
Synthesis of a Poly(3-dodecylthiophene) Bearing Aniline Groups for the Covalent Functionalization of Carbon Nanotubes

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. General Methods
2.2. Synthesis of the Monomers
2.3. Synthesis of the Polymers
2.4. Formation of the Polymer/SWNT Hybrids
3. Results and Discussion
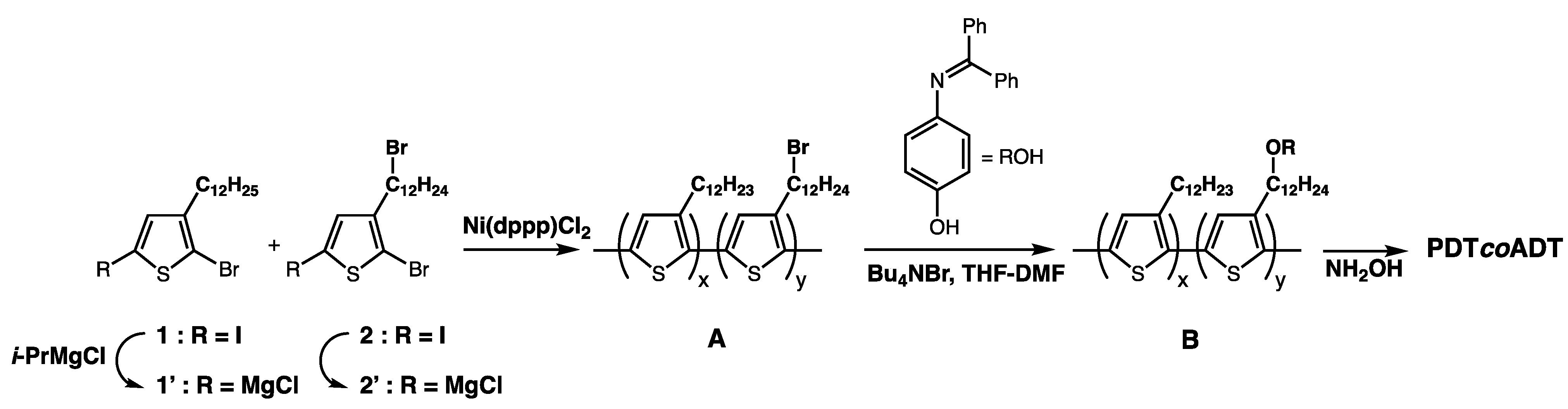
3.1. Design of the Compounds
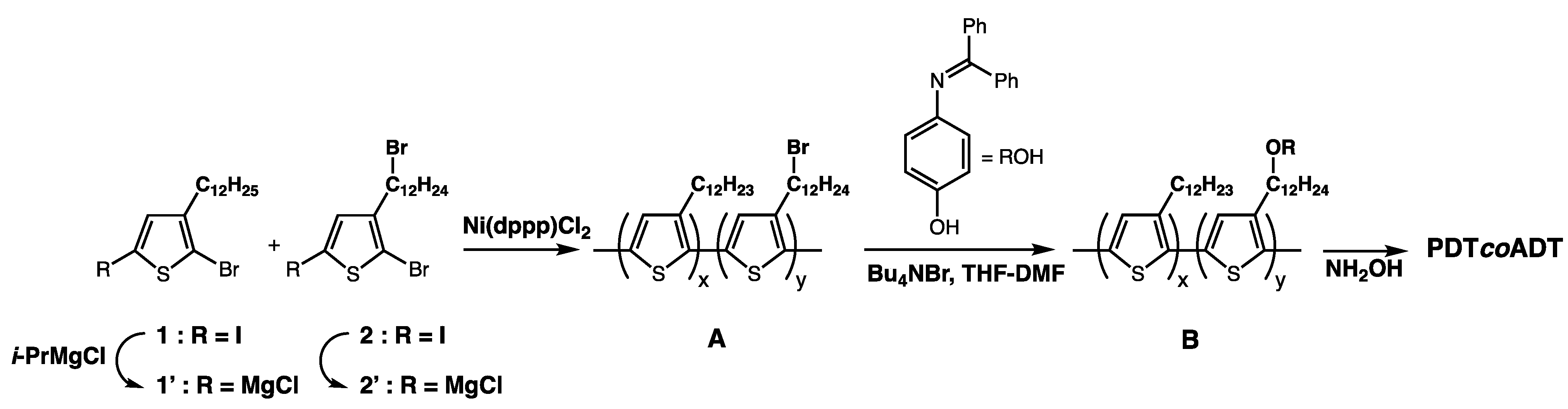
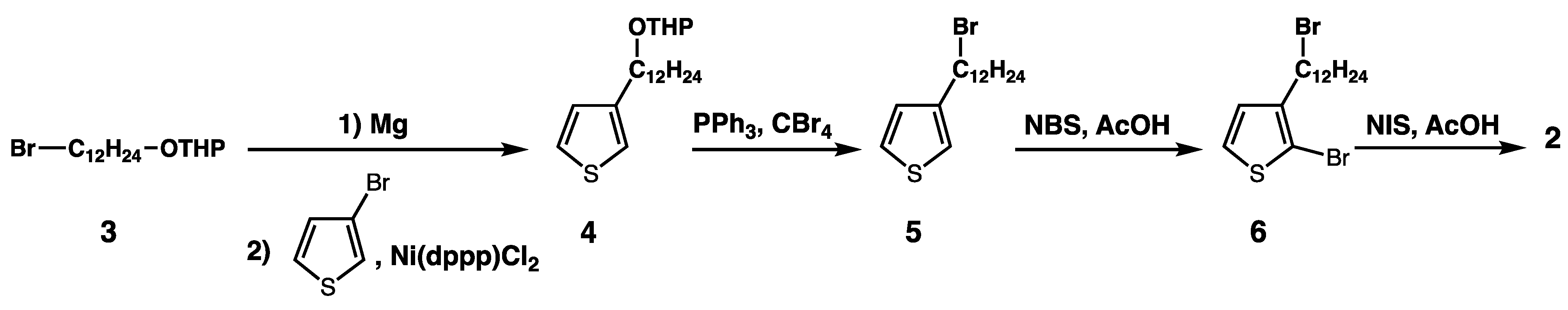
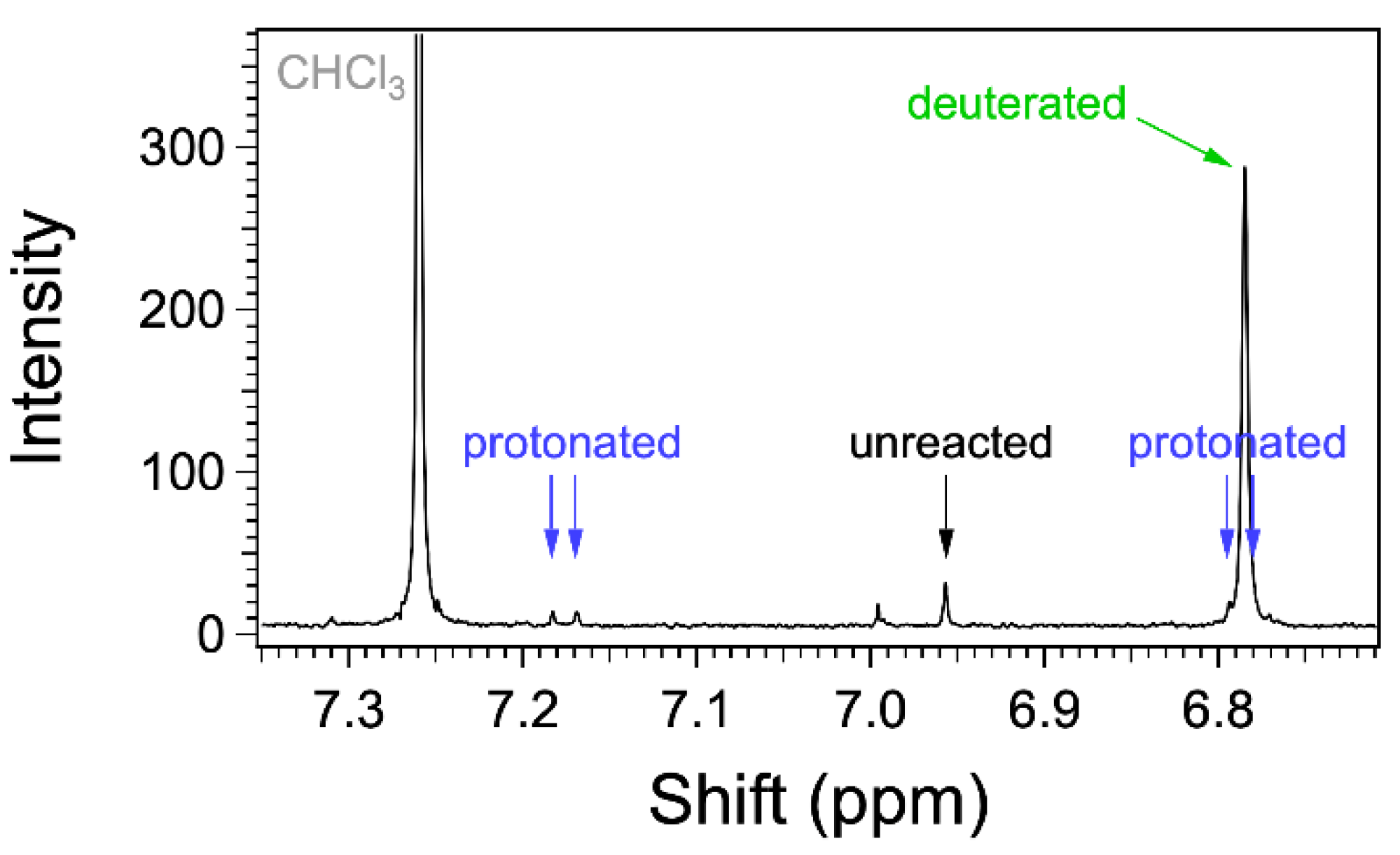
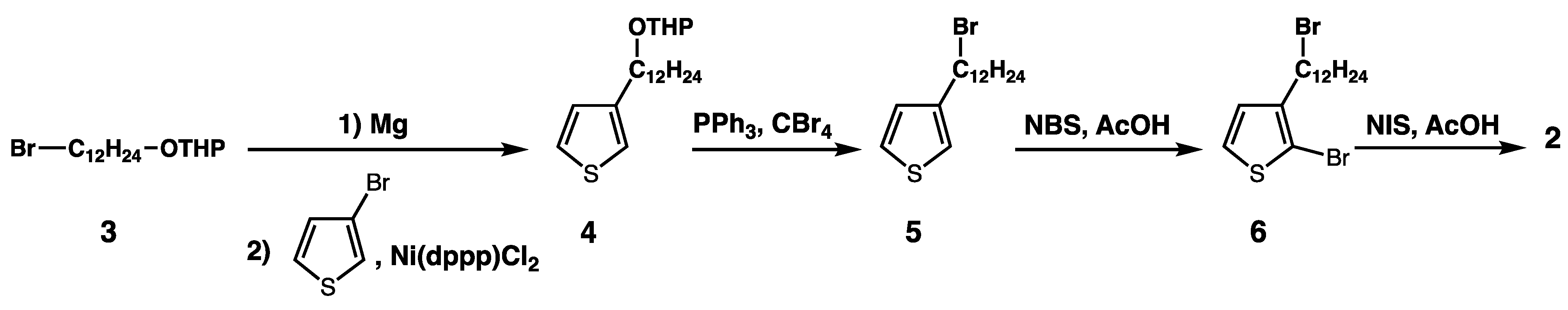
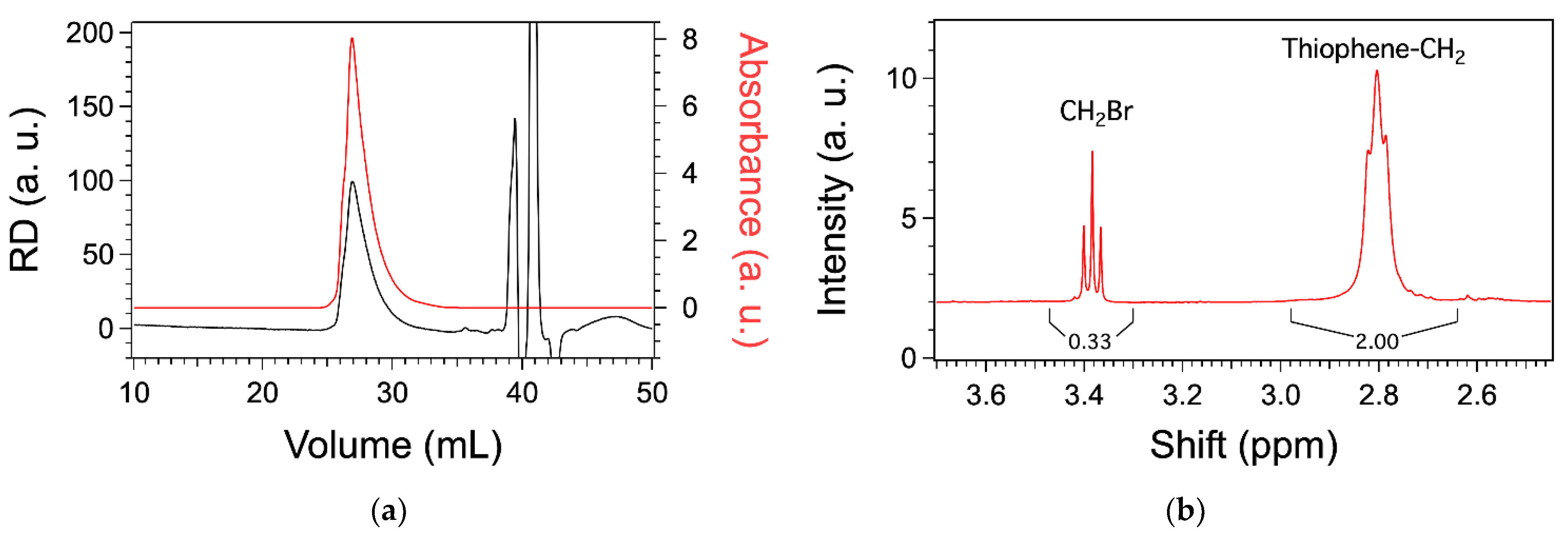
3.2. Syntheses of the Monomers and Polymers
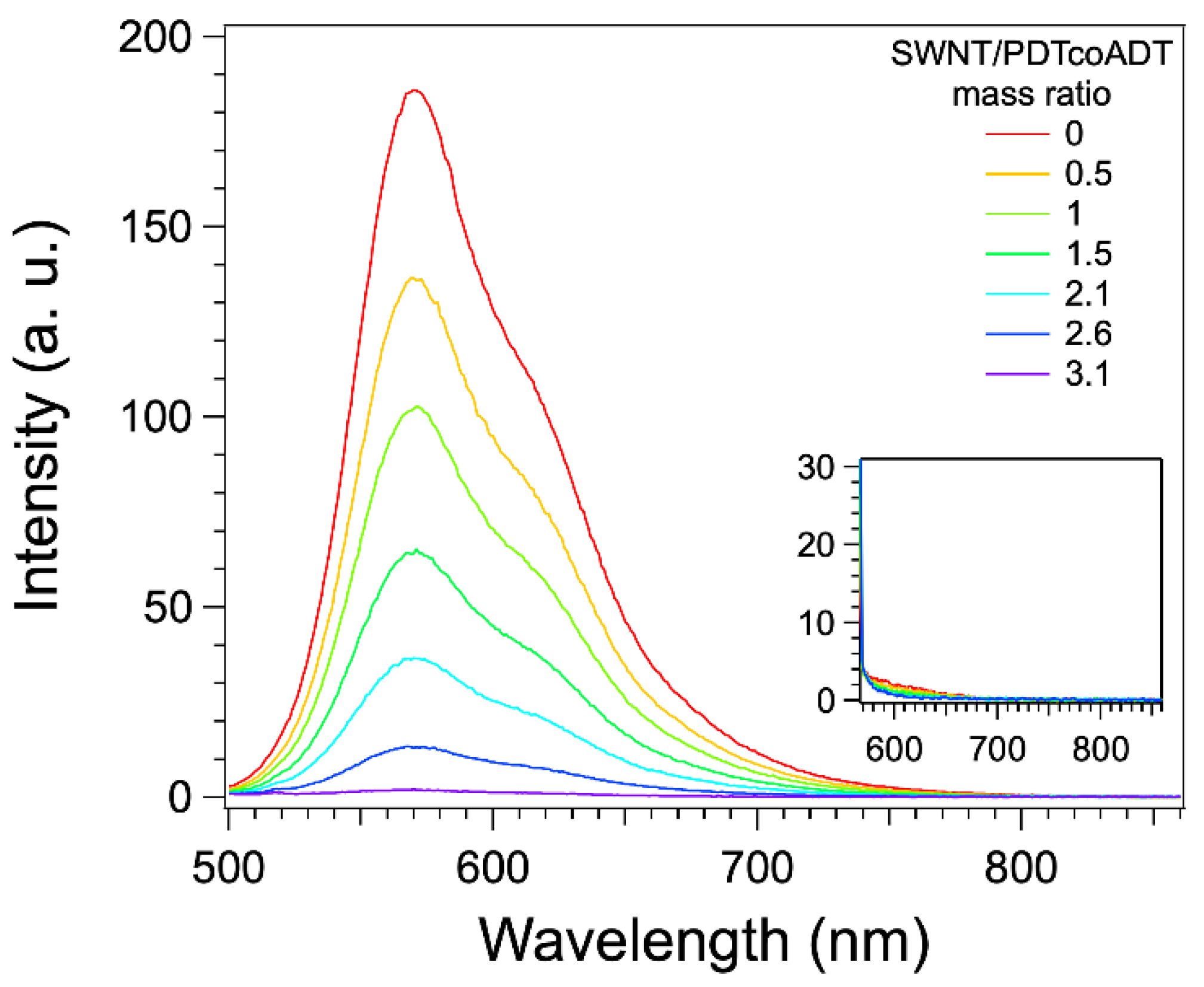
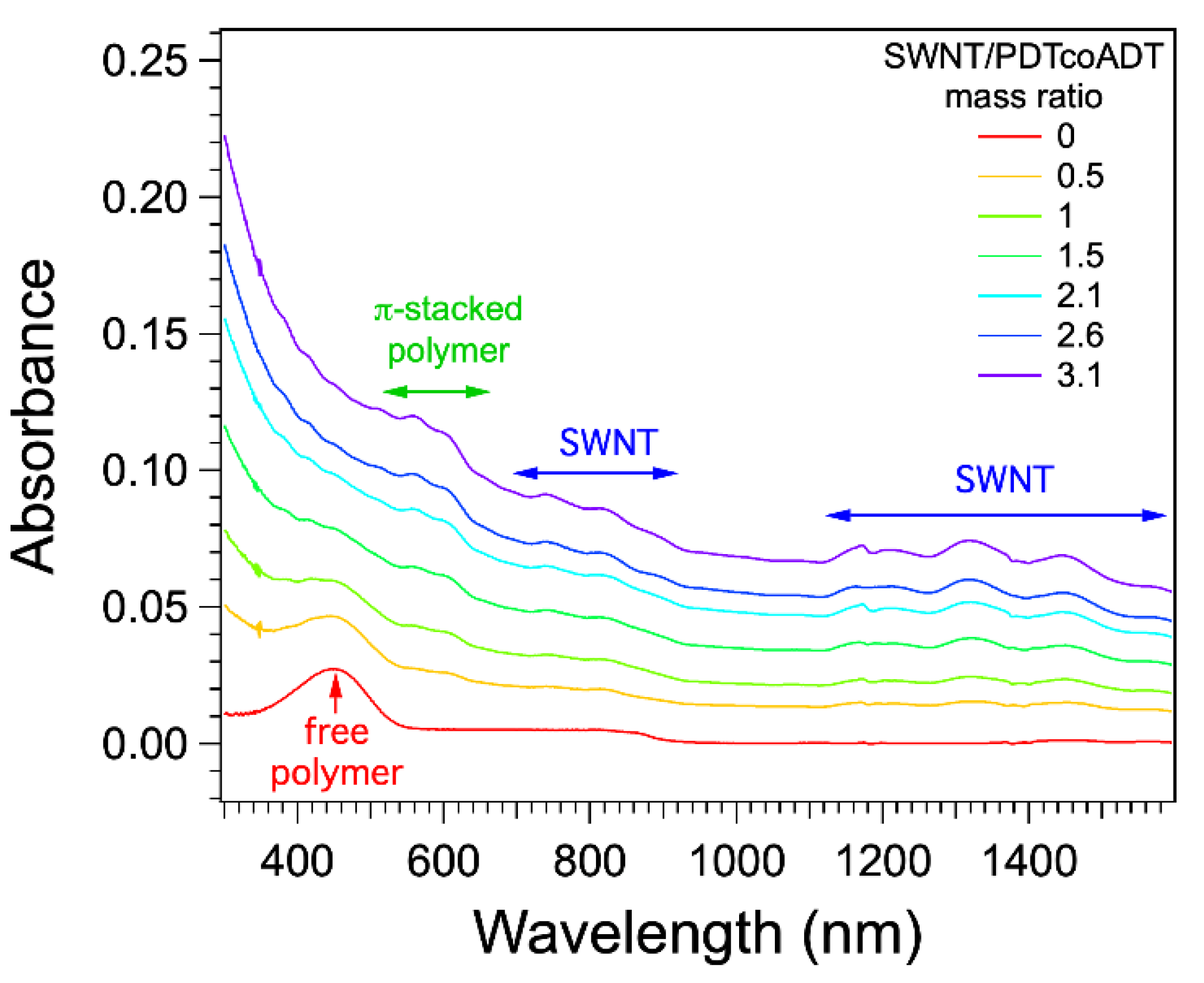
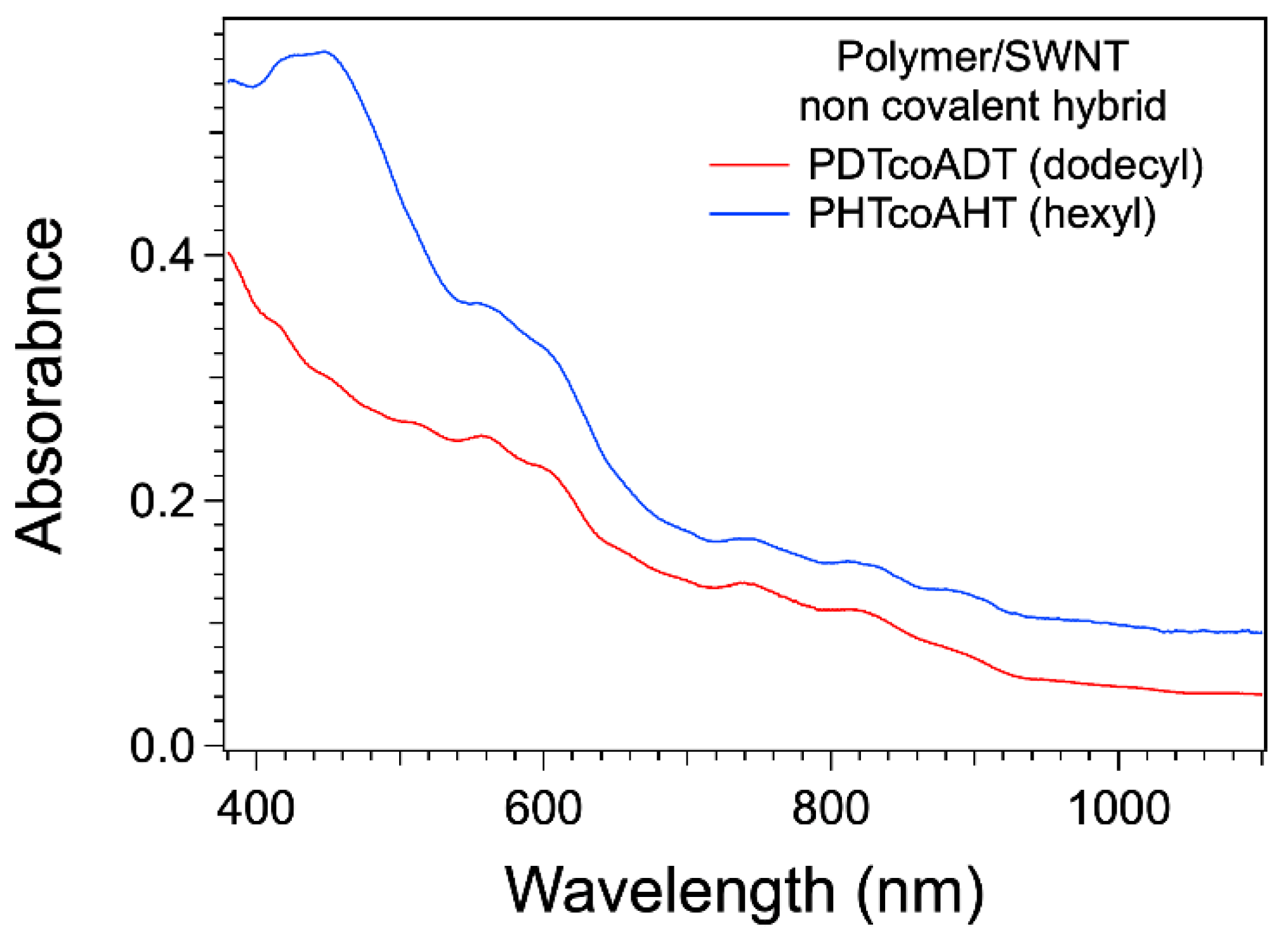
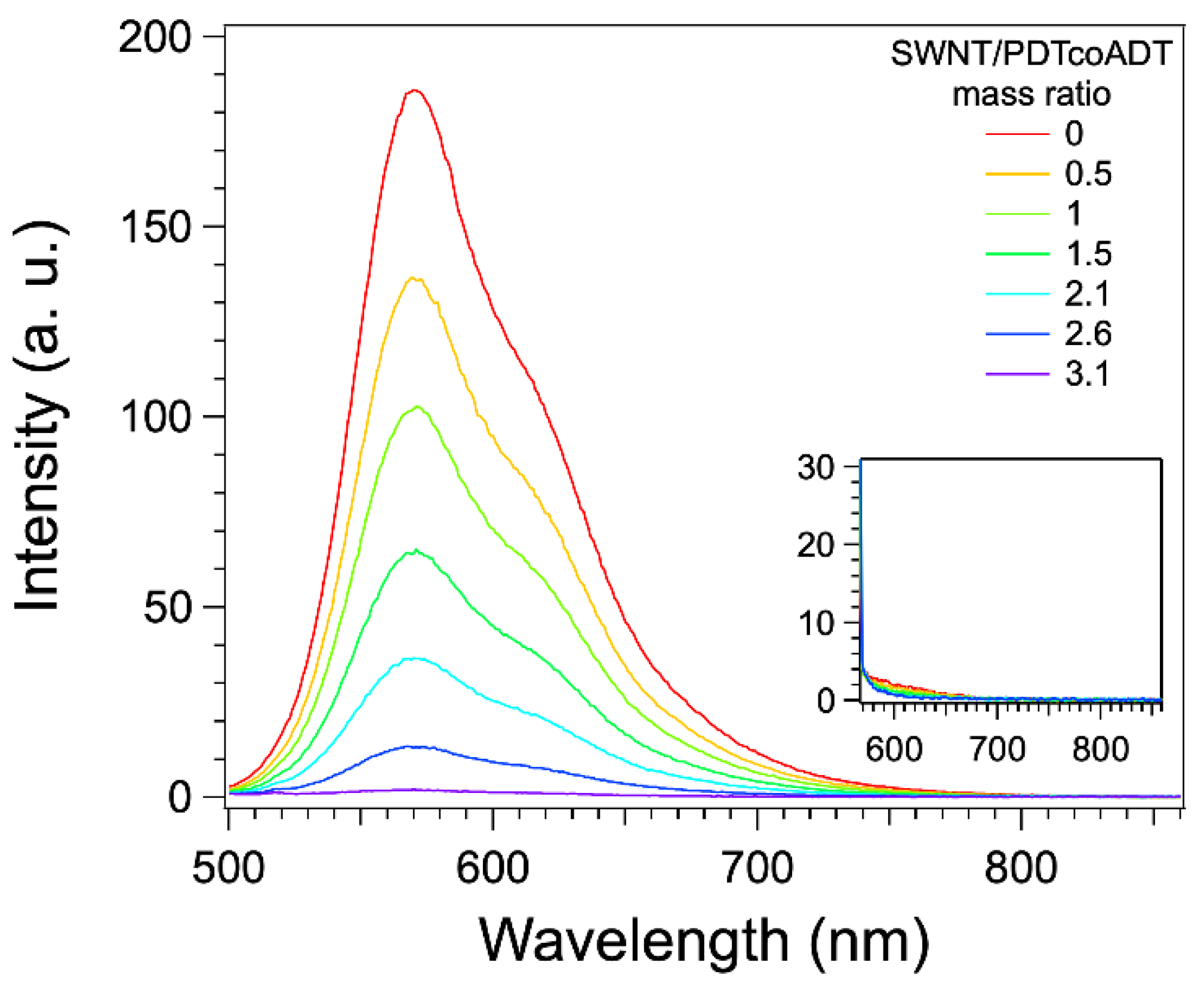
3.3. Formation and Study of the Non-Covalent Copolymer/SWNT Hybrids
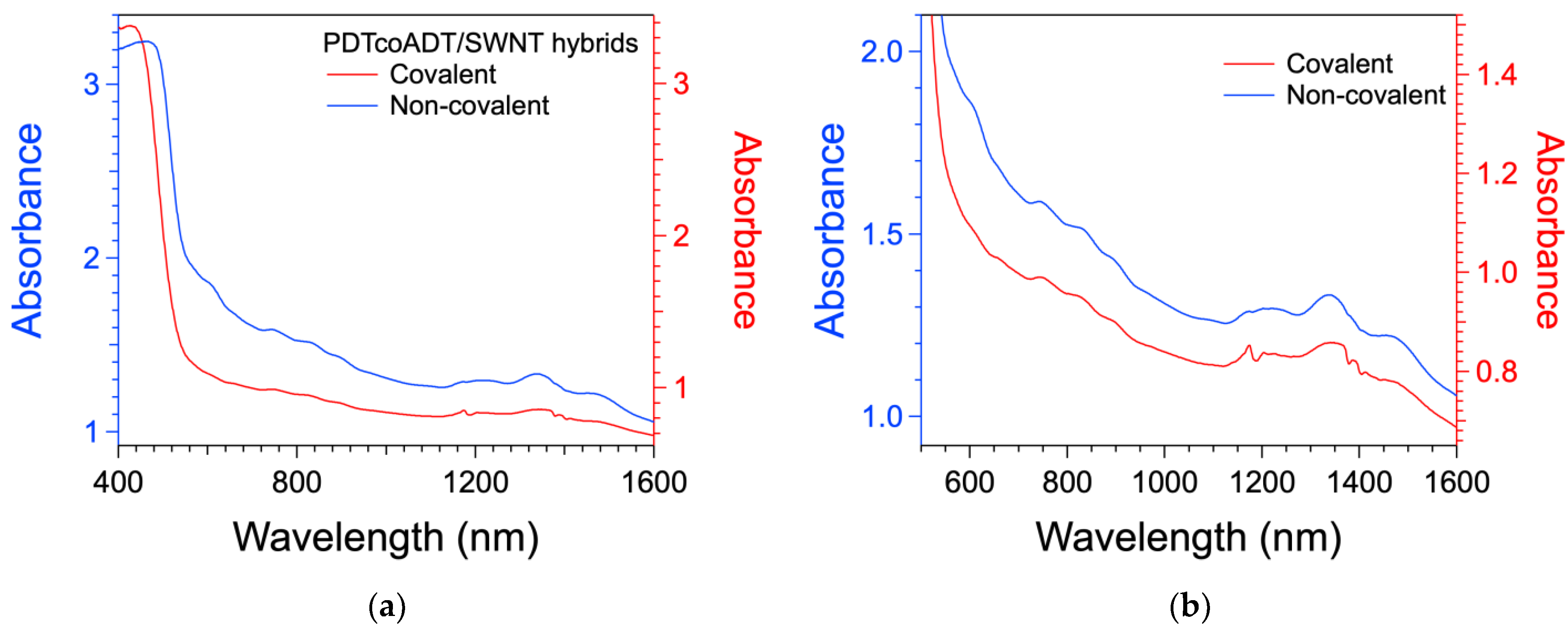
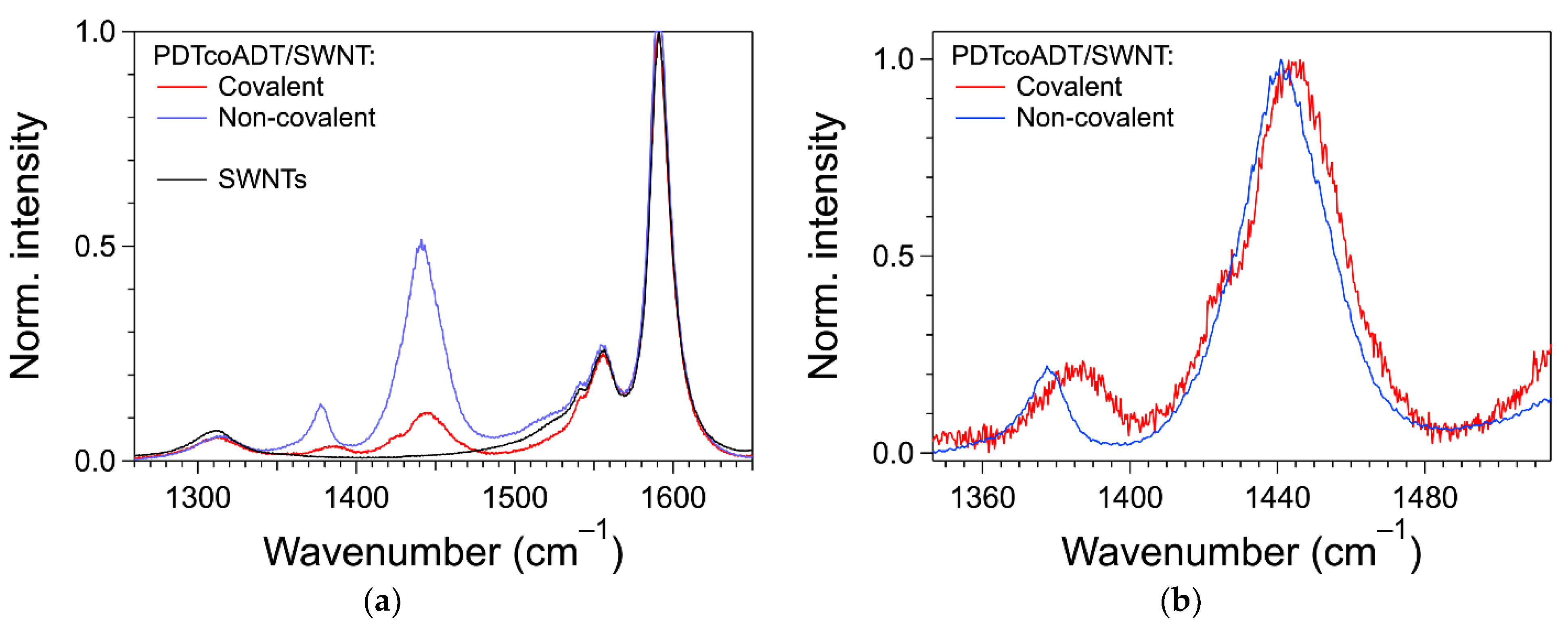
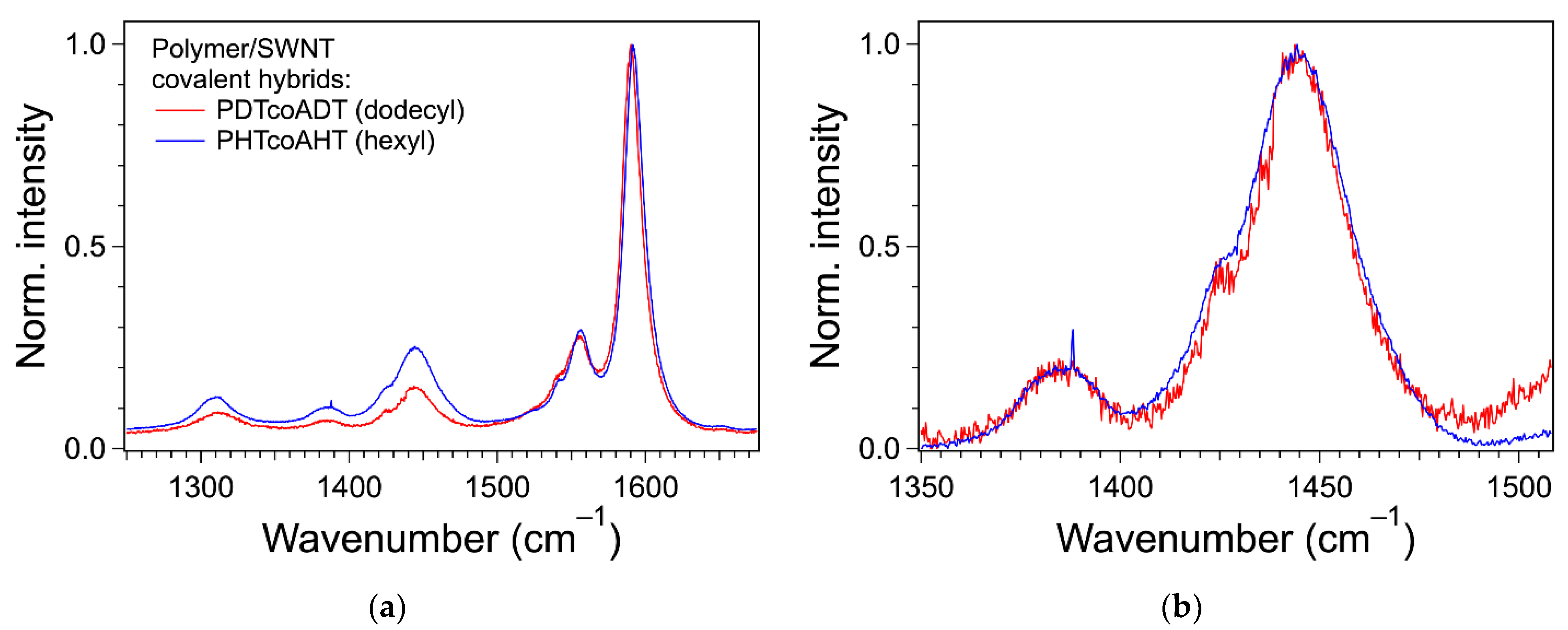
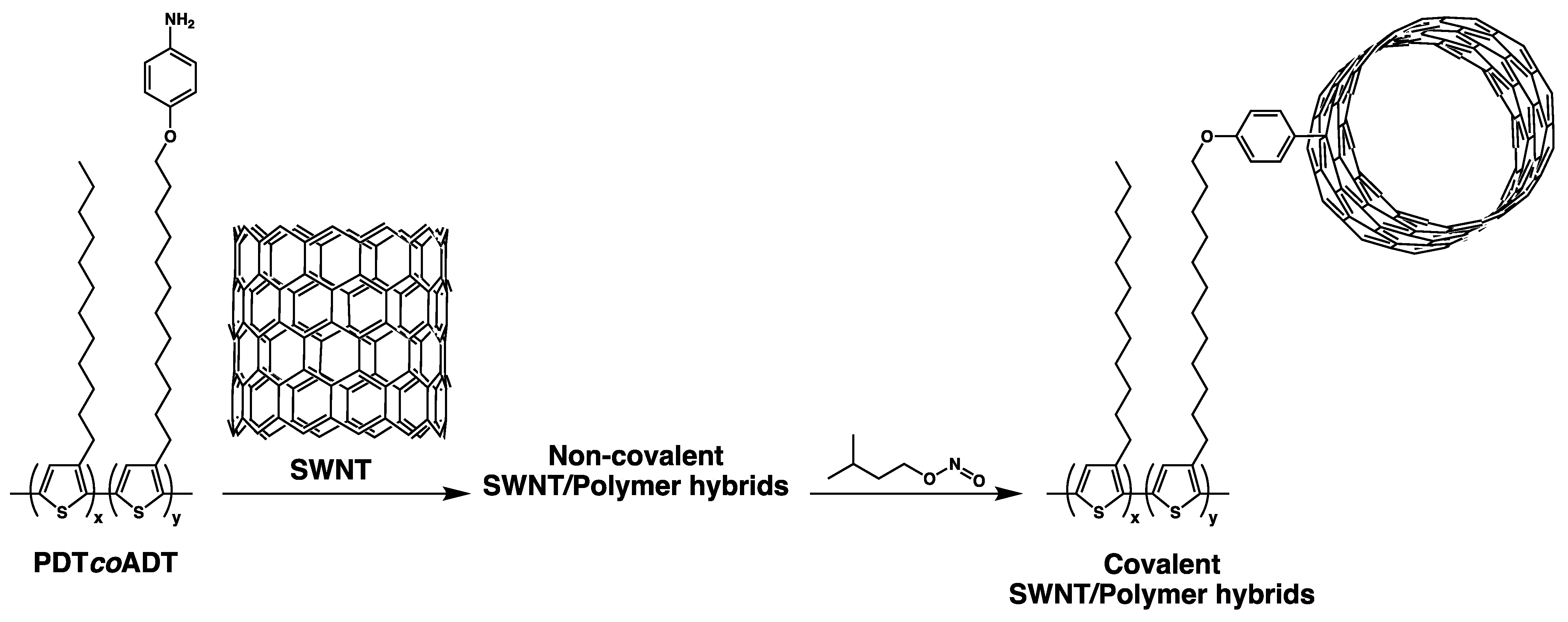
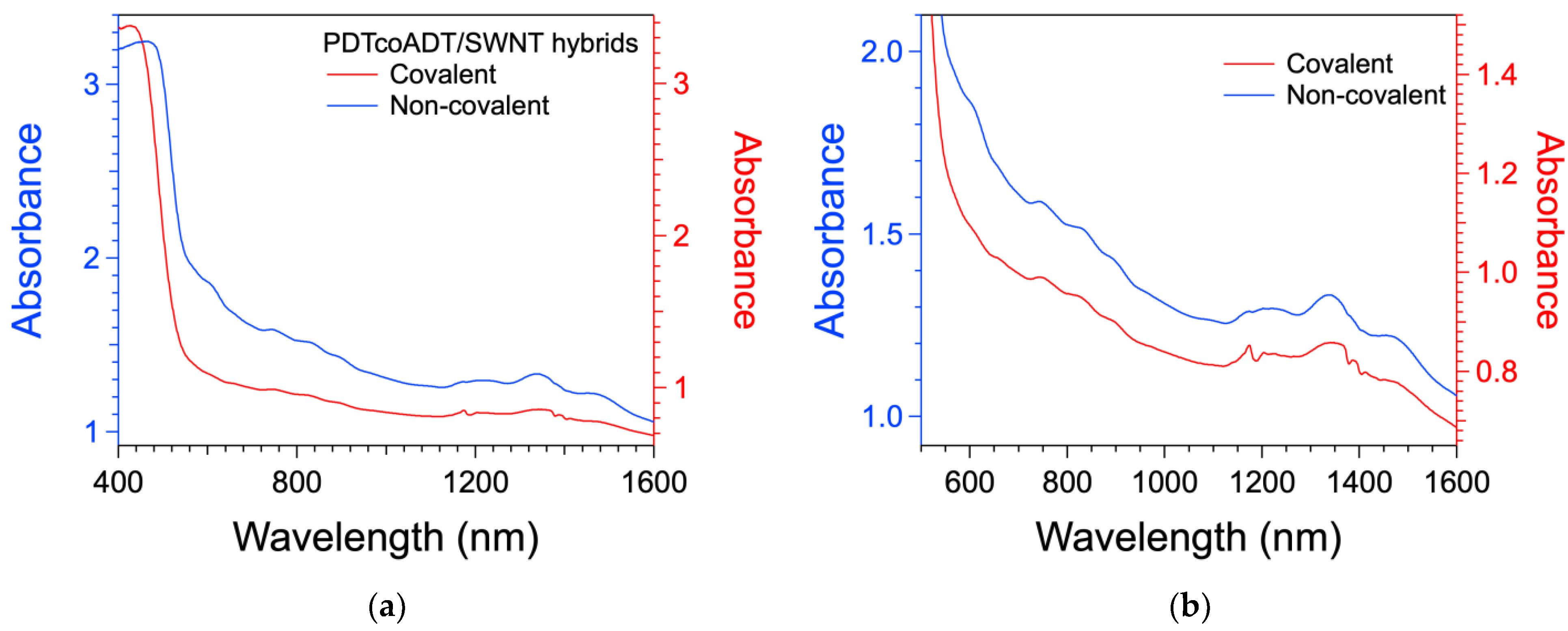
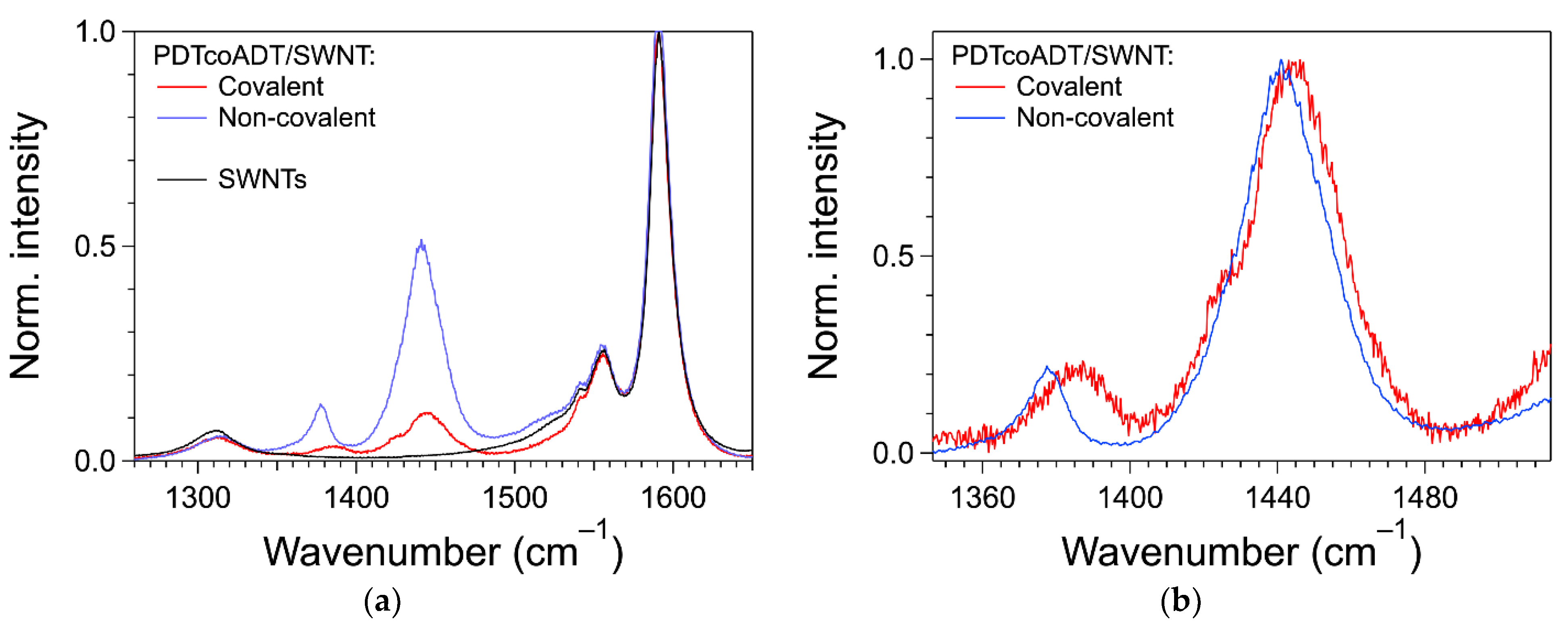
