

An Electronic Structural Analysis of O2-Binding Dicopper Complex: Insights from Spin Magnetism and Molecular Orbitals


Abstract
:
1. Introduction
2. Methods
3. Results and Discussion
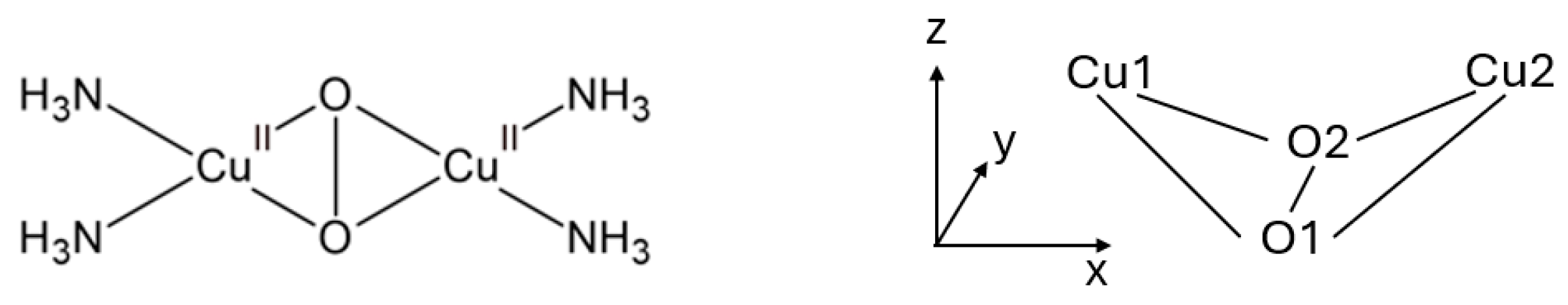
3.1. Geometry
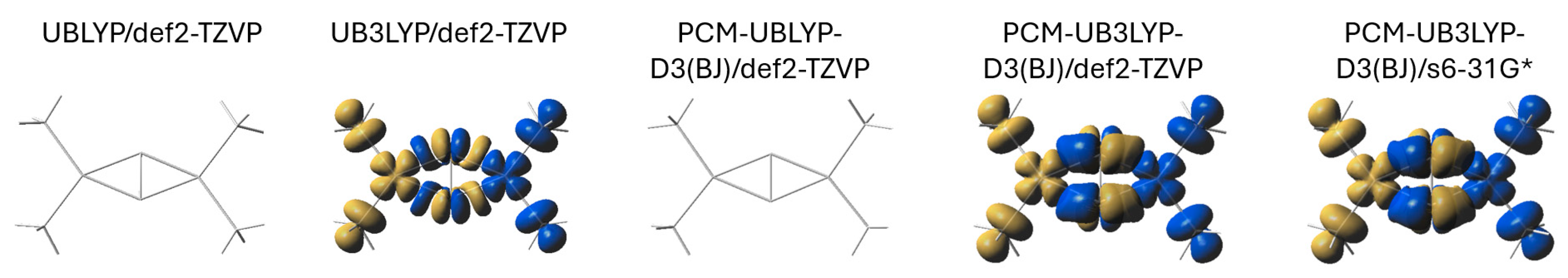
3.2. Spin Structure
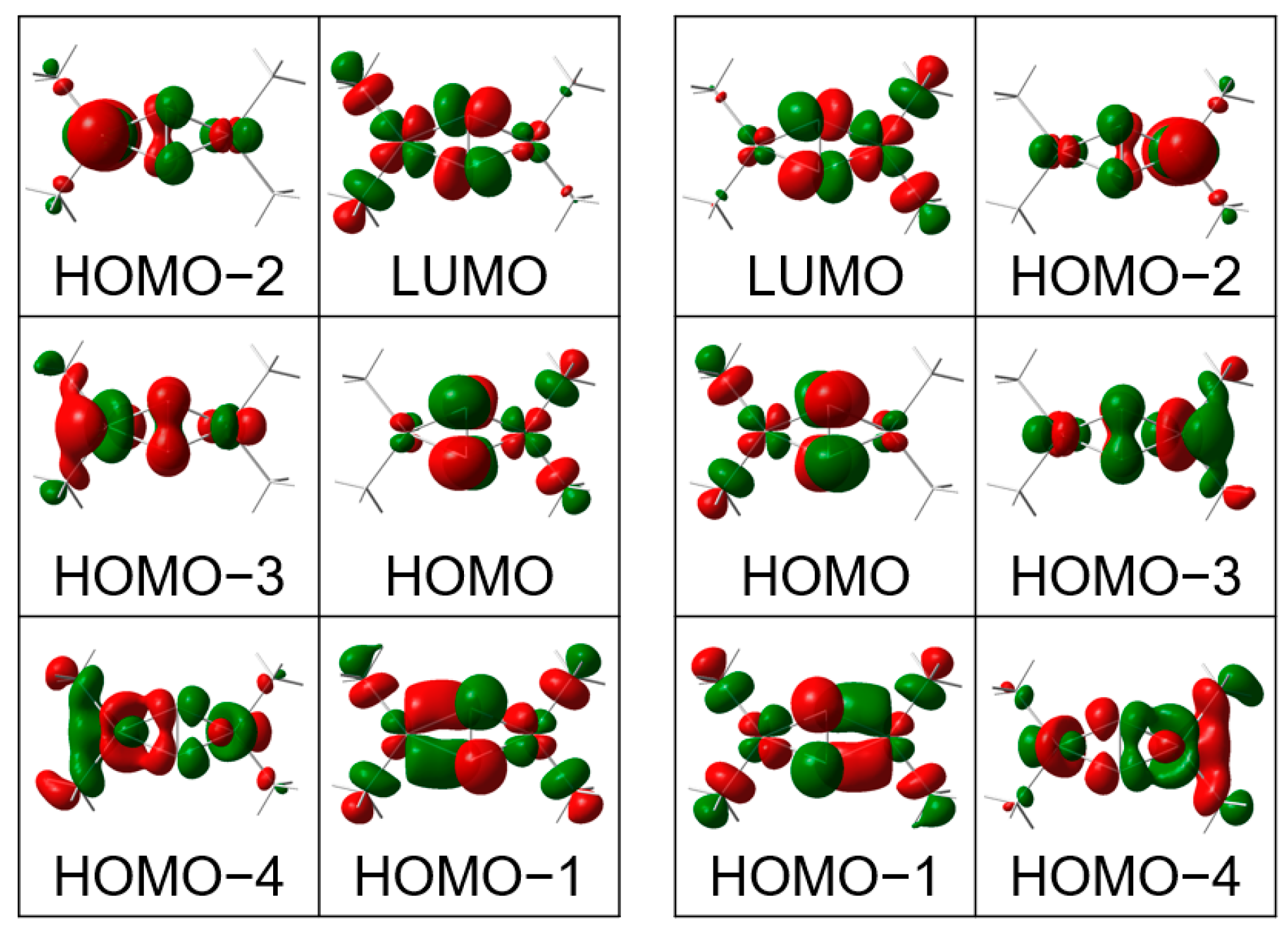
3.3. Electronic Structure
3.4. Absorption Spectrum
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kitajima, N.; Moro-oka, Y. Copper-Dioxygen Complexes. Inorganic and Bioinorganic Perspectives. Chem. Rev. 1994, 94, 737–757. [Google Scholar] [CrossRef]
- Solomon, E.I.; Heppner, D.E.; Johnston, E.M.; Ginsbach, J.W.; Cirera, J.; Qayyum, M.; Kieber-Emmons, M.T.; Kjaergaard, C.H.; Hadt, R.G.; Tian, L. Copper Active Sites in Biology. Chem. Rev. 2014, 114, 3659–3853. [Google Scholar] [CrossRef]
- Kitajima, N.; Fujisawa, K.; Morooka, Y.; Toriumi, K. .Mu.-.Eta.2:.Eta.2-Peroxo Binuclear Copper Complex, [Cu(HB(3,5-(Me2CH)2pz)3)]2(O2). J. Am. Chem. Soc. 1989, 111, 8975–8976. [Google Scholar] [CrossRef]
- Magnus, K.A.; Ton-That, H.; Carpenter, J.E. Recent Structural Work on the Oxygen Transport Protein Hemocyanin. Chem. Rev. 1994, 94, 727–735. [Google Scholar] [CrossRef]
- Kwak, J.H.; Tonkyn, R.G.; Kim, D.H.; Szanyi, J.; Peden, C.H.F. Excellent Activity and Selectivity of Cu-SSZ-13 in the Selective Catalytic Reduction of NOx with NH3. J. Catal. 2010, 275, 187–190. [Google Scholar] [CrossRef]
- Paolucci, C.; Khurana, I.; Parekh, A.A.; Li, S.; Shih, A.J.; Li, H.; Di Iorio, J.R.; Albarracin-Caballero, J.D.; Yezerets, A.; Miller, J.T.; et al. Dynamic Multinuclear Sites Formed by Mobilized Copper Ions in NOx Selective Catalytic Reduction. Science 2017, 357, 898–903. [Google Scholar] [CrossRef]
- Negri, C.; Selleri, T.; Borfecchia, E.; Martini, A.; Lomachenko, K.A.; Janssens, T.V.W.; Cutini, M.; Bordiga, S.; Berlier, G. Structure and Reactivity of Oxygen-Bridged Diamino Dicopper(II) Complexes in Cu-Ion-Exchanged Chabazite Catalyst for NH3-Mediated Selective Catalytic Reduction. J. Am. Chem. Soc. 2020, 142, 15884–15896. [Google Scholar] [CrossRef]
