1. Introduction
In 1935, Kodak launched the first chromogenic product with application in the photography and film industry. A few years later, the chromogenic process was available in a wide range of materials (negatives, prints, and positive transparencies) and formats (professional and amateur) [
1]. Its great success led to the color photography era, starting in the 1960s, when the use of color photographs surpassed that of black-and-white [
2]. Ever since, inestimable amounts of color photographs have been produced, and chromogenic photography now represents a significant part of the cultural heritage worldwide.
The chromogenic process is grounded on the color separation principle to reproduce a real scene, using superimposed emulsion layers coated on the base [
2]. Each emulsion layer is composed of color couplers, silver halides and sensitizing dyes within a gelatin binder. During processing, the color couplers, initially colorless, react with the developer, which has previously been oxidized by silver development to become dyes (
Figure 1). After processing, the blue-sensitive layer reproduces the yellow (Y) elements of the reproduced scene, the green-sensitive layer reproduces the magenta (M), and the red-sensitive layer reproduces the cyan (C). At the end of the processing, all silver is removed by a fixer [
3].
Although each manufacturer employed its own color couplers, all the industry worked with materials with similar structures [
1]. The most important classes of Y couplers are pivaloylacetanilide and benzoylacetanilide, introduced by Kodak in the 1960s. Later, other classes of Y couplers have been employed with higher stability and tinctorial strength [
4]. M couplers are normally heteroaromatic compounds. Until 1980, pyrazolones were the M couplers of choice [
5]. In the 2000s, the use of pyrazolotriazoles was widespread due to the high quality of hues produced by the correspondent dye. Most C couplers are substituted phenols or naphthols. The use of 2,5-diacylaminophenols in reversal films, disclosed by Kodak, was generalized for more than 20 years [
6]. Nevertheless, heterocyclic compounds have also been used [
6,
7].
Different functional groups can be attached to the color coupler nucleus to improve their features. Ballasting groups are usually employed to prevent dyes from wandering from one layer to another. Leaving groups are molecular fragments added to optimize the coupling rate and promote the direct formation of the chromophore [
7].
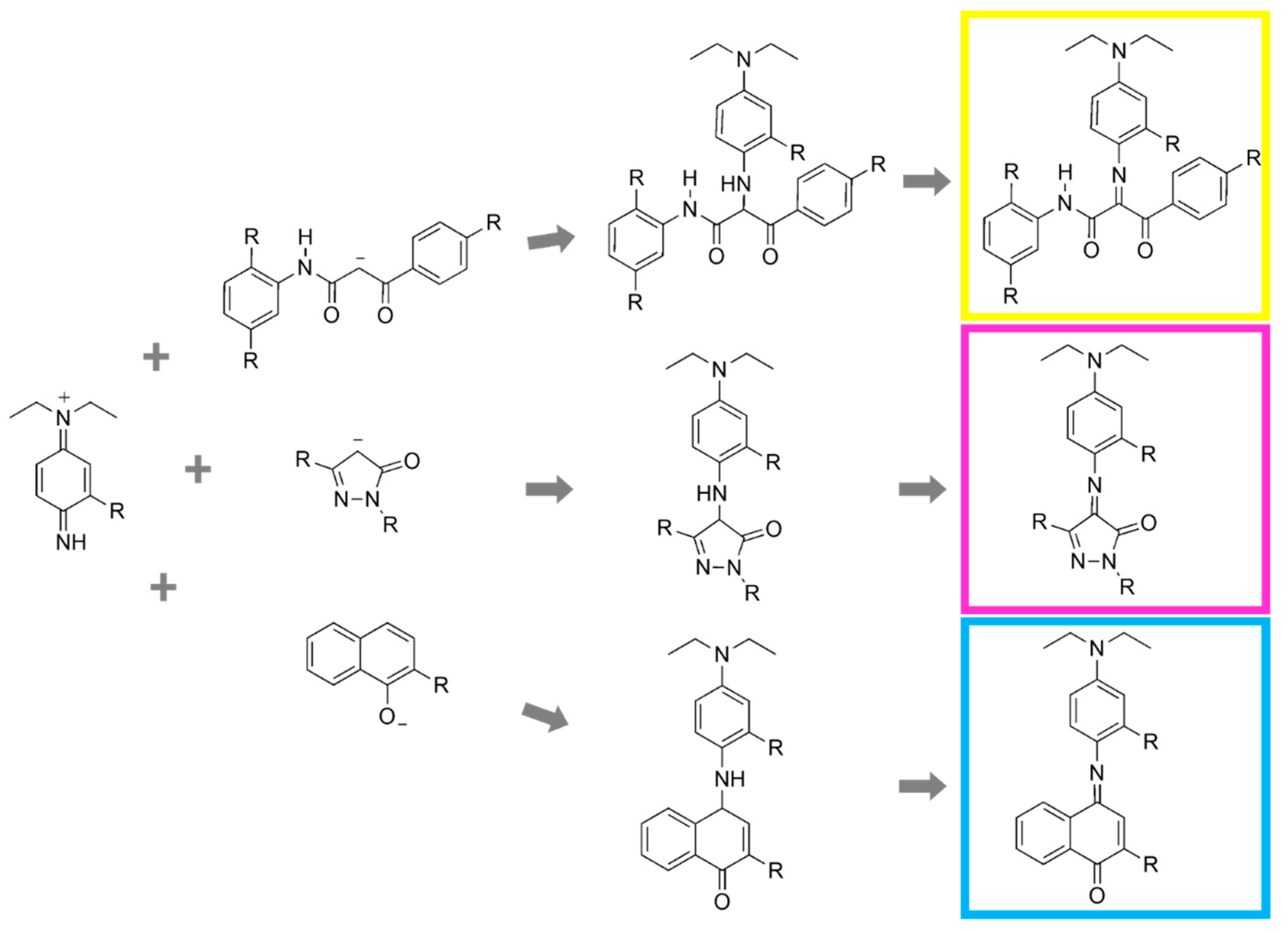
In general, the dyes resulting from the reaction of M and Y couplers with the developing agent are from the azomethine family, while those resulting from C couplers are from the indoaniline family [
8]. Schiff base is one of the most important functional groups present in the three dyes, constituting an essential link in the conjugated system responsible for the dye color [
9]. The most significant classes of couplers and the corresponding dye are summarized in
Table 1.
The precise structure of the final dyes present in a chromogenic material depends on the type of color coupler and its functional groups and on the developing agent used to process the film [