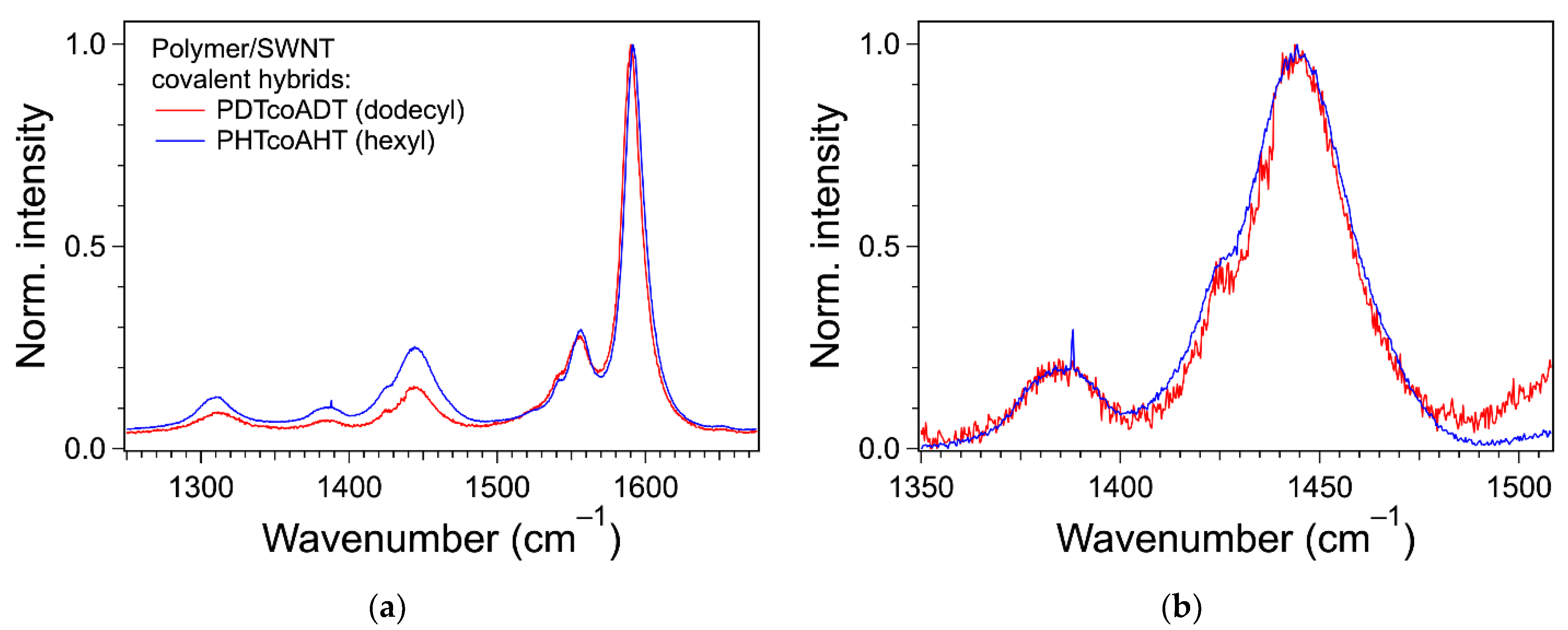
3.4. Formation of the Covalent Hybrids
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Campidelli, S. Click Chemistry for Carbon Nanotubes Functionalization. Curr. Org. Chem. 2011, 15, 1151–1159. [Google Scholar] [CrossRef]
- Li, H.; Cheng, F.; Duft, A.M.; Adronov, A. Functionalization of Single-Walled Carbon Nanotubes with Well-Defined Polystyrene by “Click” Coupling. J. Am. Chem. Soc. 2005, 127, 14518–14524. [Google Scholar] [CrossRef] [PubMed]
- Ma, P.-C.; Siddiqui, N.A.; Marom, G.; Kim, J.-K. Dispersion and functionalization of carbon nanotubes for polymer-based nanocomposites: A review. Compos. Part A Appl. Sci. Manuf. 2010, 41, 1345–1367. [Google Scholar] [CrossRef]
- Nurazzi, N.M.; Sabaruddin, F.A.; Harussani, M.M.; Kamarudin, S.H.; Rayung, M.; Asyraf, M.R.M.; Aisyah, H.A.; Norrrahim, M.N.F.; Ilyas, R.A.; Abdullah, N.; et al. Mechanical Performance and Applications of CNTs Reinforced Polymer Composites—A Review. Nanomaterials 2021, 11, 2186. [Google Scholar] [CrossRef]