- Nakamura, T.; Mason, H.S. An Electron Spin Resonance Study of Copper Valence in Oxyhemocyanin. Biochem. Biophys. Res. Commun. 1960, 3, 297–299. [Google Scholar] [CrossRef]
- Metz, M.; Solomon, E.I. Dioxygen Binding to Deoxyhemocyanin: Electronic Structure and Mechanism of the Spin-Forbidden Two-Electron Reduction of O2. J. Am. Chem. Soc. 2001, 123, 4938–4950. [Google Scholar] [CrossRef]
- Mirica, L.M.; Ottenwaelder, X.; Stack, T.D.P. Structure and Spectroscopy of Copper−Dioxygen Complexes. Chem. Rev. 2004, 104, 1013–1046. [Google Scholar] [CrossRef]
- Tolman, W.B. Making and Breaking the Dioxygen O−O Bond: New Insights from Studies of Synthetic Copper Complexes. Acc. Chem. Res. 1997, 30, 227–237. [Google Scholar] [CrossRef]
- Holland, P.L.; Tolman, W.B. Dioxygen Activation by Copper Sites: Relative Stability and Reactivity of (μ-η2:η2-Peroxo)- and Bis(μ-Oxo)Dicopper Cores. Coord. Chem. Rev. 1999, 190–192, 855–869. [Google Scholar] [CrossRef]
- Elwell, C.E.; Gagnon, N.L.; Neisen, B.D.; Dhar, D.; Spaeth, A.D.; Yee, G.M.; Tolman, W.B. Copper–Oxygen Complexes Revisited: Structures, Spectroscopy, and Reactivity. Chem. Rev. 2017, 117, 2059–2107. [Google Scholar] [CrossRef]
- Cramer, C.J.; Włoch, M.; Piecuch, P.; Puzzarini, C.; Gagliardi, L. Theoretical Models on the Cu2O2 Torture Track: Mechanistic Implications for Oxytyrosinase and Small-Molecule Analogues. J. Phys. Chem. A 2006, 110, 1991–2004. [Google Scholar] [CrossRef]
- Saito, T.; Thiel, W. Quantum Mechanics/Molecular Mechanics Study of Oxygen Binding in Hemocyanin. J. Phys. Chem. B 2014, 118, 5034–5043. [Google Scholar] [CrossRef]
- Takano, Y.; Kubo, S.; Onishi, T.; Isobe, H.; Yoshioka, Y.; Yamaguchi, K. Theoretical Studies on the Magnetic Interaction and Reversible Dioxygen Binding of the Active Site in Hemocyanin. Chem. Phys. Lett. 2001, 335, 395–403. [Google Scholar] [CrossRef]
- Abe, T.; Shiota, Y.; Itoh, S.; Yoshizawa, K. Theoretical Rationalization for the Equilibrium between (μ–η2:η2-Peroxido)CuIICuII and Bis(μ-Oxido)CuIIICuIII Complexes: Perturbational Effects from Ligand Frameworks. Dalton Trans. 2020, 49, 6710–6717. [Google Scholar] [CrossRef]