5]. Over time, the composition of developing agents and especially of color couplers has undergone many changes [
1]. According to the type of material (negative, print, reversal film), different couplers have been shaped. Moreover, due to the constant evolution of coupler technology, manufacturers often change the couplers at use [
1], and so different couplers have been applied in different stocks as well [
5]. Unfortunately, as far as it could be concluded from the conducted research, there is no way of knowing which color couplers have been employed in which products based on the available literature.
The first generation of chromogenic materials was chemically very unstable. From the late 1970s, Kodak, Fuji, and Agfa, among other industries, started to test the stability of their products on a regular basis. This has contributed to a deeper knowledge of the deterioration mechanisms associated with chromogenic dyes and led to great improvements in the stability of these materials from the 1980s onwards [
1]. Even so, chromogenic dyes are highly susceptible to oxidation and hydrolysis, both induced by light (light fading) and/or relative humidity and temperature (dark fading) [
10]. Continuous contact with environmental agents gradually disrupts the chromophore molecules, leading to the formation of colorless degradation products responsible for the fading of the image [
1]. Since the three dyes present in a film have different molecular structures, they also have different degradation rates. Therefore, these materials are prone to shift the original color balance. Residual color couplers are also vulnerable to oxidation, producing yellowish species (yellow stain) [
11]. This is especially visible in the highlight areas of the image [
10], where residual color couplers are present in higher quantities [
12]. Furthermore, the interaction between residual color couplers (nucleophiles) and dyes (electrophiles) can accelerate dye fading [
12]. Improper processing can also decrease dye stability and/or enhance staining levels [
10]. Over the years, it has been demonstrated that degradation products generated by dark and light fading can be different since the two degradation courses might lead to different disruptions of the chromophores [
11]. The same film can be very unstable when exposed to light and quite stable in the dark [
10].
During the present investigation, a gap of knowledge regarding the chromogenic materials was detected. Up to now, there are several references describing molecule structures, degradation pathways and preservation guidelines. However, at present, it is not possible to know which dyes are present in a specific artwork and how long will they last. When the stability of a specific chromogenic photograph is to be known, the material or an equivalent (same brand and model) can be induced to artificial aging to estimate its lifespan [
13]. As previously mentioned, chromogenic materials are industrial products that have been progressively upgraded. For that reason, it is nowadays difficult to find a product equivalent to the one used in the past. Thus, unless the initial condition of the artwork was documented (which is not common), there is no way to know how the colors of a specific chromogenic artwork have changed.