- Nurazzi, N.M.; Asyraf, M.R.M.; Khalina, A.; Abdullah, N.; Sabaruddin, F.A.; Kamarudin, S.H.; Ahmad, S.; Mahat, A.M.; Lee, C.L.; Aisyah, H.A.; et al. Fabrication, Functionalization, and Application of Carbon Nanotube-Reinforced Polymer Composite: An Overview. Polymers 2021, 13, 1047. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Marty, L.; Bendiab, N. New Light on Molecule–Nanotube Hybrids. Adv. Mater. 2019, 31, 1902917. [Google Scholar] [CrossRef] [PubMed]
- Piao, Y.; Meany, B.; Powell, L.R.; Valley, N.; Kwon, H.; Schatz, G.C.; Wang, Y. Brightening of carbon nanotube photoluminescence through the incorporation of sp3 defects. Nat. Chem. 2013, 5, 840–845. [Google Scholar] [CrossRef] [PubMed]
- Bahr, J.L.; Tour, J.M. Highly Functionalized Carbon Nanotubes Using in Situ Generated Diazonium Compounds. Chem. Mater. 2001, 13, 3823–3824. [Google Scholar] [CrossRef]
- Strano, M.S.; Dyke, C.A.; Usrey, M.L.; Barone, P.W.; Allen, M.J.; Shan, H.; Kittrell, C.; Hauge, R.H.; Tour, J.M.; Smalley, R.E. Electronic Structure Control of Single-Walled Carbon Nanotube Functionalization. Science 2003, 301, 1519–1522. [Google Scholar] [CrossRef] [Green Version]
- Dyke, C.A.; Tour, J.M. Covalent functionalization of single-walled carbon nanotubes for materials applications. J. Phys. Chem. A 2004, 108, 11151–11159. [Google Scholar] [CrossRef]