- Stańczak, A.; Chalupský, J.; Rulíšek, L.; Straka, M. Comprehensive Theoretical View of the [Cu2O2] Side-on-Peroxo-/Bis-μ-Oxo Equilibria. ChemPhysChem 2022, 23, e202200076. [Google Scholar] [CrossRef] [PubMed]
- Liakos, D.G.; Neese, F. Interplay of Correlation and Relativistic Effects in Correlated Calculations on Transition-Metal Complexes: The (Cu2O2)2+ Core Revisited. J. Chem. Theory Comput. 2011, 7, 1511–1523. [Google Scholar] [CrossRef]
- Samanta, K.; Jiménez-Hoyos, C.A.; Scuseria, G.E. Exploring Copper Oxide Cores Using the Projected Hartree–Fock Method. J. Chem. Theory Comput. 2012, 8, 4944–4949. [Google Scholar] [CrossRef]
- Flock, M.; Pierloot, K. Theoretical Study of the Interconversion of O2-Binding Dicopper Complexes. J. Phys. Chem. A 1999, 103, 95–102. [Google Scholar] [CrossRef]
- Rode, M.F.; Werner, H.-J. Ab Initio Study of the O2 Binding in Dicopper Complexes. Theor. Chem. Acc. 2005, 114, 309–317. [Google Scholar] [CrossRef]
- Ross, P.K.; Solomon, E.I. Electronic Structure of Peroxide Bridged Copper Dimers of Relevance to Oxyhemocyanin. J. Am. Chem. Soc. 1990, 112, 5871–5872. [Google Scholar] [CrossRef]
- Ross, P.K.; Solomon, E.I. An Electronic Structural Comparison of Copper-Peroxide Complexes of Relevance to Hemocyanin and Tyrosinase Active Sites. J. Am. Chem. Soc. 1991, 113, 3246–3259. [Google Scholar] [CrossRef]
- Malmqvist, P.Å.; Pierloot, K.; Shahi, A.R.M.; Cramer, C.J.; Gagliardi, L. The Restricted Active Space Followed by Second-Order Perturbation Theory Method: Theory and Application to the Study of CuO2 and Cu2O2 Systems. J. Chem. Phys. 2008, 128, 204109. [Google Scholar] [CrossRef]
- Yanai, T.; Kurashige, Y.; Neuscamman, E.; Chan, G.K.-L. Multireference Quantum Chemistry through a Joint Density Matrix Renormalization Group and Canonical Transformation Theory. J. Chem. Phys. 2010, 132, 024105. [Google Scholar] [CrossRef]
- Kurashige, Y.; Chalupský, J.; Lan, T.N.; Yanai, T. Complete Active Space Second-Order Perturbation Theory with Cumulant Approximation for Extended Active-Space Wavefunction from Density Matrix Renormalization Group. J. Chem. Phys. 2014, 141, 174111. [Google Scholar] [CrossRef]