To investigate the degradation mechanism of a dye, it is necessary to identify its chemical structure. Despite the pioneering studies dedicated to the characterization of chromogenic dyes, conducted by Giovanna Di Pietro [
5] and Ann Fenech [
14] and more recently by researchers from the University of Milan [
15], a sustainable methodology to identify dyes present in a specific chromogenic product is still not available today. Therefore, a research study was carried out focusing on the characterization of chromogenic dyes, taking as case studies slide-based artworks by the artist Ângelo de Sousa (1938–2011). Color slides, or chromogenic reversal films, are first-generation positive transparencies. These were widely used in photojournalism, fine arts, for commercial applications, such as advertising, fashion, industry, and academia [
16], and from the 1960s onwards, as new media of expression for artists [
16]. The pursued methodology sought to test the efficiency of some of the most common analytical techniques used in the study of cultural heritage. Both non- and micro-invasive techniques were explored. Based on the straight analysis of emulsion layers and on the isolation of the different dyes composing the chromogenic material, several procedures were proposed to molecularly describe the dyes found in chromogenic reversal films. Different techniques, such as Raman spectroscopy, thin-layer chromatography (TLC), infrared spectroscopy, high-performance liquid chromatography with a diode array detector (HPLC-DAD), and coupled with mass spectrometry (HPLC-MS), were compiled. Promising results were achieved with this approach, opening new paths for understanding these materials. Yet, this was only a preliminary study and needs further development.
2. Materials and Techniques
2.1. Case Studies
A strategy to characterize chromogenic dyes was developed based on two case studies of slide-based artworks produced by the artist Ângelo de Sousa. Both are 35 mm chromogenic reversal films from the integrated dye couplers type.
(i) Kodak Ektachrome 160T Professional (EPT)
Slides de Cavalete (1978–1979), a slide-based artwork by Ângelo de Sousa, is one of the most original photographic works produced by the artist [
17]. Knowing that chromogenic reversal films are highly susceptible to dark and light fading, that the work has been gathered in inadequate environmental conditions for more than forty years, and that the original slides have been projected in two exhibitions at least [
17], visible color change would be expected. However, due to the abstract nature of the images composing
Slides de Cavalete, the color change was not straightforwardly observed. Accordingly, this artwork was selected as a case study.
The work is composed of one hundred Kodak Ektachrome 160T. EPT was produced between 1976 and 2007, with the emulsion number 5037 [
2]. A film tip and an unprocessed film of EPT were found at the artists’ archive, probably dating from the same time of the work [
18], allowing us to conduct a series of analyses to this type of film without having to rely on the original slides. From 1976 onwards, all Ektachrome films were processed using E-6 chemistry [
2].
(ii) Fuji Fujichrome Provia 400X Professional (RXP)
A set of thirteen unexposed Fujichrome Provia 400X Professional (RXP) films was found at Ângelo de Sousa’s house. Based on the survey conducted on the artist’s photographic collection, this type of film was found to be the third most abundant model of chromogenic reversal films used by him [
18]. Although the shelf-life of the RXP films expired in 2009, considering their representativeness and availability, those films were used to conduct the present study. Fujichrome Provia 400X Professional is currently discontinued, and no references were found to understand the timeframe during which it was produced. The films used for the present study have the emulsion number 104. RXP is an E-6 processing film. In the technical datasheet of the film, a schematic cross-section is represented (
Supplementary Materials).
2.2. Cross-Sections
Cross-sections were prepared from both EPT and RXP samples, processed and unprocessed films, to help us to characterize the films under study. To do so, a small sample was removed from the border of the films (about 2 × 2 mm). In chromogenic reversal films, due to the inversion of the image during processing, the borders of the films are black, i.e., the emulsion layer has a high concentration of Y, M, and C dyes. These areas can be used for analysis without compromising the image. This fragment was taped over a small piece of a compact disc (CD) to hold it steady. The CD coating was previously removed mechanically, and the fragment was fixed to the piece of CD plastic with tape. Cross-sections were prepared by using a Leica RM 2155 rotary microtome equipped with low-profile blades Leica DB 80 LX. The assembly previously set was fixed in the specimen clamping system. Slices of 15 μm were cut by using a clean portion of the blade for each cut and making a quick but relatively gentle motion (to obtain a clean cut and avoid ridges and fractures). The cuts were controlled by using a stereomicroscope. The sample slices were placed over a microscope slide and covered with a cover slip. The collected samples were observed under the optical microscope.
2.3. Optical Microscopy
To observe the stratigraphic layers composing EPT and RXP films, microscopy images were acquired using a Zeiss Axioplan 2 Imaging system (HAL 100) coupled to Nikon DXM1200F digital camera and ACT-1 software. A white card was placed on the condenser under the slides to obtain a uniform background in the image. Before photographing the samples, the white spirit was carefully added between the cover glass and the slide with a Pasteur pipette. Reflected polarized light was used.
