
Main Group Catalysis: Cationic Si(II) and Ge(II) Compounds as Catalysts in Organosilicon Chemistry


Abstract
:1. Introduction
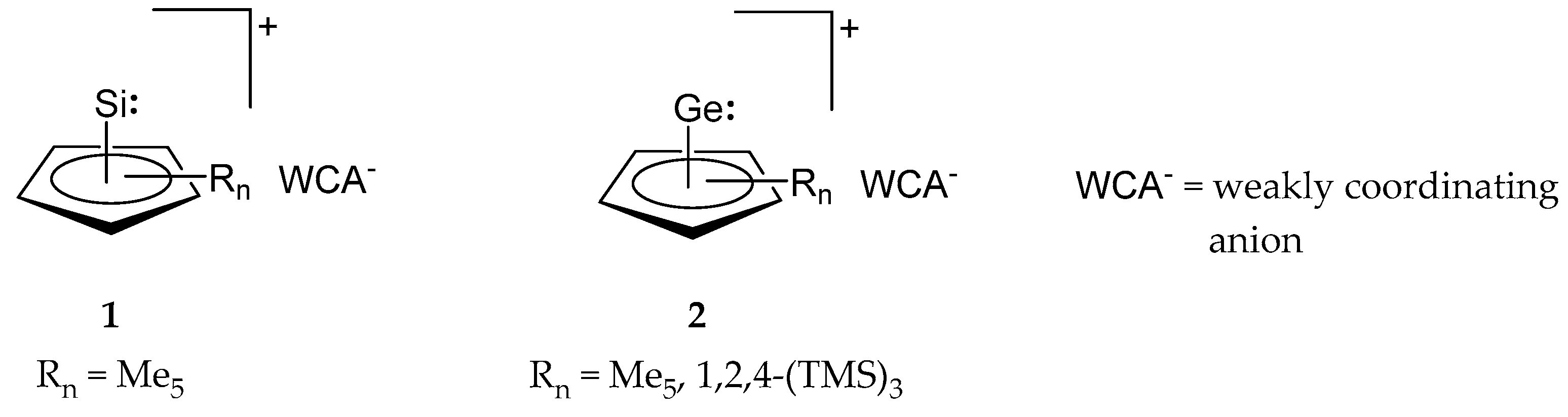
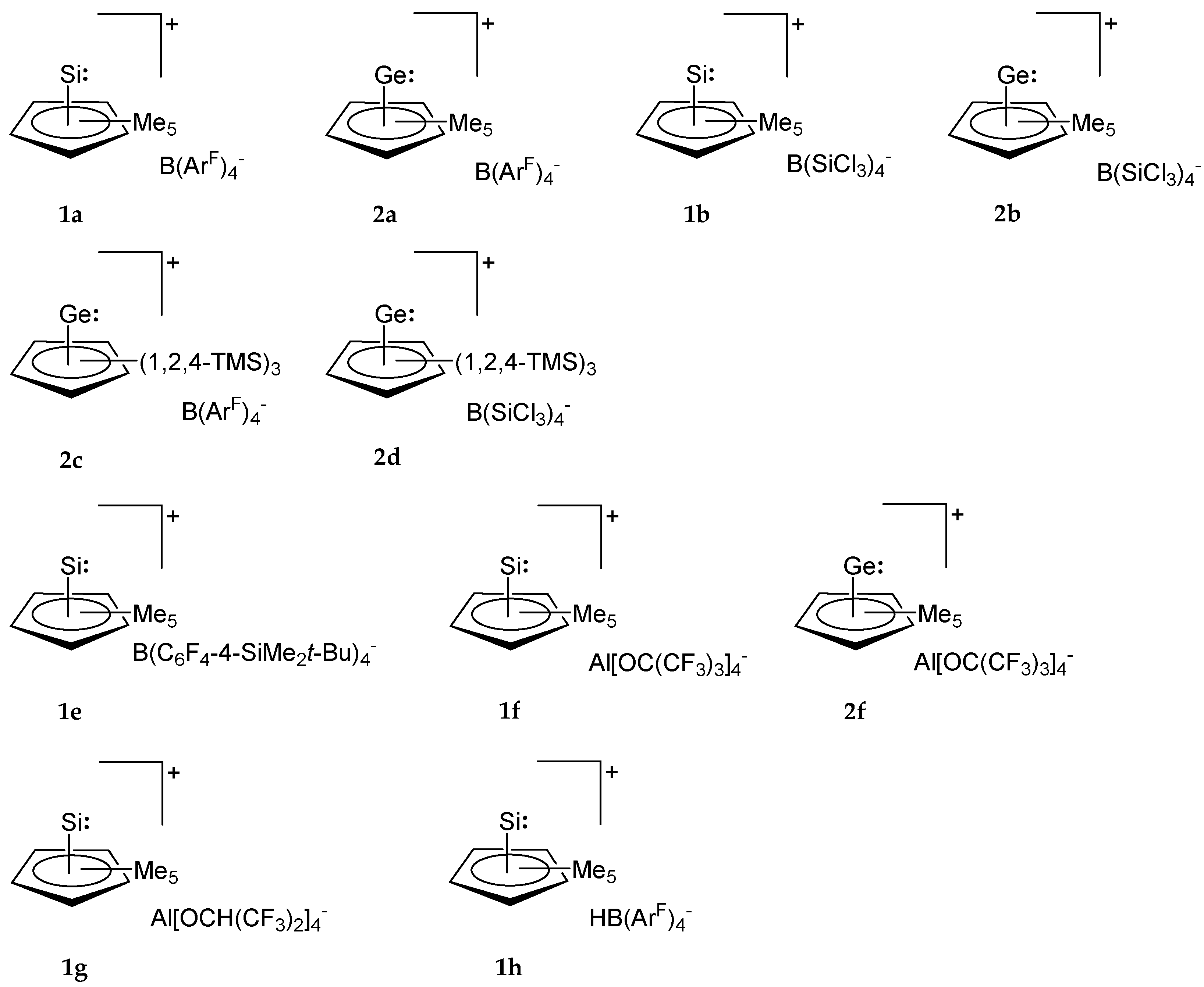
2. Synthesis and Properties of Cp-Coordinated Cationic Si(II)+ (1) and Ge(II)+ Compounds (2)


3. Cp-Coordinated Si(II) and Ge(II) Compounds 1 and 2 as Catalysts in Organosilicon Chemistry
3.1. Hydrosilylation of Carbon-Carbon Double Bonds
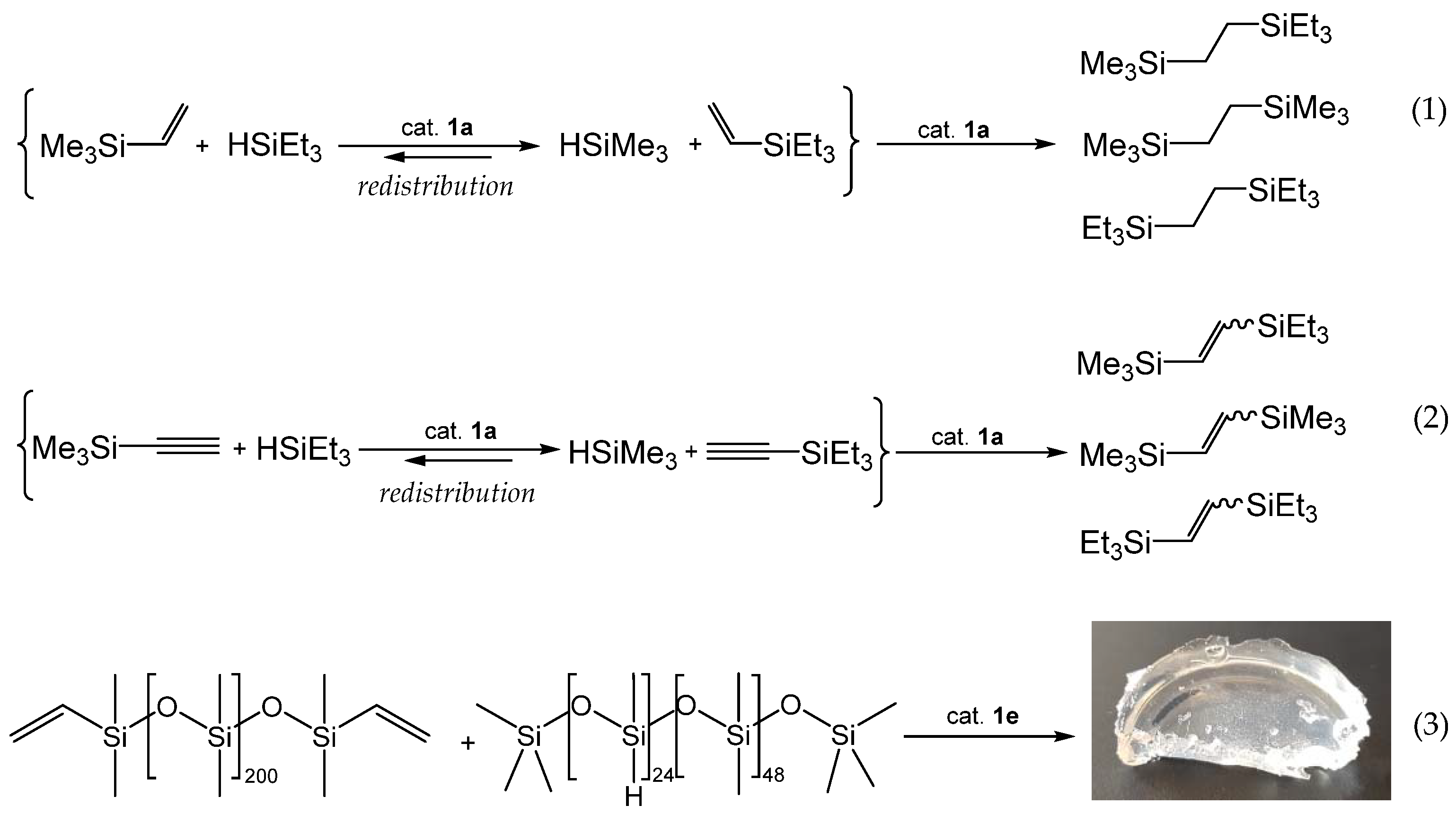
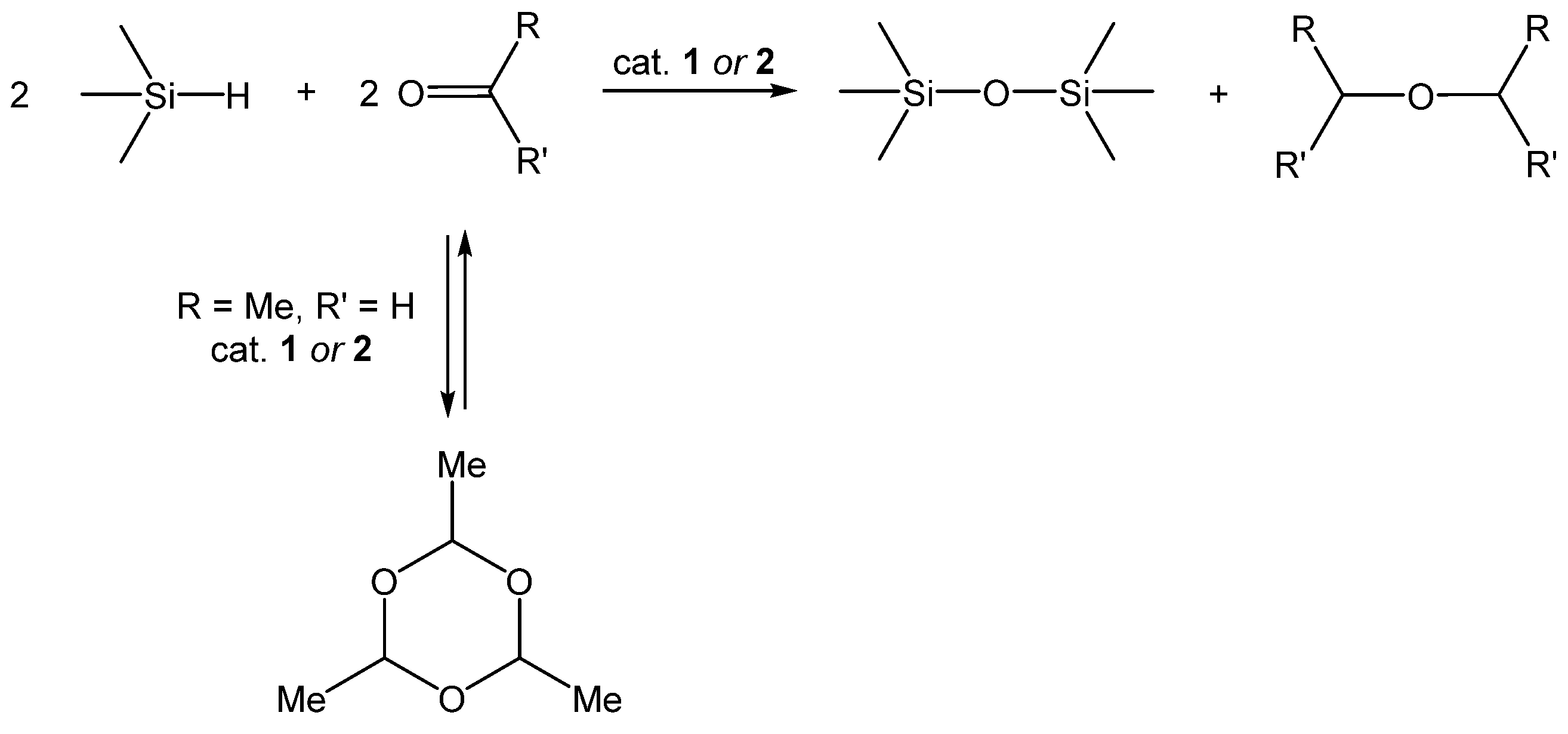
3.2. Redistribution Reactions of Hydridosil(ox)anes
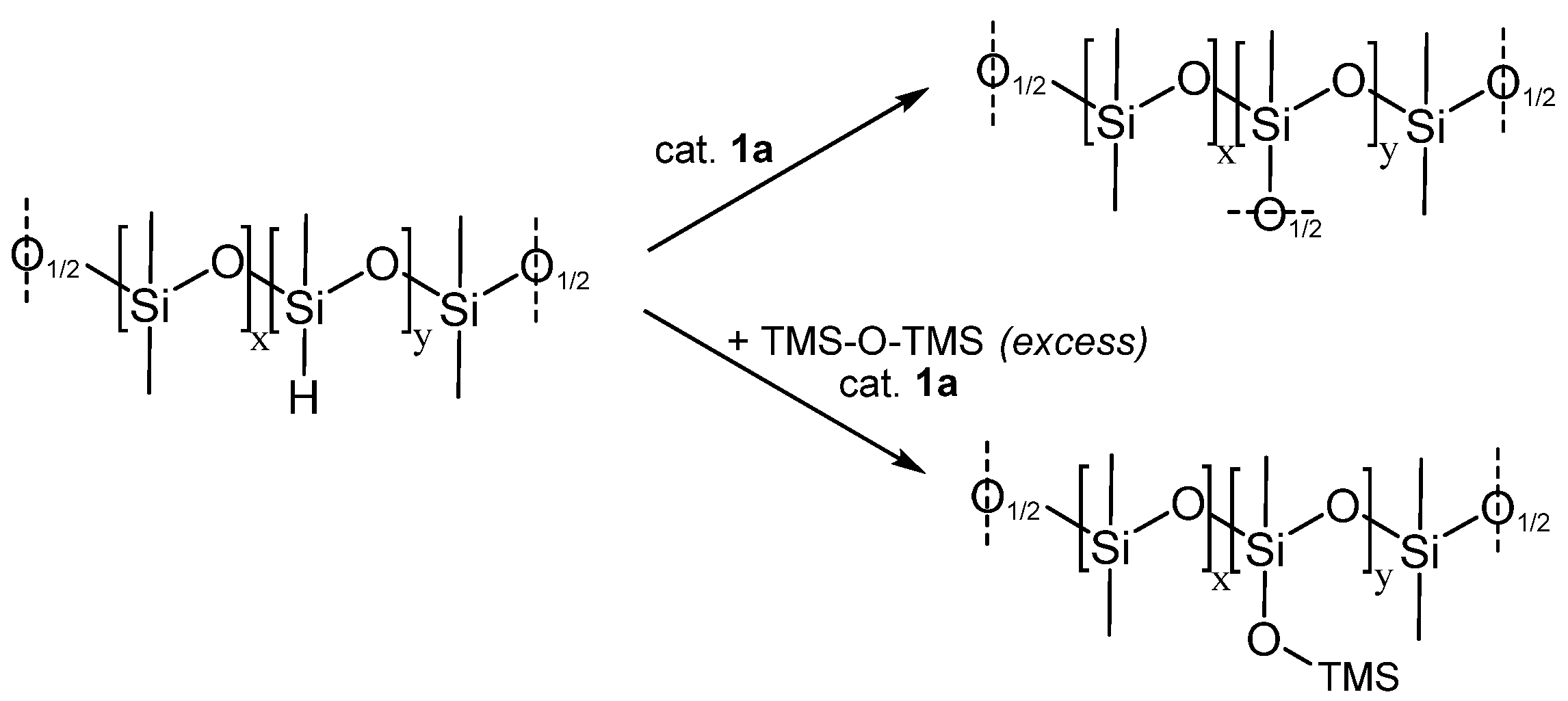
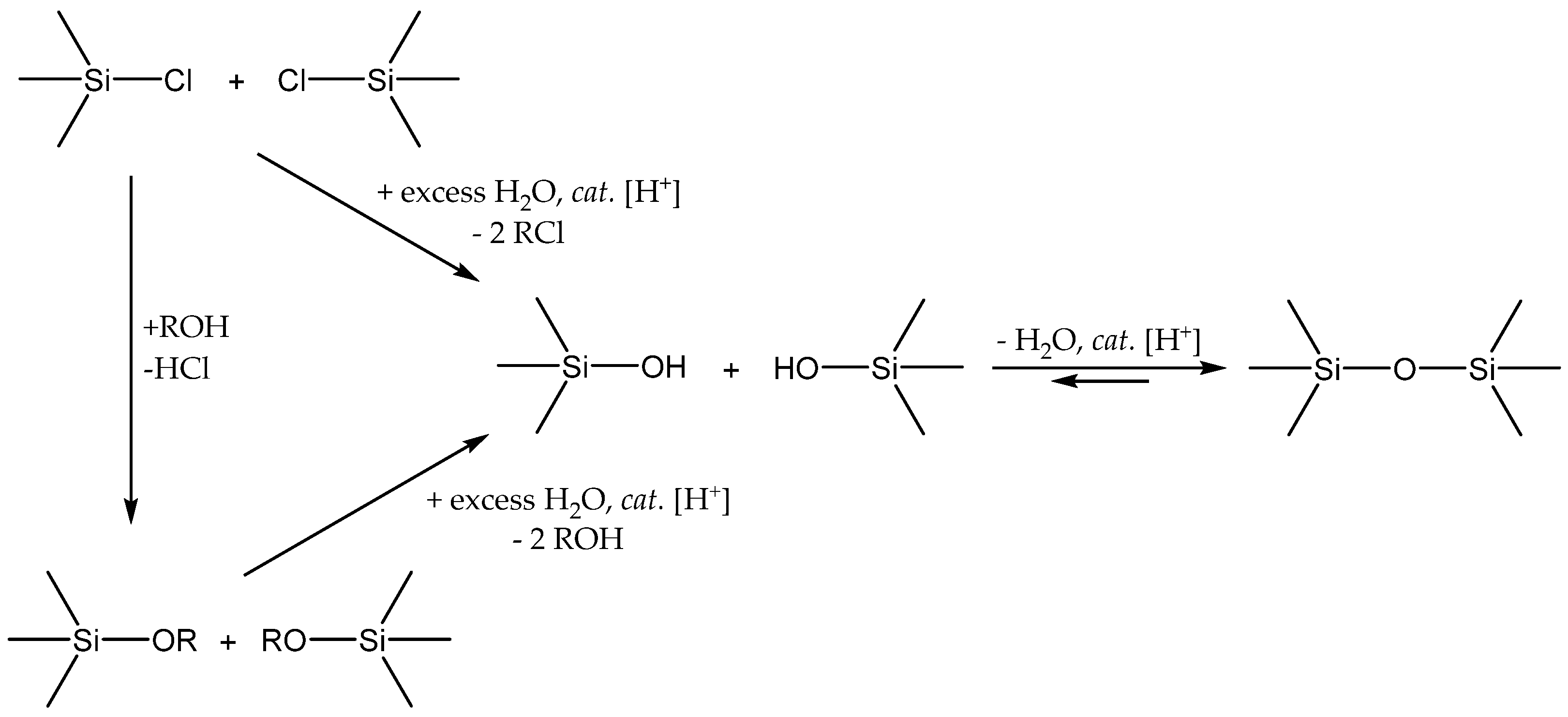
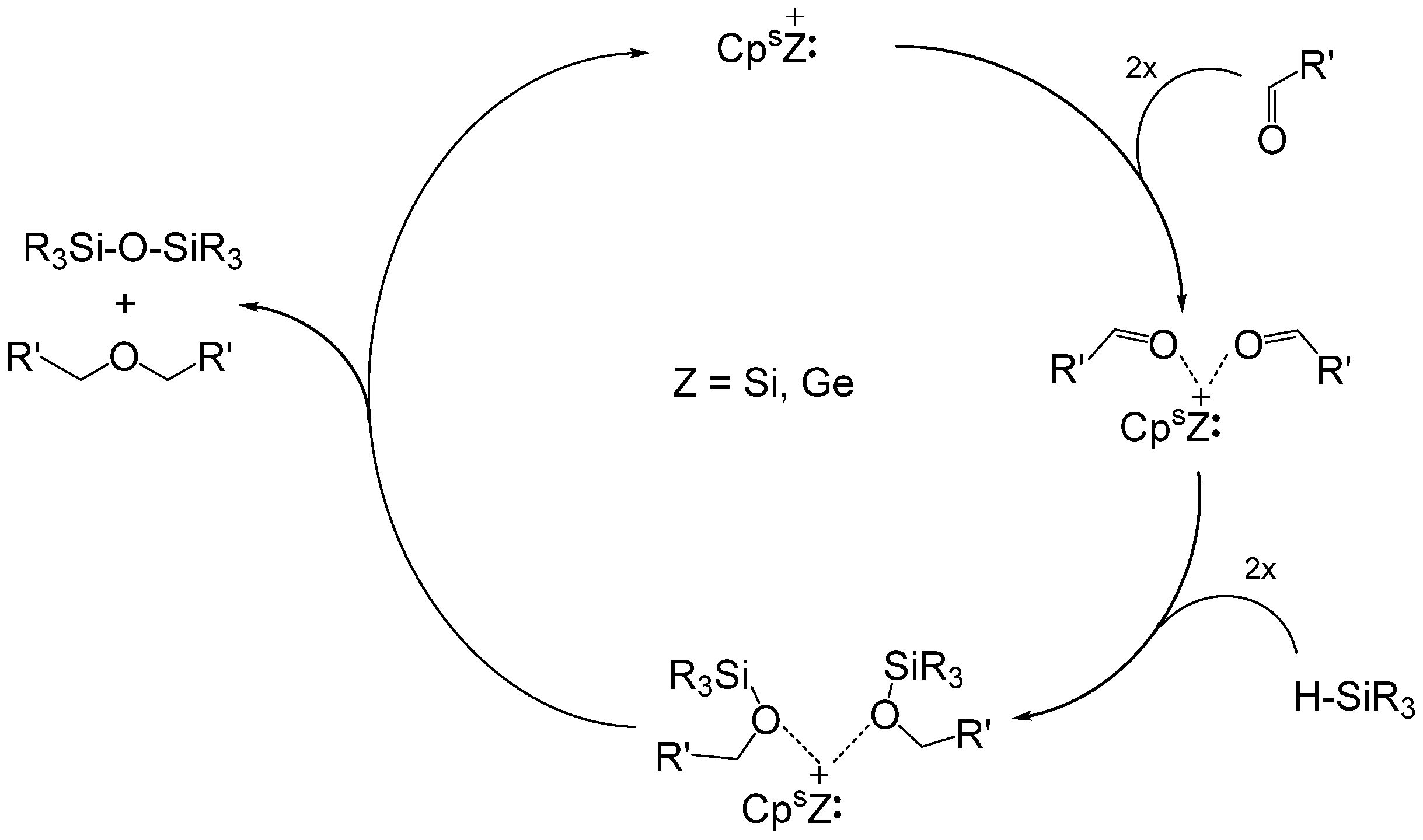
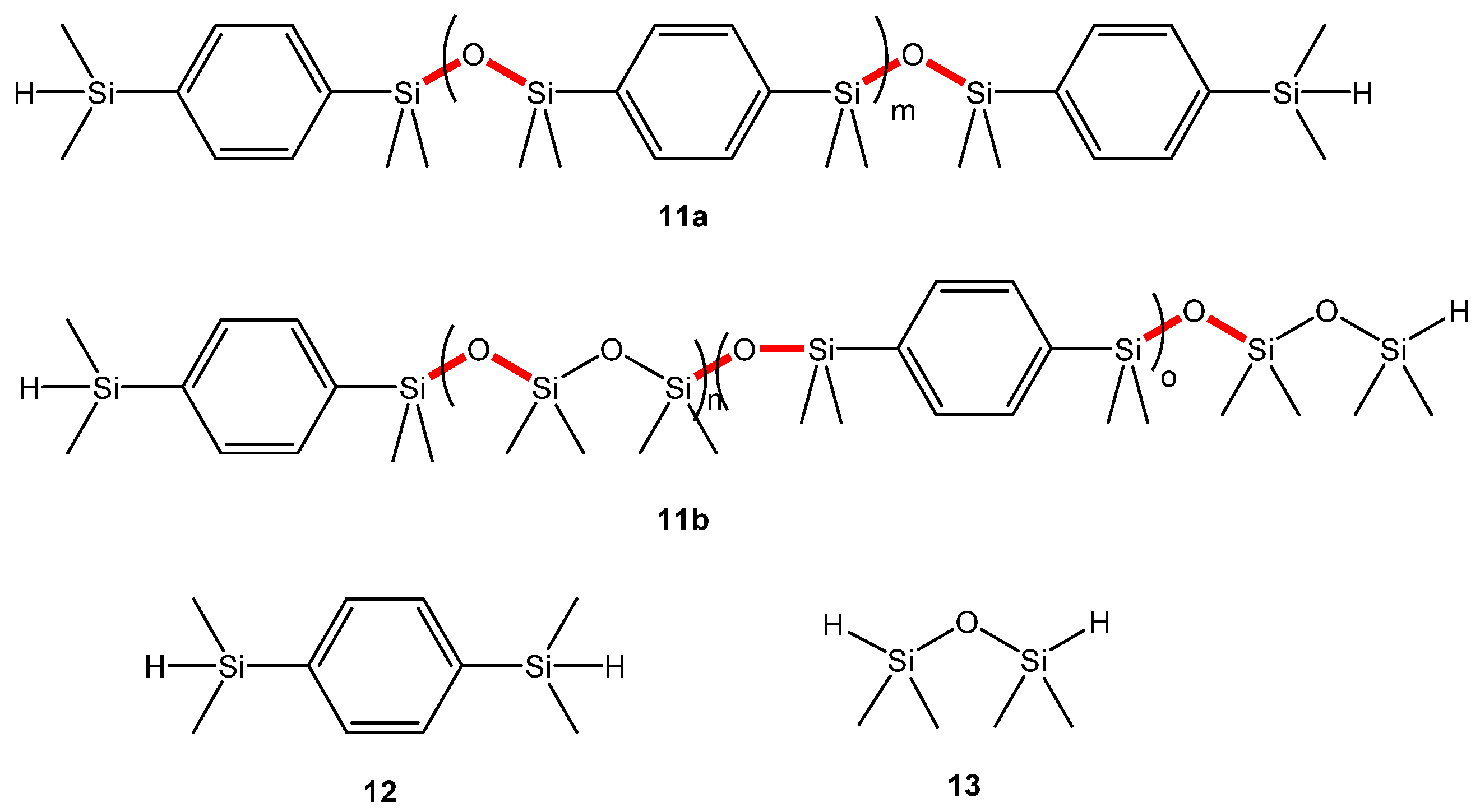
3.3. Siloxane Coupling Reactions
4. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References and Notes
- Chu, T.; Nikonov, G.I. Oxidative Addition and Reductive Elimination at Main-Group Element Centers. Chem. Rev. 2018, 118, 3608–3680. [Google Scholar] [CrossRef]
- Weetman, C.; Inoue, S. The Road Travelled: After Main-Group Elements as Transition Metals. Chem. Cat. Chem. 2018, 10, 4213–4228. [Google Scholar] [CrossRef]