- Waigum, A.; Black, J.A.; Köhn, A. A Generalized Hybrid Scheme for Multireference Methods. J. Chem. Phys. 2021, 155, 204106. [Google Scholar] [CrossRef] [PubMed]
- De Moraes, M.M.F.; Tecmer, P. Towards Reliable and Efficient Modeling of [Cu2O2]2+-Based Compound Electronic Structures with the Partially Fixed Reference Space Protocols. Phys. Chem. Chem. Phys. 2024, 26, 19742–19754. [Google Scholar] [CrossRef]
- Gao, F.; Mei, D.; Wang, Y.; Szanyi, J.; Peden, C.H.F. Selective Catalytic Reduction over Cu/SSZ-13: Linking Homo- and Heterogeneous Catalysis. J. Am. Chem. Soc. 2017, 139, 4935–4942. [Google Scholar] [CrossRef]
- Liu, C.; Kubota, H.; Toyao, T.; Maeno, Z.; Shimizu, K. Mechanistic Insights into the Oxidation of Copper(I) Species during NH3-SCR over Cu-CHA Zeolites: A DFT Study. Catal. Sci. Technol. 2020, 10, 3586–3593. [Google Scholar] [CrossRef]
- Chen, L.; Janssens, T.V.W.; Vennestrøm, P.N.R.; Jansson, J.; Skoglundh, M.; Grönbeck, H. A Complete Multisite Reaction Mechanism for Low-Temperature NH3-SCR over Cu-CHA. ACS Catal. 2020, 10, 5646–5656. [Google Scholar] [CrossRef]
- Millan, R.; Cnudde, P.; van Speybroeck, V.; Boronat, M. Mobility and Reactivity of Cu+ Species in Cu-CHA Catalysts under NH3-SCR-NOx Reaction Conditions: Insights from AIMD Simulations. JACS Au 2021, 1, 1778–1787. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian; 16 Rev. C.01 2016; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Becke, A.D. Density-Functional Exchange-Energy Approximation with Correct Asymptotic Behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti Correlation-Energy Formula into a Functional of the Electron Density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef]
- Staroverov, V.N.; Scuseria, G.E.; Tao, J.; Perdew, J.P. Comparative Assessment of a New Nonempirical Density Functional: Molecules and Hydrogen-Bonded Complexes. J. Chem. Phys. 2003, 119, 12129–12137. [Google Scholar] [CrossRef]
- Stephens, P.J.; Devlin, F.J.; Chabalowski, C.F.; Frisch, M.J. Ab Initio Calculation of Vibrational Absorption and Circular Dichroism Spectra Using Density Functional Force Fields. J. Phys. Chem. 1994, 98, 11623–11627. [Google Scholar] [CrossRef]