2.4. Raman Spectroscopy
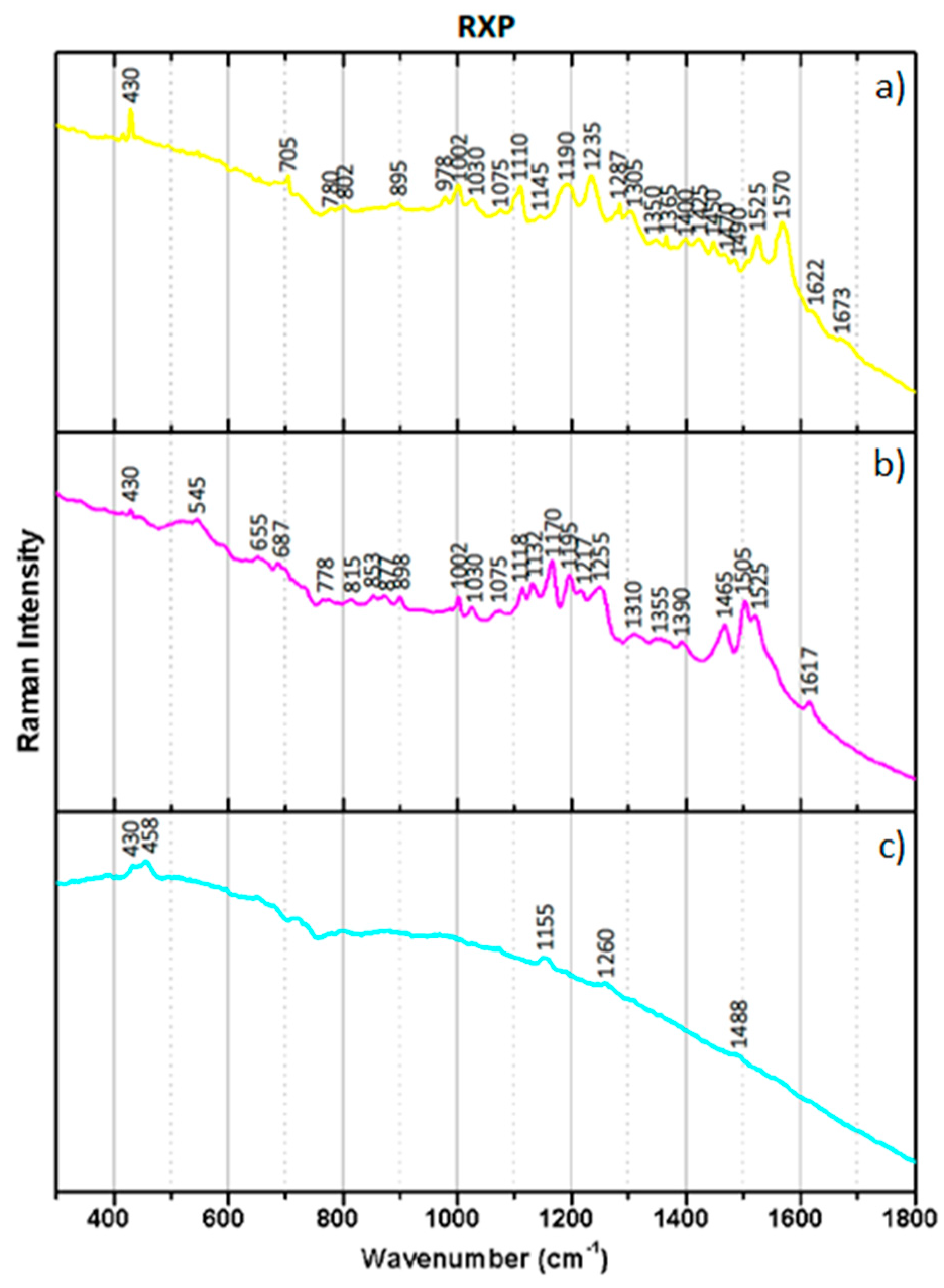
Raman microscopy was carried out using a Horiba Jobin Yvon LabRAM 300 spectrometer, equipped with a He-Ne laser 17 mW operating at 632.8 nm and coupled to the confocal microscope with high stability Olympus BX41. Micro-samples were collected from both the emulsion of EPT and RXP samples (directly from the film surface) using a micro-tool under a stereomicroscope. An attempt to remove only Y, M, or C emulsion layers was pursued for individual analysis of each dye. The collected micro-samples were placed over a microscope slide. Raman spectra were recorded as extended scans, and the laser beam was focused either with a 50× or a 100× Olympus objective lens or with a 50× Olympus ultra-long working distance (ULWD) objective for depth probing. The Raman microscope allowed for the selection of precise areas of analysis, i.e., of each colored layer individually. The laser power at the surface of the samples was varied with the aid of a set of neutral density filters. Raman spectra were collected between 200 and 2000 cm−1. All spectra are presented without baseline correction.