- Berger, F.J.; Lüttgens, J.; Nowack, T.; Kutsch, T.; Lindenthal, S.; Kistner, L.; Müller, C.C.; Bongartz, L.M.; Lumsargis, V.A.; Zakharko, Y.; et al. Brightening of Long, Polymer-Wrapped Carbon Nanotubes by sp 3 Functionalization in Organic Solvents. ACS Nano 2019, 13, 9259–9269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Ye, Y.; Xue, Y.; Xie, X.; Mai, Y.-W. Recent advances in covalent functionalization of carbon nanomaterials with polymers: Strategies and perspectives. J. Polym. Sci. Part A Polym. Chem. 2017, 55, 622–631. [Google Scholar] [CrossRef] [Green Version]
- Abousalman-Rezvani, Z.; Eskandari, P.; Roghani-Mamaqani, H.; Salami-Kalajahi, M. Functionalization of carbon nanotubes by combination of controlled radical polymerization and “grafting to” method. Adv. Colloid Interface Sci. 2020, 278, 102126. [Google Scholar] [CrossRef] [PubMed]
- Díez-Pascual, A.M. Chemical Functionalization of Carbon Nanotubes with Polymers: A Brief Overview. Macromol 2021, 1, 64–83. [Google Scholar] [CrossRef]