- Heyd, J.; Scuseria, G.E.; Ernzerhof, M. Erratum: “Hybrid Functionals Based on a Screened Coulomb Potential” [J. Chem. Phys. 118, 8207 (2003)]. J. Chem. Phys. 2006, 124, 219906. [Google Scholar] [CrossRef]
- Adamo, C.; Barone, V. Toward Reliable Density Functional Methods without Adjustable Parameters: The PBE0 Model. J. Chem. Phys. 1999, 110, 6158–6170. [Google Scholar] [CrossRef]
- Szilagyi, R.K.; Metz, M.; Solomon, E.I. Spectroscopic Calibration of Modern Density Functional Methods Using [CuCl4]2-. J. Phys. Chem. A 2002, 106, 2994–3007. [Google Scholar] [CrossRef]
- Atanasov, M.; Comba, P.; Martin, B.; Müller, V.; Rajaraman, G.; Rohwer, H.; Wunderlich, S. DFT Models for Copper(II) Bispidine Complexes: Structures, Stabilities, Isomerism, Spin Distribution, and Spectroscopy. J. Comput. Chem. 2006, 27, 1263–1277. [Google Scholar] [CrossRef] [PubMed]
- Becke, A.D. A New Mixing of Hartree–Fock and Local Density-functional Theories. J. Chem. Phys. 1993, 98, 1372–1377. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. Density Functional for Spectroscopy: No Long-Range Self-Interaction Error, Good Performance for Rydberg and Charge-Transfer States, and Better Performance on Average than B3LYP for Ground States. J. Phys. Chem. A 2006, 110, 13126–13130. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. Comparative DFT Study of van Der Waals Complexes: Rare-Gas Dimers, Alkaline-Earth Dimers, Zinc Dimer, and Zinc-Rare-Gas Dimers. J. Phys. Chem. A 2006, 110, 5121–5129. [Google Scholar] [CrossRef] [PubMed]
- Swart, M.; Güell, M.; Luis, J.M.; Solà, M. Spin-State-Corrected Gaussian-Type Orbital Basis Sets. J. Phys. Chem. A 2010, 114, 7191–7197. [Google Scholar] [CrossRef]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the Damping Function in Dispersion Corrected Density Functional Theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef]
- Yanai, T.; Tew, D.P.; Handy, N.C. A new hybrid exchange–correlation functional using the Coulomb-attenuating method (CAM-B3LYP). Chem. Phys. Lett. 2004, 393, 51–57. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F.-W. Calculation of Molecular Orbital Composition. Acta Chim. Sin. 2011, 69, 2393. [Google Scholar]
