammation in Parkinson’s Disease. J. Parkinsons Dis. 2021, 11, 619–631. [Google Scholar] [CrossRef] [PubMed]
- Veselý, B.; Dufek, M.; Thon, V.; Brozman, M.; Királová, S.; Halászová, T.; Koriťáková, E.; Rektor, I. Interleukin 6 and complement serum level study in Parkinson’s disease. J. Neural Transm. 2018, 125, 875–881. [Google Scholar] [CrossRef]
- Li, N.; Wang, J.X.; Huo, T.T.; Zhao, J.R.; Wang, T.J. Associations of IL-1β and IL-6 gene polymorphisms with Parkinson’s disease. Eur. Rev. Med. Pharmacol. Sci. 2021, 25, 890–897. [Google Scholar] [CrossRef]
- Ramírez, J.; Cañete, J.D. Anakinra for the treatment of rheumatoid arthritis: A safety evaluation. Expert Opin. Drug Saf. 2018, 17, 727–732. [Google Scholar] [CrossRef]
- Stojakovic, A.; Paz-Filho, G.; Arcos-Burgos, M.; Licinio, J.; Wong, M.L.; Mastronardi, C.A. Role of the IL-1 Pathway in Dopaminergic Neurodegeneration and Decreased Voluntary Movement. Mol. Neurobiol. 2017, 54, 4486–4495. [Google Scholar] [CrossRef]
- Sivapalasingam, S.; Lederer, D.J.; Bhore, R.; Hajizadeh, N.; Criner, G.; Hosain, R.; Mahmood, A.; Giannelou, A.; Somersan-Karakaya, S.; O’brien, M.P.; et al. Efficacy and Safety of Sarilumab in Hospitalized Patients With Coronavirus Disease 2019: A Randomized Clinical Trial. Clin. Infect. Dis. 2022, 75, e380–e388. [Google Scholar] [CrossRef]
- Miliukhina, I.V.; Usenko, T.S.; Senkevich, K.A.; Nikolaev, M.A.; Timofeeva, A.A.; Agapova, E.A.; Semenov, A.V.; Lubimova, N.E.; Totolyan, A.A.; Pchelina, S.N. Plasma Cytokines Profile in Patients with Parkinson’s Disease Associated with Mutations in GBA Gene. Bull. Exp. Biol. Med. 2020, 168, 423–426. [Google Scholar] [CrossRef] [PubMed]
- Boyman, O.; Sprent, J. The role of interleukin-2 during homeostasis and activation of the immune system. Nat. Rev. Immunol. 2012, 12, 180–190. [Google Scholar] [CrossRef]
- Si, Y.; Zhang, Y.; Zuloaga, K.; Yang, Q. The role of innate lymphocytes in regulating brain and cognitive function. Neurobiol. Dis. 2023, 179, 106061. [Google Scholar] [CrossRef]
- Noble, S.; Goa, K.L. Aldesleukin (recombinant interleukin-2). BioDrugs 1997, 7, 394–422. [Google Scholar] [CrossRef] [PubMed]
- Tomasovic, L.M.; Liu, K.; VanDyke, D.; Fabilane, C.S.; Spangler, J.B. Molecular Engineering of Interleukin-2 for Enhanced Therapeutic Activity in Autoimmune Diseases. BioDrugs 2024, 38, 227–248. [Google Scholar] [CrossRef]
- Muthuraman, M.; Koirala, N.; Ciolac, D.; Pintea, B.; Glaser, M.; Groppa, S.; Tamás, G.; Groppa, S. Deep Brain Stimulation and L-DOPA Therapy: Concepts of Action and Clinical Applications in Parkinson’s Disease. Front. Neurol. 2018, 9, 711. [Google Scholar] [CrossRef] [PubMed]
- Calabresi, P.; Mechelli, A.; Natale, G.; Volpicelli-Daley, L.; Di Lazzaro, G.; Ghiglieri, V. Alpha-synuclein in Parkinson’s disease and other synucleinopathies: From overt neurodegeneration back to early synaptic dysfunction. Cell Death Dis. 2023, 14, 176. [Google Scholar] [CrossRef]
- Abbott, A. Fetal-cell revival for Parkinson’s. Nature 2014, 510, 195–196. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Chen, Q.; Chen, X.; Han, F.; Chen, Z.; Wang, Y. The blood–brain barrier: Structure, regulation and drug delivery. Sig. Transduct. Target. Ther. 2023, 8, 1–27. [Google Scholar] [CrossRef]
- Cha, Y.; Park, T.Y.; Leblanc, P.; Kim, K.S. Current Status and Future Perspectives on Stem Cell-Based Therapies for Parkinson’s Disease. J. Mov. Disord. 2023, 16, 22–41. [Google Scholar] [CrossRef]
- Jiang, S.; Wang, H.; Yang, C.; Feng, F.; Xu, D.; Zhang, M.; Xie, M.; Cui, R.; Zhu, Z.; Jia, C.; et al. Phase 1 study of safety and preliminary efficacy of intranasal transplantation of human neural stem cells (ANGE-S003) in Parkinson’s disease. J. Neurol. Neurosurg. Psychiatry 2024, 95, 1102–1111. [Google Scholar] [CrossRef] [PubMed]
- Madrazo, I.; Kopyov, O.; Ávila-Rodríguez, M.A.; Ostrosky, F.; Carrasco, H.; Kopyov, A.; Avendaño-Estrada, A.; Jiménez, F.; Magallón, E.; Zamorano, C.; et al. Transplantation of Human Neural Progenitor Cells (NPC) into Putamina of Parkinsonian Patients: A Case Series Study, Safety and Efficacy Four Years after Surgery. Cell Transplant. 2019, 28, 269–285. [Google Scholar] [CrossRef] [PubMed]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ebrahim, G.; Hutchinson, H.; Gonzalez, M.; Dagra, A. Central and Peripheral Immunity Responses in Parkinson’s Disease: An Overview and Update. Neuroglia 2025, 6, 17. https://doi.org/10.3390/neuroglia6020017
Ebrahim G, Hutchinson H, Gonzalez M, Dagra A. Central and Peripheral Immunity Responses in Parkinson’s Disease: An Overview and Update. Neuroglia. 2025; 6(2):17. https://doi.org/10.3390/neuroglia6020017
Chicago/Turabian StyleEbrahim, Ghaidaa, Hunter Hutchinson, Melanie Gonzalez, and Abeer Dagra. 2025. "Central and Peripheral Immunity Responses in Parkinson’s Disease: An Overview and Update" Neuroglia 6, no. 2: 17. https://doi.org/10.3390/neuroglia6020017
APA StyleEbrahim, G., Hutchinson, H., Gonzalez, M., & Dagra, A. (2025). Central and Peripheral Immunity Responses in Parkinson’s Disease: An Overview and Update. Neuroglia, 6(2), 17. https://doi.org/10.3390/neuroglia6020017