2.5. Extraction of the Dyes
In order to characterize the different dyes present in the films under study, a sample of each emulsion layer was collected by scratching a portion of the black borders from the film (ca. 0.002 g). The collected sample was then added to a volumetric flask containing 2 mL of ethanol and distilled water (3:1, v:v) and left for about 48 h. The extraction process was aided by heating (circa 35 °C) and stirring. After extraction, the solution was filtered through a 0.45 μm Teflon membrane filter and then dried in a round-bottom flask under a stream of nitrogen until completely dry. The extract was then re-dissolved in the desired solvent according to the analysis to be performed.
2.6. Thin-Layer Chromatography (TLC)
Pre-coated silica TLC sheets (ALUGRAM® Xtra SIL G/UV254, Machery-Nagel, Düren, Germany), cut in ca. 2 × 9 cm, were used for the separation of the dyes extracted from both EPT and RXP films. The extracts were dissolved in ethyl acetate (just enough to acquire a concentrated solution) for the elution. Both mixtures from the EPT and RXP were applied on the TLC plate, by putting several drops (ca. 5) on the same spot at the bottom of the TLC plate, using a glass capillary. The TLC plate was then immersed in a beaker containing the mobile phase, covered with a watch glass, to be developed. Based on previous tests, diethyl ether:ethyl acetate (90:10) mobile phase was selected to develop the EPT sample, and ethyl acetate to develop the RXP sample. A filter paper was used to cover the inside walls of the beaker and homogeneously spread the solvent onto the overall volume. The retardation value (Rf) was calculated for the different dyes of both types of samples.