- Wasem Klein, F.; Lamps, J.-P.; Raoui, M.; Paillet, M.; Sauvajol, J.-L.; Mésini, P.J.; Petit, P. Design and synthesis of aniline-appended P3HT for single step covalent functionalisation of carbon nanotubes. Polym. Chem. 2020, 11, 6319–6327. [Google Scholar] [CrossRef]
- Gomulya, W.; Costanzo, G.D.; De Carvalho, E.J.F.; Bisri, S.Z.; Derenskyi, V.; Fritsch, M.; Fröhlich, N.; Allard, S.; Gordiichuk, P.; Herrmann, A.; et al. Semiconducting single-walled carbon nanotubes on demand by polymer wrapping. Adv. Mater. 2013, 25, 2948–2956. [Google Scholar] [CrossRef] [PubMed]
- Fong, D.; Adronov, A. Recent developments in the selective dispersion of single-walled carbon nanotubes using conjugated polymers. Chem. Sci. 2017, 8, 7292–7305. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.W.; Yoon, Y.; Park, S.; Oh, J.H.; Hong, S.; Liyanage, L.S.; Wang, H.; Morishita, S.; Patil, N.; Park, Y.J.; et al. Selective dispersion of high purity semiconducting single-walled carbon nanotubes with regioregular poly(3-alkylthiophene)s. Nat. Commun. 2011, 2, 541–548. [Google Scholar] [CrossRef]