- Glendening, E.D.; Landis, C.R.; Weinhold, F. NBO 7.0: New Vistas in Localized and Delocalized Chemical Bonding Theory. J. Comput. Chem. 2019, 40, 2234–2241. [Google Scholar] [CrossRef]
- Rybicki, M.; Sauer, J. Acid Strength of Zeolitic Brønsted Sites—Dependence on Dielectric Properties. Catal. Today 2019, 323, 86–93. [Google Scholar] [CrossRef]
- Yamaguchi, K.; Takahara, Y.; Fueno, T. Ab-Initio Molecular Orbital Studies of Structure and Reactivity of Transition Metal-OXO Compounds. In Applied Quantum Chemistry; Springer: Dordrecht, The Netherlands, 1986; pp. 155–184. ISBN 978-94-009-4746-7. [Google Scholar]
- Kitagawa, Y.; Saito, T.; Yamaguchi, K.; Kitagawa, Y.; Saito, T.; Yamaguchi, K. Approximate Spin Projection for Broken-Symmetry Method and Its Application. In Symmetry (Group Theory) and Mathematical Treatment in Chemistry; IntechOpen: London, UK, 2018; ISBN 978-1-78923-315-5. [Google Scholar]
- Solomon, E.I.; Dooley, D.M.; Wang, R.-H.; Gray, H.B.; Cerdonio, M.; Mogno, F.; Romani, G.L. Susceptibility Studies of Laccase and Oxyhemocyanin Using an Ultrasensitive Magnetometer. Antiferromagnetic Behavior of the Type 3 Copper in Rhus Laccase. J. Am. Chem. Soc. 1976, 98, 1029–1031. [Google Scholar] [CrossRef]
- Bernardi, F.; Bottoni, A.; Casadio, R.; Fariselli, P.; Rigo, A. An Ab Initio Study of the Dioxygen Binding Site of Hemocyanin: A Comparison between CASSCF, CASPT2, and DFT Approaches. Int. J. Quantum Chem. 1996, 58, 109–119. [Google Scholar] [CrossRef]
- Bernardi, F.; Bottoni, A.; Casadio, R.; Fariselli, P.; Rigo, A. Ab Initio Study of the Mechanism of the Binding of Triplet O2 to Hemocyanin. Inorg. Chem. 1996, 35, 5207–5212. [Google Scholar] [CrossRef]
- Karlin, K.D.; Tyeklár, Z.; Farooq, A.; Jacobson, R.R.; Sinn, E.; Lee, D.W.; Bradshaw, J.E.; Wilson, L.J. Peroxide (O22−) as a Bridging Ligand for Copper(II): Strong Exchange Coupling in Complexes Derived from Copper(I) and Dioxygen. Inorganica Chim. Acta 1991, 182, 1–3. [Google Scholar] [CrossRef]
- al-Badri, M.A.; Linscott, E.; Georges, A.; Cole, D.J.; Weber, C. Superexchange Mechanism and Quantum Many Body Excitations in the Archetypal Di-Cu Oxo-Bridge. Commun. Phys. 2020, 3, 4. [Google Scholar] [CrossRef]