The dyes were also separated with a preparative TLC, to enable their collection and further analysis with other techniques. Silica sheets measuring 20 × 20 cm (DC-Alufolien Kieselgel 60, Merck, New York, NY, USA) were used. EPT and RXP solutions were applied in a line using a piece of cotton placed inside a Pasteur pipette, to which the tip has been removed. The same mobile phases previously described were utilized for the development of the preparative TLC sheets. A glass tray was used for the elution of the TLC sheets. The areas of the silica where the different dyes adsorbed were then scraped with a scalpel, transferred to flasks with ethyl acetate and stirred to allow extraction of the dyes (ca. 30 min). Only the C dye from the RXP sample could not be extracted in ethyl acetate, which was substituted by ethanol. After complete extraction, the mixture was filtered through a paper filter. Each dye from both EPT and RXP samples was gathered separately. The obtained solutions were then dried in a round-bottom flask under a stream of nitrogen.
2.7. Fourier-Transform Infrared Spectroscopy (FTIR)
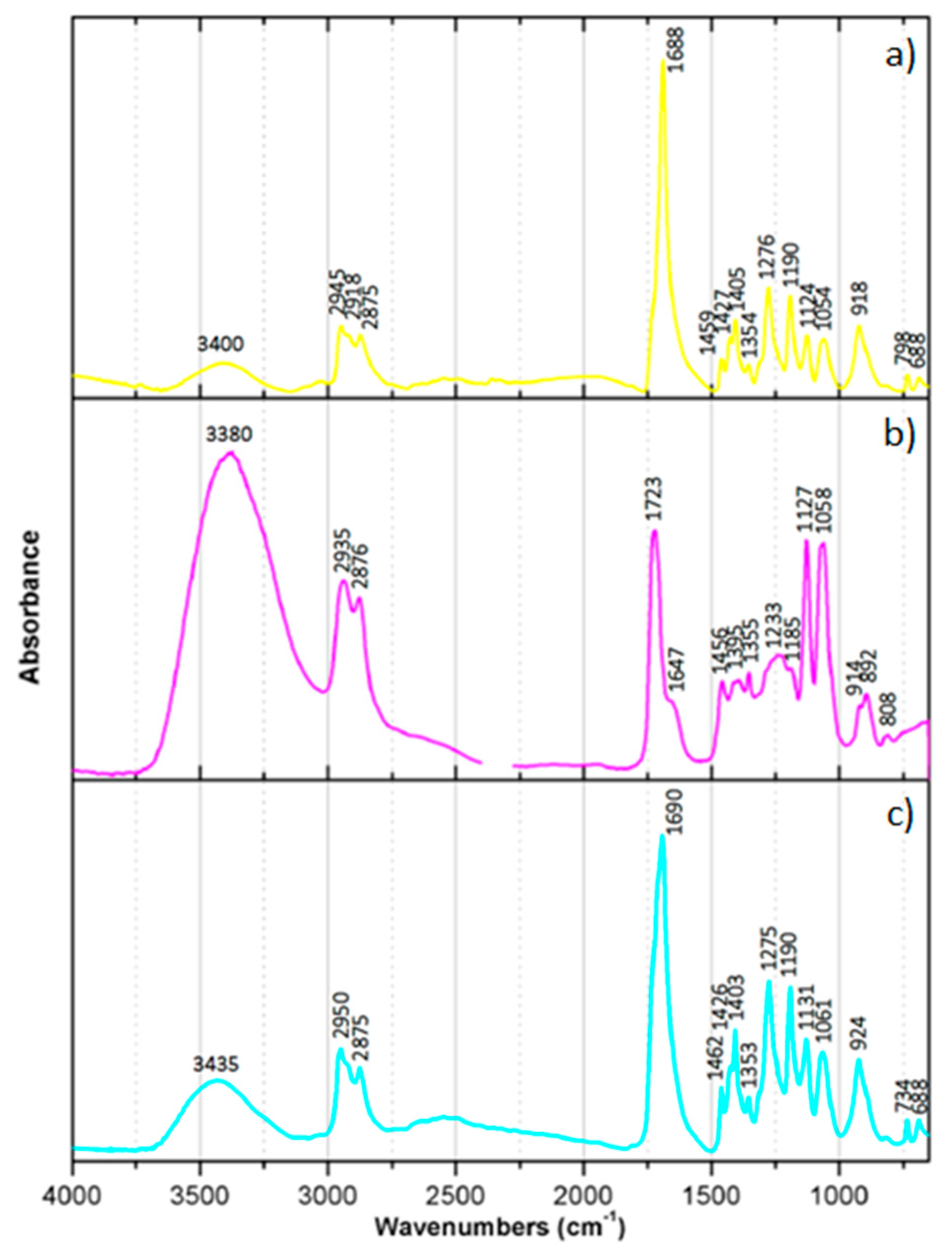
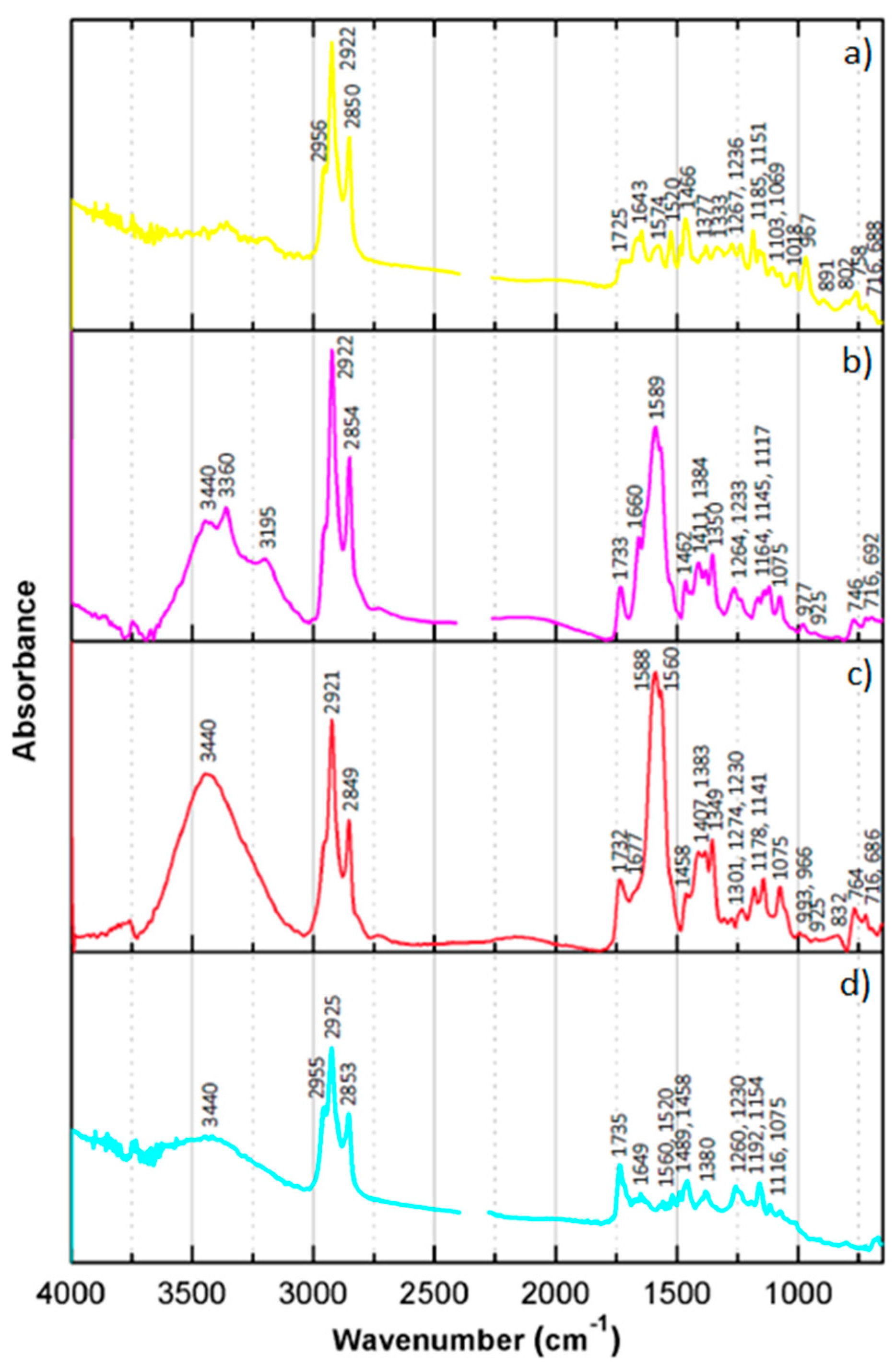
Infrared analyses were performed with a Nicolet Nexus spectrophotometer. The isolated dyes were examined by placing small droplets of each dye in acetyl acetate solution over a KBr disc (Spectra-Tech Inc., Oak Ridge, TN, USA, 25 × 4 mm), separately. Between each drop placed in the disc the solvent was left to evaporate. Infrared spectra were acquired in transmission mode, from 4000 to 650 cm−1, with 64 scans and 4 cm−1 spectral resolution. Spectral analysis was performed using Omnic E.S.P. 5.2 and OriginPro 8 software. All spectra of the dyes presented were not baseline corrected.
2.8. High-Performance Liquid Chromatography Coupled with Diode Array Detector (HPLC-DAD)
EPT and RXP extracts were also separated with HPLC, and the isolated dyes were characterized with DAD. The tests were carried out in an analytical Thermo Electron, FinniganTM Surveyor® HPLC-DAD system with a Thermo Electron, FinniganTM Surveyor® LC pump, autosampler, and PDA detector, and using a reversed-phase RP18 analytic column (Chromolith, 100 × 4.6 mm). The wavelength range of the detector was 200–800 nm, with a 0.8 s sampling