- Harder, S. Early Main Group Metal Catalysis: Concepts and Reactions; John Wiley & Sons Inc.: Hoboken, NJ, USA, 2020. [Google Scholar]
- Stephan, D.W. Dogma breaking catalysis. Nature 2018, 553, 160–162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janssen-Müller, D.; Oestreich, M. Transition-Metal-Like Catalysis with a Main-Group Element: Bismuth-Catalyzed CF Coupling of Aryl Boronic Esters. Angew. Chem. Int. Ed. 2020, 59, 8328–8330. [Google Scholar] [CrossRef] [PubMed]
- Power, P.P. Main-group elements as transition metals. Nature 2010, 463, 171–177. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Lou, S.-J.; Hou, Z. Electron-deficient boron-based catalysts for C-H bond functionalization. Chem. Soc. Rev. 2021, 50, 1945–1967. [Google Scholar] [CrossRef]
- Chardon, A.; Osi, A.; Mahaut, D.; Saida, A.B.; Berionni, G. Non-planar Boron Lewis Acids Taking the Next Step: Development of Tunable Lewis Acids, Lewis Superacids and Bifunctional Catalysts. Synlett 2020, 31, 1639–1648. [Google Scholar] [CrossRef]
- Brook, M.A. New Control Over Silicone Synthesis using SiH Chemistry: The Piers-Rubinsztajn Reaction. Chem. Eur. J. 2018, 24, 8458–8469. [Google Scholar] [CrossRef]