- Qayyum, M.F.; Sarangi, R.; Fujisawa, K.; Stack, T.D.P.; Karlin, K.D.; Hodgson, K.O.; Hedman, B.; Solomon, E.I. L-Edge X-Ray Absorption Spectroscopy and DFT Calculations on Cu2O2 Species: Direct Electrophilic Aromatic Attack by Side-on Peroxo Bridged Dicopper(II) Complexes. J. Am. Chem. Soc. 2013, 135, 17417–17431. [Google Scholar] [CrossRef]
- Oda, A.; Shionoya, H.; Hotta, Y.; Takewaki, T.; Sawabe, K.; Satsuma, A. Spectroscopic Evidence of Efficient Generation of Dicopper Intermediate in Selective Catalytic Reduction of NO over Cu-Ion-Exchanged Zeolites. ACS Catal. 2020, 10, 12333–12339. [Google Scholar] [CrossRef]
- Eickman, N.C.; Himmelwright, R.S.; Solomon, E.I. Geometric and Electronic Structure of Oxyhemocyanin: Spectral and Chemical Correlations to Met Apo, Half Met, Met, and Dimer Active Sites. Proc. Natl. Acad. Sci. USA 1979, 76, 2094–2098. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Model | Symmetry | rO-O (Å) | rCu-Cu (Å) | rCu-O (Å) | ∠Cu-O-O-Cu (°) |
|---|---|---|---|---|---|
| UBLYP/def2-TZVP | D2 | 1.47 | 3.64 | 1.96 | 180.0 |
| UBLYP/s6-31G* | D2 | 1.49 | 3.61 | 1.95 | 180.0 |
| UB3LYP/def2-TZVP | D2 | 1.46 | 3.59 | 1.94 | 180.0 |
| UB3LYP/s6-31G* | C2 | 1.46 | 3.58 | 1.94 | 169.6 |
| UBLYP-D3(BJ)/def2-TZVP | D2 | 1.48 | 3.62 | 1.95 | 180.0 |
| UB3LYP-D3(BJ)/def2-TZVP | D2 | 1.45 | 3.59 | 1.94 | 180.0 |
| UB3LYP-D3(BJ)/s6-31G* | C2 | 1.46 | 3.57 | 1.93 | 171.9 |
| PCM-UBLYP/def2-TZVP | D2 | 1.48 | 3.62 | 1.95 | 180.0 |
| PCM-UB3LYP/def2-TZVP | C2 | 1.45 | 3.52 | 1.95 | 153.5 |
| PCM-UB3LYP/s6-31G* | C2 | 1.46 | 3.55 | 1.94 | 161.8 |
| PCM-UBLYP-D3(BJ)/def2-TZVP | C2 | 1.49 | 3.60 | 1.94 | 180.0 |
| PCM-UBLYP-D3(BJ)/s6-31G* | C2 | 1.51 | 3.57 | 1.93 | 180.0 |
| PCM-UB3LYP-D3(BJ)/def2-TZVP | C2 | 1.45 | 3.50 | 1.94 | 151.4 |
| PCM-UB3LYP-D3(BJ)/s6-31G* | C2 | 1.46 | 3.41 | 1.94 | 141.3 |
| Expt. [7] | 3.40 ± 0.05 | 1.911 ± 0.009 |
| Model | ⟨S2⟩ | −2J (cm−1) | |
|---|---|---|---|
| S0 | T1 | ||
| UBLYP/def2-TZVP | 0.00 | 2.00 | 7850 |
| UBLYP/s6-31G* | 0.00 | 2.00 | 8043 |