interval and 4 nm resolution. Samples were injected into the column via a Rheodyne injector with a 25 μL loop. The elution gradient used at a flow rate of 1.0 mL min−1 consisted of HPLC-grade methanol (A) and Millipore water containing 0.3% perchloric acid (B). The elution gradient was set as follows (A:B, v:v): 7:93 from 0 to 2 min; 15:85 from 8 to 25 min; 75:25 from 25 to 27 min; 80:20 from 27 to 29 min; and 100:0 from 29 to 40 min.
2.9. High-Performance Liquid Chromatography Coupled to Diode Array Detection and Mass Spectrometry (HPLC-DAD-MS) and Liquid Chromatography–High-Resolution Mass Spectrometry (LC-HRMS)
In order to gather information about the chemical structure of the dyes present in the films, experiments were carried out with mass spectrometry. Due to budget and time constraints, only one of the samples, EPT, was analyzed. The EPT sample was previously analyzed in a low-resolution mass spectrometer coupled to an HPLC-DAD to identify the dyes in the DAD chromatogram through the absorption bands and to correlate their retention time with the corresponding signals observed in the total ion chromatograms. Thereafter, the sample was analyzed in a high-resolution mass spectrometer to identify the m/z values on the high-resolution total ion chromatograms.
HPLC-DAD-MS analysis was performed on a Dionex Ultimate 3000SD system with a diode array detector coupled online to an LCQ Fleet ion trap mass spectrometer (Thermo Scientific, Waltham, MA, USA), equipped with an ESI source. The mass spectrometer was operated in the ESI-positive and negative ion modes, with the following optimized parameters: ion spray voltage, ±4.5 kV; capillary voltage, 16/−18 V; tube lens offset, −70/58 V, sheath gas (N2), 80 arbitrary units; auxiliary gas (N2), 5 arbitrary units; capillary temperature, 270 °C. Spectra typically correspond to the average of 20–35 scans, and were recorded in the range between 100 and 1000 Da. Data acquisition and processing were performed using the software Xcalibur 2.2.