- Rao, B.; Kinjo, R. Boron-Based Catalysts for C-C Bond-Formation Reactions. Chem. Asian J. 2018, 13, 1279–1292. [Google Scholar] [CrossRef]
- Empel, C.; Nguyen, T.V.; Koenigs, R.M. Tropylium-Catalyzed O-H Insertion Reactions of Diazoalkanes with Carboxylic Acids. Org. Lett. 2021, 23, 548–553. [Google Scholar] [CrossRef]
- Omoregbee, K.; Luc, K.N.H.; Dinh, A.H.; Nguyen, T.V. Tropylium-promoted prenylation reactions of phenols in continuous flow. J. Flow Chem. 2020, 10, 161–166. [Google Scholar] [CrossRef]
- Shang, W.; Duan, D.; Liu, Y.; Lv, J. Carbocation Lewis Acid TrBF4-Catalyzed 1,2-Hydride Migration: Approaches to (Z)-α, β-Unsaturated Esters and α-Branched β-Ketocarbonyls. Org. Lett. 2019, 21, 8013–8017. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Lv, J.; Luo, S. Enantioselective Diels-Alder reaction of anthracene by chiral tritylium catalysis. Beilstein J. Org. Chem. 2019, 15, 1304–1312. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Liu, H.; Guo, H. Recent Advances in Phosphonium Salt Catalysis. Adv. Synth. Catal. 2021, 363, 2023–2036. [Google Scholar] [CrossRef]