| UB3LYP/def2-TZVP | 0.54 | 2.01 | 5863 |
| UB3LYP/s6-31G* | 0.55 | 2.01 | 6096 |
| UB3LYP-D3(BJ)/s6-31G* | 0.53 | 2.01 | 6686 |
| PCM-UB3LYP/s6-31G* | 0.63 | 2.01 | 5099 |
| PCM-UBLYP-D3(BJ)/def2-TZVP | 0.00 | 2.00 | 8736 |
| PCM-UBLYP-D3(BJ)/s6-31G* | 0.00 | 2.00 | 8942 |
| PCM-UB3LYP-D3(BJ)/def2-TZVP | 0.74 | 2.01 | 2379 |
| PCM-UB3LYP-D3(BJ)/s6-31G* | 0.81 | 2.01 | 1373 |
| VBCI [24] | 2250 | ||
| CASSCF(12e, 8o) [56] | 832 | ||
| CASSCF(14e, 12o) [57] | 1148 | ||
| CASPT2(12e, 14o) [21] | 4288 | ||
| Expt. [55,58] | ≥600 | ||
| Model | y |
|---|---|
| UBLYP/def2-TZVP | 0.00 |
| UB3LYP/def2-TZVP | 0.07 |
| UB3LYP/s6-31G* | 0.07 |
| PCM-UBLYP-D3(BJ)/def2-TZVP | 0.00 |
| PCM-UB3LYP-D3(BJ)/def2-TZVP | 0.18 |
| PCM-UB3LYP-D3(BJ)/s6-31G* | 0.26 |
| Orbital | Energy (eV) | Cu1 d (%) | Cu2 d (%) | O1 p (%) | O2 p (%) |
|---|---|---|---|---|---|
| LUMO | 1.74 | 1.6 | 48.1 | 18.2 | 18.0 |
| HOMO | −1.74 | 13.3 | 1.9 | 37.2 | 37.2 |
| HOMO−1 | −2.65 | 13.8 | 18.5 | 20.0 | 20.5 |
| HOMO−2 | −3.87 | 6.5 | 75.9 | 4.5 | 4.5 |
| HOMO−3 | −4.01 | 14.8 | 62.1 | 7.6 | 7.4 |
| HOMO−4 | −4.13 | 16.0 | 56.1 | 7.0 | 6.8 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Morimoto, R.; Sugiyama, K.; Higashi, M.; Sato, H. An Electronic Structural Analysis of O2-Binding Dicopper Complex: Insights from Spin Magnetism and Molecular Orbitals. Chemistry 2025, 7, 44. https://doi.org/10.3390/chemistry7020044
Morimoto R, Sugiyama K, Higashi M, Sato H. An Electronic Structural Analysis of O2-Binding Dicopper Complex: Insights from Spin Magnetism and Molecular Orbitals. Chemistry. 2025; 7(2):44. https://doi.org/10.3390/chemistry7020044
Chicago/Turabian StyleMorimoto, Ryusei, Kanami Sugiyama, Masahiro Higashi, and Hirofumi Sato. 2025. "An Electronic Structural Analysis of O2-Binding Dicopper Complex: Insights from Spin Magnetism and Molecular Orbitals" Chemistry 7, no. 2: 44. https://doi.org/10.3390/chemistry7020044
APA StyleMorimoto, R., Sugiyama, K., Higashi, M., & Sato, H. (2025). An Electronic Structural Analysis of O2-Binding Dicopper Complex: Insights from Spin Magnetism and Molecular Orbitals. Chemistry, 7(2), 44. https://doi.org/10.3390/chemistry7020044