- Miyakoshi, R.; Yokoyama, A.; Yokozawa, T. Catalyst-Transfer Polycondensation. Mechanism of Ni-Catalyzed Chain-Growth Polymerization Leading to Well-Defined Poly(3-hexylthiophene). J. Am. Chem. Soc. 2005, 127, 17542–17547. [Google Scholar] [CrossRef]
- Iovu, M.C.; Sheina, E.E.; Gil, R.R.; McCullough, R.D. Experimental Evidence for the Quasi-“Living” Nature of the Grignard Metathesis Method for the Synthesis of Regioregular Poly(3-alkylthiophenes). Macromolecules 2005, 38, 8649–8656. [Google Scholar] [CrossRef]
- Tahar-Djebbar, I.; Nekelson, F.; Heinrich, B.; Donnio, B.; Guillon, D.; Kreher, D.; Mathevet, F.; Attias, A.-J. Lamello-Columnar Mesophase Formation in a Side-Chain Liquid Crystal π-Conjugated Polymer Architecture. Chem. Mater. 2011, 23, 4653–4656. [Google Scholar] [CrossRef]
- Vallat, P.; Lamps, J.P.; Schosseler, F.; Rawiso, M.; Catala, J.M. Quasi-Controlled Polymerization through a Nickel Catalyst Process of a Functionalized Thiophene Monomer: Kinetic Studies and Application to the Synthesis of Regioregular Poly(thiophene-3-acetic acid). Macromolecules 2007, 40, 2600–2602. [Google Scholar] [CrossRef]