- Waked, A.E.; Chitnis, S.S.; Stephan, D.W. P(V) dications: Carbon-based Lewis acid initiators for hydrodefluorination. Chem. Commun. 2019, 55, 8971–8974. [Google Scholar] [CrossRef] [PubMed]
- Bayne, J.M.; Stephan, D.W. C-F Bond Activation Mediated by Phosphorus Compounds. Chem. Eur. J. 2019, 25, 9350–9357. [Google Scholar] [CrossRef]
- Chitnis, S.S.; Krischer, F.; Stephan, D.W. Catalytic Hydrodefluorination of C-F Bonds by an Air-Stable PIII Lewis Acid. Chem. Eur. J. 2018, 24, 6543–6546. [Google Scholar] [CrossRef]
- Dasgupta, A.; Richards, E.; Melen, R.L. Frustrated Radical Pairs: Insights from EPR Spectroscopy. Angew. Chem. Int. Ed. 2021, 60, 53–65. [Google Scholar] [CrossRef]
- Jupp, A.R.; Stephan, D.W. New Directions for Frustrated Lewis Pair Chemistry. Trends Chem. 2019, 1, 35–48. [Google Scholar] [CrossRef]
- Lam, J.; Szkop, K.M.; Mosaferi, E.; Stephan, D.W. FLP catalysis: Main group hydrogenations of organic unsaturated substrates. Chem. Soc. Rev. 2019, 48, 3592–3612. [Google Scholar] [CrossRef]
- Liberman-Martin, A.L.; Bergman, R.G.; Don Tilley, T. Lewis Acidity of Bis(perfluorocatecholato)silane: Aldehyde Hydrosilylation Catalyzed by a Neutral Silicon Compound. J. Am. Chem. Soc. 2015, 137, 5328. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panisch, R.; Bolte, M.; Müller, T. Hydrogen- and Fluorine-Bridged Disilyl Cations and Their Use in Catalytic C-F Activation. J. Am. Chem. Soc. 2006, 128, 9676–9682. [Google Scholar] [CrossRef] [PubMed]
- Lühmann, N.; Panisch, R.; Müller, T. A catalytic C-C bond-forming reaction between aliphatic fluorohydrocarbons and arylsilanes. Appl. Organomet. Chem. 2010, 24, 533–537. [Google Scholar] [CrossRef]
- Sinhababu, S.; Singh, D.; Sharma, M.K.; Siwatch, R.K.; Mahawar, P.; Nagendran, S. Ge(II) cation catalyzed hydroboration of aldehydes and ketones. Dalton Trans. 2019, 48, 4094–4100. [Google Scholar] [CrossRef] [PubMed]
- Roy, M.M.D.; Fujimoro, S.; Ferguson, M.J.; McDonald, R.; Tokitoh, N.; Rivard, E. Neutral, Cationic and Hydride-substituted Siloxygermylenes. Chem. Eur. J. 2018, 24, 14392–14399. [Google Scholar] [CrossRef] [PubMed]
- Fritz-Langhals, E.; Gowans, S. Process for metal-free hydrosilylation of unsaturated compounds using silicon(IV) cationic complexes as catalysts. WO 2017-EP83669 (19 December 2017). Chem. Abstr. 2019, 171, 1249217. [Google Scholar]
- Fritz-Langhals, E.; Gowans, S. Noble-metal free hydrosilylatable mixture. PCT/EP2020/060581, 4 April 2020.