High-resolution mass spectra were acquired on a quadrupole-time-of-flight (QqToF) mass spectrometer (Impact II, Bruker Daltonics, Bremen, Germany) equipped with an ESI source. The spectrometric parameters were set as follows: end plate offset, 500 V; capillary voltage, +4.5/−2,7 kV; nebulizer, 4 bars; dry gas, 8 L.min−1; dry temperature, 200 °C. Internal calibration was achieved with an ammonium formate solution introduced to the ion source via a 20 μL loop at the beginning of each analysis, using a six-port valve. Acquisition was performed in a full scan mode in the m/z 100–1000 range and in a data-dependent MS/MS mode with an acquisition rate of 3 Hz using a dynamic method with a fixed cycle time of 3 s and an m/z-dependent isolation window of 0.03 Da. Data acquisition and processing were performed using Data Analysis 4.2 software.
In both equipments, the separation was achieved on a Cortes C18 column (150 × 2.1 mm, 2.7 μm particle size, Waters) at 35 °C, using a flow rate of 0.3 mL min−1. The mobile phase was 0.1% of acid formic in water (v/v, eluent A) and acetonitrile (eluent B), and the elution gradient (A:B, v:v) was as follows: 70:30 from 0 to 2 min; 0:100 at 20 min to 33 min; 70:30 at 35 min and 10 min of re-equilibration time. The wavelength was monitored between 250 and 700 nm.
4. Conclusions
Within the present study, different spectroscopic and chromatographic techniques have been tested with the aim of outlining suitable procedures for the characterization of chromogenic dyes and paving the way for the molecular identification of dyes present in chromogenic reversal films.
Raman spectroscopy has shown the ability to characterize chromogenic dyes. Micro-samples were collected from the borders of the chromogenic reversal films under study in order to analyze C, M, and Y emulsions separately. The confocal microscope of the equipment was used to select the desired colored areas. The dyes from EPT and RXP samples produced different Raman spectra.
Chromatographic techniques also proved to be powerful tools for isolating and characterizing chromogenic dyes. As a major disadvantage, all tested chromatographic techniques require the collection and destruction of a small sample. However, considering that the borders of chromogenic reversal films can be used to remove a sample without compromising the image and that the analysis of one sample can bring insights into several works (of the same model/batch), collecting a sample can be easily justified. TLC has the advantage of giving immediate and easy-to-interpret information by displaying colored spots in the TLC sheet. By calculating the Rf of the separated dyes, additional information about the compounds under study was gathered. Knowing that every molecule has a specific Rf value for a specific solvent and solvent concentration, TLC analysis provides evidence of the identity of the compound. Additionally, the time and cost associated with this type of analysis are substantially lower when compared with liquid chromatography. Most importantly, by using preparative TLC, the separated compounds can be isolated and used for further analysis. In the present study, the isolated dyes were characterized with IR spectroscopy. The collected IR spectra provided fingerprint spectra of C, M, and Y dyes from EPT and RXP samples. HPLC has also been traced as a useful technique for the characterization of chromogenic dyes. As in TLC, the Rt associated with each dye in a specific HPLC equipment separated with a given elution program offers additional information about the molecules. Coupled with DAD, the absorbance spectra of each dye can be observed. The shape and maximum of the absorbance spectrum of a dye in the visible region can provide a clue to differentiate and characterize the different samples. Based on the shape of the M dyes, it is proposed that M from EPT is from the pyrazolone family. The absorbance spectra collected in DAD also worked as background information for the analysis with MS. Thereby, the peaks of interest (dyes) were selected and analyzed with MS. The collected data demonstrated that M dye displays an isotopic distribution containing three chloride atoms in the ionic structure and a sulfur atom, suggesting that the dye may be derived from the pyrazolone four-equivalent-type coupler. The isotopic distribution pattern of the Y dye clearly indicates that only a chloride atom is present in the ionic structure of the (de)protonated molecule, suggesting that it might be a pivaloylacetanilide-type coupler. Finally, C dye has an isotopic distribution pattern confirming that no chloride or bromide atoms are present in its molecular structure, leading us to propose that it might be from the diacylaminophenols coupler type.
The main difficulty encountered within the investigation was the inexistence of references to support the assignment of the obtained results to specific families of chromogenic dyes. Although the tested analytical techniques have shown promising results, building databases of chromogenic dyes for Raman and IR spectroscopies, as well as MS, might be the key to the identification of these materials.


 ,
, 










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
