- Yang, Y.-L.; Lee, Y.-H.; Lee, Y.-P.; Chiang, C.-J.; Shen, C.; Wu, C.-C.; Ohta, Y.; Yokozawa, T.; Dai, C.-A. Synthesis and characterization of poly(3-hexylthiophene)–poly(3-hexyloxythiophene) random copolymers with tunable band gap via Grignard metathesis polymerization. Polym. Int. 2014, 63, 2068–2075. [Google Scholar] [CrossRef]
- Spano, F.C.; Silva, C. H- and J-Aggregate Behavior in Polymeric Semiconductors. Annu. Rev. Phys. Chem. 2014, 65, 477–500. [Google Scholar] [CrossRef] [PubMed]
- Rumbles, G.; Samuel, I.D.W.; Magnani, L.; Murray, K.A.; DeMello, A.J.; Crystall, B.; Moratti, S.C.; Stone, B.M.; Holmes, A.B.; Friend, R.H. Chromism and luminescence in regioregular poly(3-dodecylthiophene). Synth. Met. 1996, 76, 47–51. [Google Scholar] [CrossRef]
- Giulianini, M.; Waclawik, E.R.; Bell, J.M.; Crescenzi, M.D.; Castrucci, P.; Scarselli, M.; Diociauti, M.; Casciardi, S.; Motta, N. Evidence of Multiwall Carbon Nanotube Deformation Caused by Poly(3-hexylthiophene) Adhesion. J. Phys. Chem. C 2011, 115, 6324–6330. [Google Scholar] [CrossRef]
- Wood, S.; Hollis, J.R.; Kim, J.-S. Raman spectroscopy as an advanced structural nanoprobe for conjugated molecular semiconductors. J. Phys. D Appl. Phys. 2017, 50, 073001. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wasem Klein, F.; Lamps, J.-P.; Paillet, M.; Petit, P.; Mésini, P.J. Synthesis of a Poly(3-dodecylthiophene) Bearing Aniline Groups for the Covalent Functionalization of Carbon Nanotubes. Reactions 2021, 2, 473-485. https://doi.org/10.3390/reactions2040030
Wasem Klein F, Lamps J-P, Paillet M, Petit P, Mésini PJ. Synthesis of a Poly(3-dodecylthiophene) Bearing Aniline Groups for the Covalent Functionalization of Carbon Nanotubes. Reactions. 2021; 2(4):473-485. https://doi.org/10.3390/reactions2040030
Chicago/Turabian StyleWasem Klein, Felipe, Jean-Philippe Lamps, Matthieu Paillet, Pierre Petit, and Philippe J. Mésini. 2021. "Synthesis of a Poly(3-dodecylthiophene) Bearing Aniline Groups for the Covalent Functionalization of Carbon Nanotubes" Reactions 2, no. 4: 473-485. https://doi.org/10.3390/reactions2040030
APA StyleWasem Klein, F., Lamps, J.-P., Paillet, M., Petit, P., & Mésini, P. J. (2021). Synthesis of a Poly(3-dodecylthiophene) Bearing Aniline Groups for the Covalent Functionalization of Carbon Nanotubes. Reactions, 2(4), 473-485. https://doi.org/10.3390/reactions2040030