- Jutzi, P.; Mix, A.; Rummel, B.; Schoeller, W.W.; Neumann, B.; Stammler, H.-G. The (Me5C5)Si+ Cation: A Stable Derivative of HSi+. Science 2004, 305, 849–851. [Google Scholar] [CrossRef]
- Fritz-Langhals, E.; Werge, S.; Kneissl, S.; Piroutek, P. Novel Si(II)+ and Ge(II)+ Compounds as Efficient Catalysts in Organosilicon Chemistry: Siloxane Coupling Reaction. Org. Process Res. Dev. 2020, 24, 1484–1495. [Google Scholar] [CrossRef]
- Leszczynska, K.; Mix, A.; Berger, R.J.F.; Rummel, B.; Neumann, B.; Stammler, H.-G.; Jutzi, P. The Pentamethylcyclopentadienylsilicon(II) Cation as a Catalyst for the Specific Degradation of Oligo(ethyleneglycol) Diethers. Angew. Chem. Int. Ed. 2011, 50, 6843–6846. [Google Scholar] [CrossRef]
- Kühler, T.; Jutzi, P. Decamethylsilicocene: Synthesis, Structure, Bonding and Chemistry. Adv. Organomet. Chem. 2003, 49, 1–34. [Google Scholar] [CrossRef]
- Ghana, P.; Arz, M.I.; Schnakenburg, G.; Straßmann, M.; Filippou, A.C. Metal−Silicon Triple Bonds: Access to [Si(η5 - C5Me5)]+ from SiX2(NHC) and its Conversion to the Silylidyne Complex [TpMe(CO)2MoSi(η3 -C5Me5)] (TpMe = κ3 -N,N′,N″- hydridotris(3,5-dimethyl-1-pyrazolyl)borate). Organometallics 2018, 37, 772–780. [Google Scholar] [CrossRef]
- Schöpper, A.; Saak, W.; Weidenbruch, M. Molecular structure of decamethylgermanocene in the solid state. J. Organomet. Chem. 2006, 691, 809–810. [Google Scholar] [CrossRef]
- Jutzi, P.; Schlüter, E.; Hursthouse, M.B.; Arif, A.M.; Short, R.L. Synthesis of trimethylsilylated germanocenes; X-ray structure of and steric effects in hexakis(trimethylsilyl)germanocene. J. Organomet. Chem. 1986, 299, 285–295. [Google Scholar] [CrossRef]
- Bochmann, S.; Böhme, U.; Brendler, E.; Friebel, M.; Gerwig, M.; Gründler, F.; Günther, B.; Kroke, E.; Lehnert, R.; Ruppel, L. Unexpected Formation of the Highly Symmetric Borate Ion [B(SiCl3)4]−. Eur. J. Inorg. Chem. 2021, 2021, 2583–2594. [Google Scholar] [CrossRef]
- Fritz-Langhals, E. Silicon(II) Cation Cp*Si:+ X-: A New Class of Efficient Catalysts in Organosilicon Chemistry. Org. Process Res. Dev. 2019, 23, 2369–2377. [Google Scholar] [CrossRef] [Green Version]
- Trögel, D.; Stohrer, J. Recent advances and actual challenges in late transition metal catalyzed hydrosilylation of olefins from an industrial point of view. Coord. Chem. Rev. 2011, 255, 1440–1459. [Google Scholar] [CrossRef]
- Marciniec, B. Comprehensive Handbook of Hydrosilylation, 1st ed.; Pergamon Press: Oxford, UK, 1992. [Google Scholar]
- Matisons, J. Hydrosilylation in Advances in Silicon Science, 1st ed.; Springer: Dordrecht, The Netherlands, 2009; Volume 1, pp. 175–179. [Google Scholar]
- Marciniec, B. Functionalisation and Cross-Linking of Organosilicon Polymers in: Hydrosilylation, 1st ed.; Marciniec, B., Ed.; Springer: Dordrecht, The Netherlands, 2009; pp. 159–189. [Google Scholar]
- Duarte de Almeida, A.; Wang, H.; Junge, K.; Cui, X.; Beller, M. Recent Advances in Catalytic Hydrosilylations: Developments beyond Traditional Platinum Catalysts. Angew. Chem. Int. Ed. 2021, 60, 550–565. [Google Scholar] [CrossRef] [PubMed]
- Obligation, J.V.; Chirik, P.J. Earth-abundant transition metal catalysts for alkene hydrosilylation and hydroboration. Nat. Chem. Rev. 2018, 2, 15–34. [Google Scholar] [CrossRef] [PubMed]
- Rubin, M.; Schwier, T.; Gevorgyan, V. Highly Efficient B(C6F5)3-Catalyzed Hydrosilylation of Olefins. J. Org. Chem. 2002, 67, 1936–1940. [Google Scholar] [CrossRef]
- Oestreich, M.; Hermeke, J.; Mohr, J. A unified survey of Si-H and H-H bond activation catalysed by electron-deficient boranes. Chem. Soc. Rev. 2015, 44, 2202–2220. [Google Scholar] [CrossRef] [Green Version]
- Andrews, R.J.; Chitnis, S.S.; Stephan, D.W. Carbonyl and olefin hydrosilylation mediated by an air-stable phosphorus(III) dication under mild conditions. Chem. Commun. 2019, 55, 5599–5602. [Google Scholar] [CrossRef] [PubMed]
- Rubinsztajn, S.; Chojnowski, J.; Cypryk, M.; Mizerska, U.; Fortuniak, W.; Bak-Sypien, I.I. Kinetic and mechanistic studies of the transformation of the catalyst, tris(pentafluorophenyl)borane, in the presence of silyl and germyl hydrides. J. Catal. 2019, 379, 90–99. [Google Scholar] [CrossRef]
- Picture of silicone elastomer taken by the author for illustration.
- Parks, D.J.; Blackwell, J.M.; Piers, W.E. Studies on the mechanism of B(C6F5)3-catalyzed hydrosilylation of carbonyl functions. J. Org. Chem. 2000, 65, 3090–3098. [Google Scholar] [CrossRef] [PubMed]
- Fritz-Langhals, E.; Weidner, R.; Werge, S. The oxygen effect is described experimentally in: Cationic Ge(II) compounds, process for preparing same, and their use as catalysts in hydrosilylation. WO 2020 228922 (10 May 2019). Chem. Abstr. 2019, 174, 8196. [Google Scholar]
- Fritz-Langhals, E. Process for producing hydridosilanes by disproportionation of hydrosiloxanes in the presence of Si(II) cationic catalyst. WO 2018 103864 (9 December 2016). Chem. Abstr. 2018, 169, 63102. [Google Scholar]
- Chojnowski, J.; Fortuniak, W.; Kurjata, J.; Rubinsztajn, S.; Cella, J.A. Oligomerization of Hydrosiloxanes in the Presence of Tris(pentafluorphenyl)borane. Macromolecules 2006, 39, 3802–3807. [Google Scholar] [CrossRef]
- Fritz-Langhals, E. Crosslinking of hydridosiloxanes with silicon(II) compounds. WO 2018215056 (23 May 2017). Chem. Abstr. 2018, 170, 31073. [Google Scholar]
- Chojnowski, J.; Kurjata, J.; Fortuniak, W.; Rubinsztajn, S.; Cella, J.A.; Trzebicka, B. Hydride Transfer Ring-Opening Polymerization of a Cyclic Oligomethylhydrosiloxane. Route to a Polymer of Closed Multicyclic Structure. Macromolecules 2012, 45, 2654–2661. [Google Scholar] [CrossRef]
- Fritz-Langhals, E. Process for producing organosiloxanes comprising triorgansilyloxy groups. WO 2020 025144 (3 August 2018). Chem. Abstr. 2020, 172, 221746. [Google Scholar]
- Cervantes, J.; Zárraga, R.; Salazar-Hernández, C. Organotin catalysts in organosilicon chemistry. Appl. Organometal. Chem. 2012, 26, 157–163. [Google Scholar] [CrossRef]
- Wang, D.; Klein, J.; Mejia, E. Catalytic Systems for the Cross-Linking of Organosilicon Polymers. Chem. Asian J. 2017, 12, 1180–1197. [Google Scholar] [CrossRef] [PubMed]
- Lukevics, E.; Ignatovich, L. Biological activity of organogermanium compounds, chapter 23. In The Chemistry of Organic Germanium, Tin and Lead Compounds, 1st ed.; Rappoport, Z., Ed.; Wiley: New York, NY, USA, 2002; Volume 2. [Google Scholar]
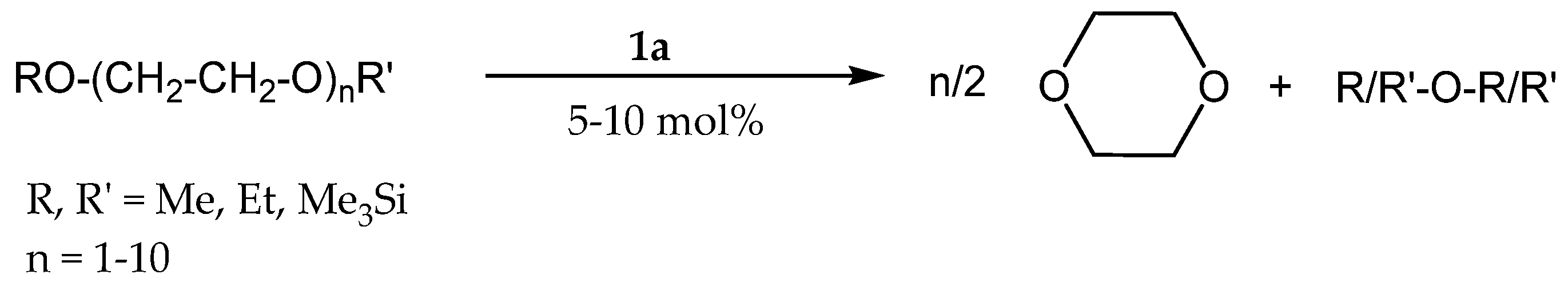
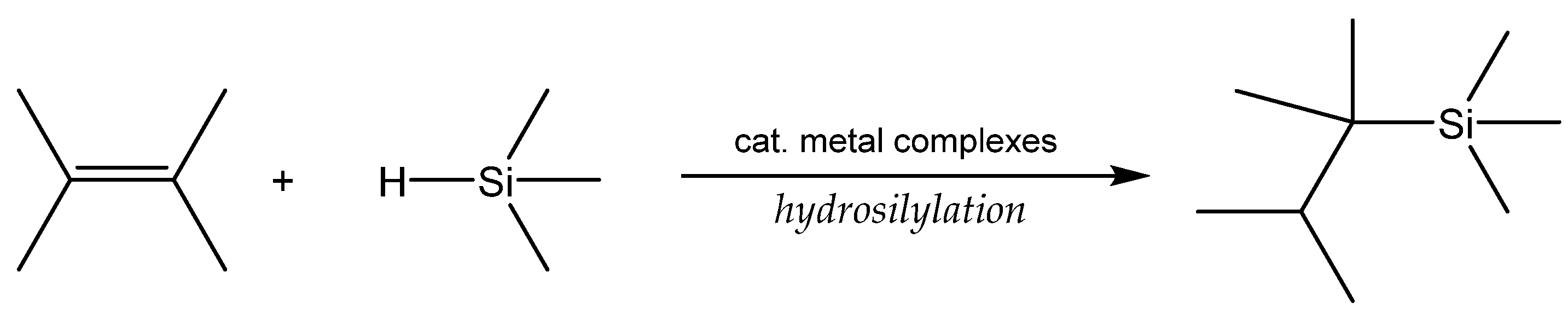
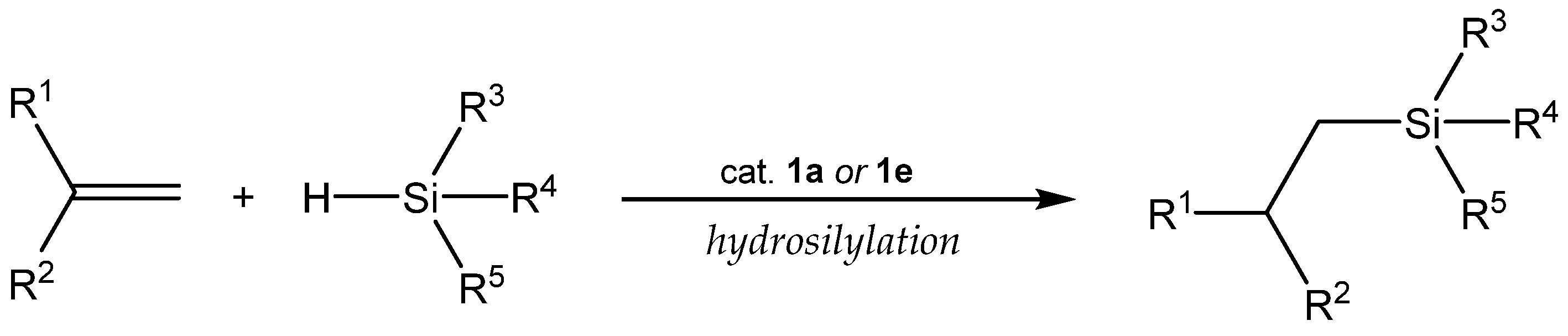
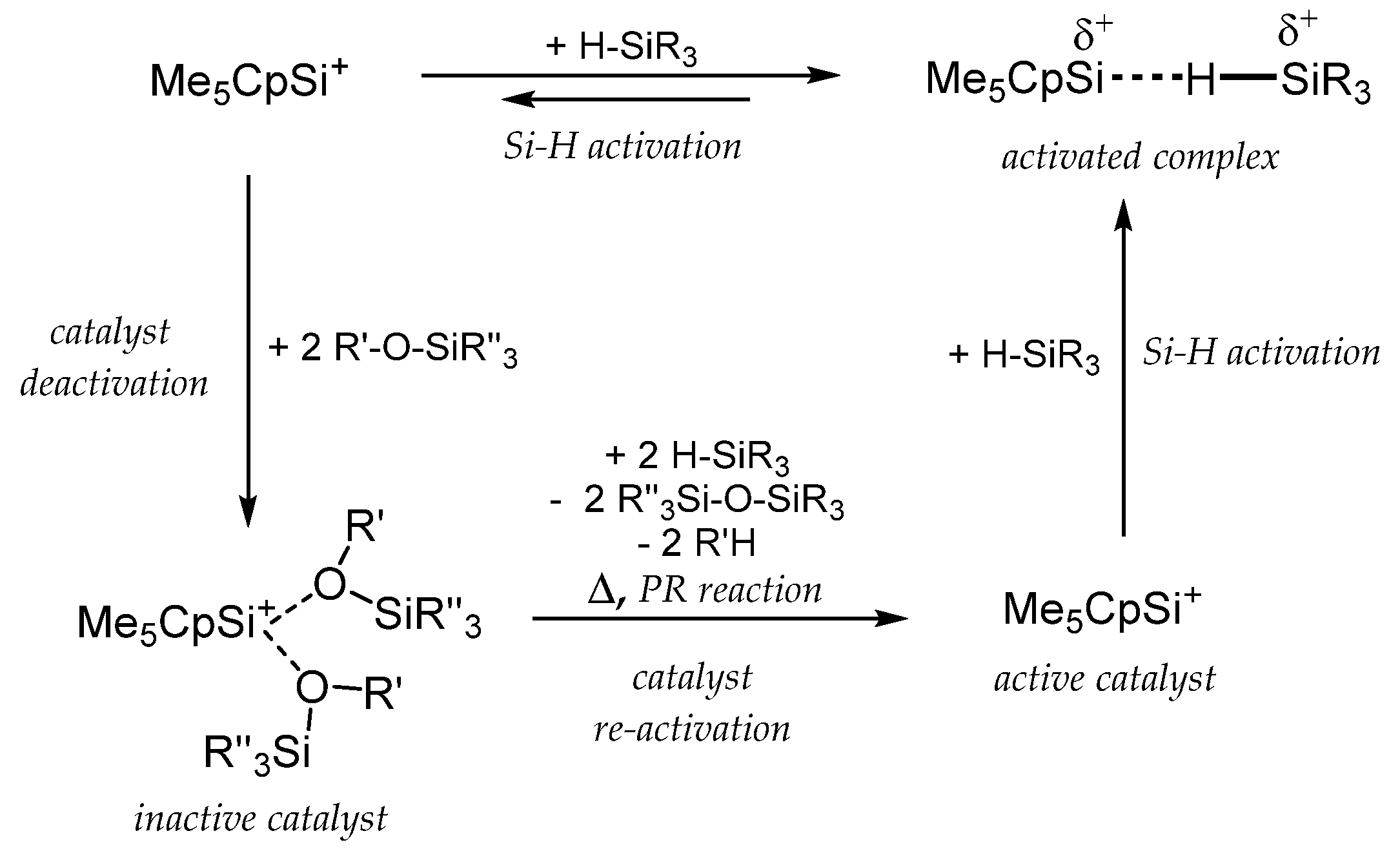




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Educt | Rn | H+[WCA]− | Product | Number | Ref. |
|---|---|---|---|---|---|---|
| 1 | 3 | Me5 | H+(OEt)2B(ArF)4− | Me5CpSi+B(ArF)4− | 1a 1 | [33] |
| 2 | 3 | Me5 | H+B(SiCl3)4− 2 | Me5CpSi+B(SiCl3)4− | 1b | [30] |
| 3 | 4 | Me5 | H+B(SiCl3)4− | Me5CpGe+B(SiCl3)4− | 2b | [30] |
| 4 | 4 | 1,2,4-TMS3 | H+(OEt)2B(ArF)4− | (1,2,4-TMS)3CpGe+B(ArF)4− | 2c | [30] |
| 5 | 4 | 1,2,4-TMS3 | H+B(SiCl3)4− | (1,2,4-TMS)3CpGe+B(SiCl3)4− | 2d | [30] |
| Entry | Educt | Electrophile | Product | Number | Ref. |
|---|---|---|---|---|---|
| 1 | Si | Ph3C+B(ArF)4− | Me5CpSi+B(ArF)4− | 1a | [37] |
| 2 | Ge | Ph3C+B(ArF)4− | Me5CpGe+B(ArF)4− | 2a | [30] |
| 3 | Si | Ph3C+B(C6F4-4-SiMe2t-Bu)4− | Me5CpSi+(C6F4-4-SiMe2t-Bu)4− | 1e | [37] |
| 4 | Si | Ph3C+Al[OC(CF3)3]4− | Me5CpSi+Al[OC(CF3)3]4− | 1f | [37] |
| 5 | Ge | Ph3C+Al[OC(CF3)3]4− | Me5CpGe+Al[OC(CF3)3]4− | 2f | [30] |
| 6 | Si | Ph3C+Al[OCH(CF3)2]4− | Me5CpSi+Al[OCH(CF3)2]4− | 1g | [37] |
| 7 | Si | B(ArF)3 | Me5CpSi+HB(ArF)3− | 1h | [37] |
| Entry | R1 | R2 | R3 | R4 | R5 | Mol% 1a | T (°C) | Yield (%) |
|---|---|---|---|---|---|---|---|---|
| 1 | Ph | Me | Et | Et | Et | 0.1 | 50 | >98 |
| 2 | Ph | Me | Me | Me | Ph | 0.06 | 50 | >98 |
| 3 | Ph | Me | Me | OSiMe3 | OSiMe3 | 0.2 | 25 | >98 |
| 4 | Ph | Me | Me | Me | Cl | 0.1 | 25 | 90 |
| 5 | n-C4H9 | H | Me | Me | OSiMe3 | 0.09 | 25 | 98 |
| 6 | n-C4H9 | H | Me | OSiMe3 | OSiMe3 | 0.05 | 25 | 81 |
| 7 | n-C4H9 | H | Me | Me | Cl | 0.1 | 50 | >90 |
| 8 | Ph | Me | Me | Me | OSiMe3 | 0.0013 | 25 | >97 |
| 9 | Ph | Me | Me | Me | OSiMe3 | 0.012 | 25 | >97 1 |
| 10 2 | Ph | Me | Me | Me | OSiMe3 | 0.012 | 25 | >90 |
| 11 2 | Ph | Me | Me | Me | OSiMe3 | 0.0096 | 25 | >98 3 |
| Product | Si-OEt | Si-H | Mol% 1e | T (°C)/t (h) | Yield (%) |
|---|---|---|---|---|---|
| (PMDSO)2SiPh2 1 | Ph2Si(OEt)2 | TMS-O-SiMe2-H | 0.0014 | 60/4 | >95 |
| (ClMe2SiO)2SiPh2 | Ph2Si(OEt)2 | Me2ClSi-H | 0.13 | 60/2 | >95 |
| 82 | Ph2Si(OEt)2 | 1,4-(SiMe2H)benzene | 0.11 | 60/2 | >95 |
| 93 | Ph2Si(OEt)2 | H-SiMe2-O-SiMe2-H | 0.10 | 60/2 | >95 |
| 104 | Me3SiOEt | TMS-O-D9-DH24-TMS | 0.033 | 60/1 | >95 |
| Entry | Si-H | Oxidant (Equiv.) | Catalyst (Mol%) | Yield (%) 1 |
|---|---|---|---|---|
| 1 | Et3Si-H | hexanal (1.0) | 1a (0.059) | 96/96 |
| 2 | Et3Si-H | hexanal (1.0) | 1f (0.061) | 97/97 |
| 3 | Et3Si-H | hexanal (1.0) | 2a (0.062) | >95/>95 |
| 4 | Et3Si-H | hexanal (1.0) | 2f (0.062) | 97/97 |
| 5 | Et3Si-H | hexanal (1.1) | 2c (0.059) | 97/97 |
| 6 | Et3Si-H | hexanal (1.0) | 1b(0.065) | 97/97 |
| 7 | Et3Si-H | hexanal (1.0) | 2b(0.065) | 98/98 |
| 8 | Et3Si-H | hexanal (1.1) | 2d(0.059) | 98/98 |
| 9 | Et3Si-H | methylethylketone (1.0) | 1a (0.050) | 79/79 2 |
| 10 | Et3Si-H | cyclobutanone (0.9) | 2a (0.01) | 95/>90 |
| 11 | Ph 3Me2Si-H | paraldehyde (1.1) | 2a (0.0055) | 97 4 |
| 12 | Ph2MeSi-H | paraldehyde (1.1) | 2a (0.0065) | 96 4 |
| 13 | PhVi 5MeSi-H | paraldehyde (1.1) | 2a (0.0029) | 95 4 |
| 14 | (i-C3H7)3Si-H | paraldehyde (1.1) | 2a (0.0058) | 6 |
| 15 | Et3Si-H | paraldehyde (1.1) | 2d (0.0050) | 98 |
| 16 | PhMe2Si-H | paraldehyde (1.1) | 2d (0.0027) | 97 4 |
| 17 | PhMe2Si-H | paraldehyde (1.1) | 2d (0.0001) | 98 4 |
| 18 | PhViMeSi-H | paraldehyde (1.1) | 2d (0.0054) | 94 4 |
| 19 | PhViMeSi-H | paraldehyde (1.1) | 2d (0.0027) | 95 4 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fritz-Langhals, E. Main Group Catalysis: Cationic Si(II) and Ge(II) Compounds as Catalysts in Organosilicon Chemistry. Reactions 2021, 2, 442-456. https://doi.org/10.3390/reactions2040028
Fritz-Langhals E. Main Group Catalysis: Cationic Si(II) and Ge(II) Compounds as Catalysts in Organosilicon Chemistry. Reactions. 2021; 2(4):442-456. https://doi.org/10.3390/reactions2040028
Chicago/Turabian StyleFritz-Langhals, Elke. 2021. "Main Group Catalysis: Cationic Si(II) and Ge(II) Compounds as Catalysts in Organosilicon Chemistry" Reactions 2, no. 4: 442-456. https://doi.org/10.3390/reactions2040028
APA StyleFritz-Langhals, E. (2021). Main Group Catalysis: Cationic Si(II) and Ge(II) Compounds as Catalysts in Organosilicon Chemistry. Reactions, 2(4), 442-456. https://doi.org/10.3390/reactions2040028