
Adult-Onset Case of Female Idiopathic Hypogonadotropic Hypogonadism and Ataxia: Genetic Background

 , ,
, ,  and
and 
Abstract
1. Introduction
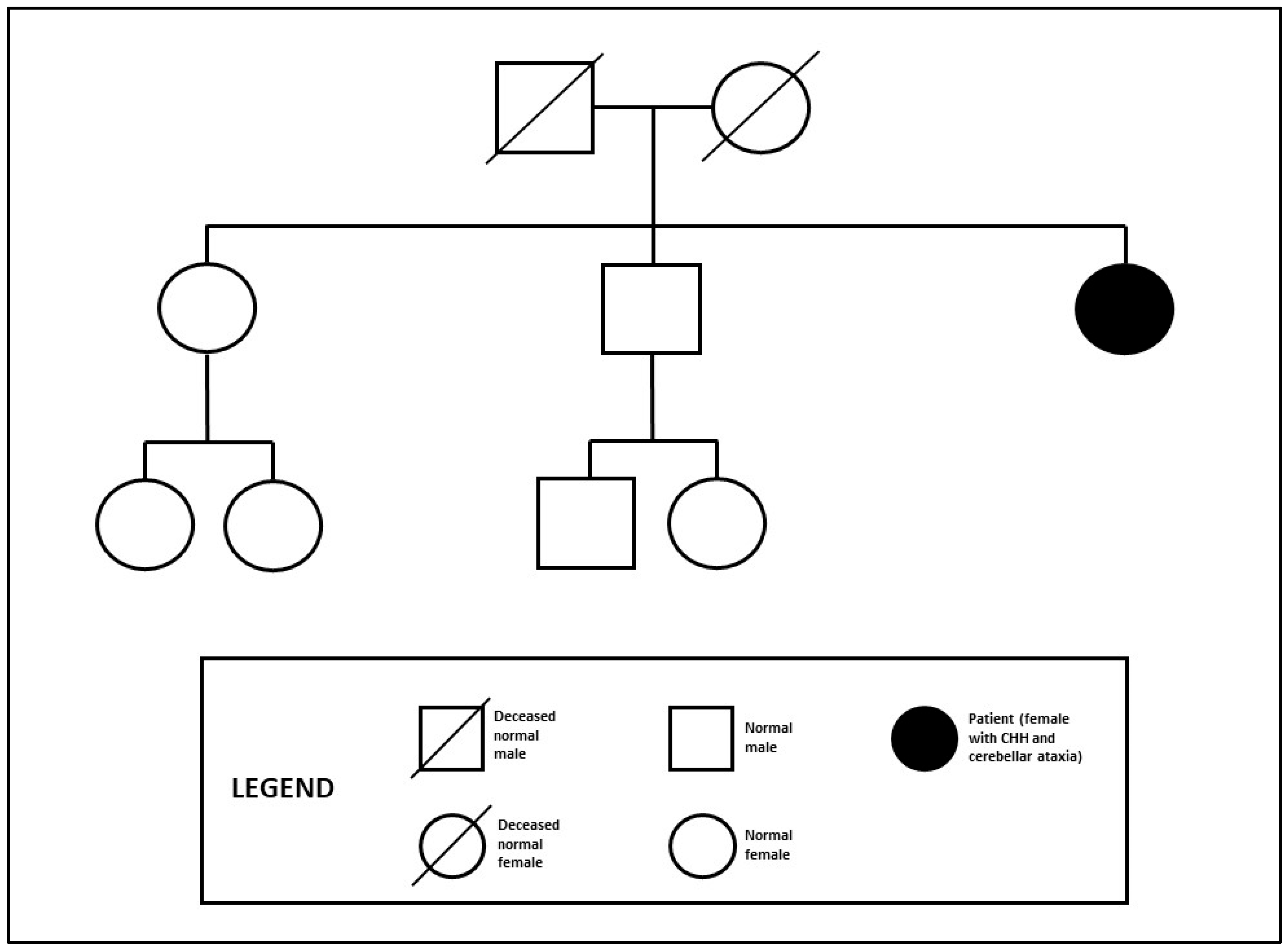
2. Case History
3. Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Pitteloud, N.; Durrani, S.; Raivio, T.; Sykiotis, G.P. Complex genetics of congenital hypogonadotropic hypogonadism. Front. Horm. Res. 2010, 39, 142–153. [Google Scholar] [CrossRef] [PubMed]
- Dodè, C.; Hardelin, J.P. Kallmann Syndrome. Eur. J. Hum. Genet. 2009, 17, 139–146. [Google Scholar] [CrossRef] [PubMed]
- Serafini, T.; Colamarino, S.A.; Leonardo, E.D.; Wng, H.; Beddington, R.; Skarnes, W.C.; Tessier-Lavigne, M. Netrin-1 is required for commissural axon guidance in the developing vertebrate nervous system. Cell 1996, 87, 1001–1014. [Google Scholar] [CrossRef] [PubMed]
- Srour, M.; Riviere, J.B.; Pham, J.M.; Dube, M.P.; Girard, S.; Morin, S.; Dion, P.A.; Asselin, G.; Rochefort, D.; Hince, P.; et al. Mutations in DCC gene cause congenital mirror movements. Science 2010, 328, 592. [Google Scholar] [CrossRef] [PubMed]
- Marsh, A.P.; Heron, D.; Edwards, T.J.; Quartier, A.; Galea, C.; Nava, C.; Rastetter, A.; Moutard, M.L.; Anderson, V.; Bitoun, P.; et al. Mutations in DCC cause isolated agenesis of the corpus callosus with incomplete penetrance. Nat. Genet. 2017, 49, 511–514. [Google Scholar] [CrossRef] [PubMed]
- Bouilly, J.; Messina, A.; Papadakis, G.; Cassatella, D.; Xu, C.; Acierno, J.S.; Tata, B.; Sykiotis, G.; Santini, S.; Sidis, Y.; et al. DCC/NTN1 complex mutations in patients with congenital hypogonadotropic hypogonadism impair GnRH neuron development. Hum. Mol. Genet. 2018, 27, 359–372. [Google Scholar] [CrossRef] [PubMed]
- Stamou, M.I.; Georgopoulos, N.A. Kallmann syndrome: Phenotype and genotype of hypogonadotropic hypogonadism. Metabolism 2017, 86, 124–134. [Google Scholar] [CrossRef] [PubMed]
- Hafiza, N.; Mariam, L.; Cheryl, F. Management of congenital hypogonadotropic hypogonadism in females. Hum. Fertil. 2021, 26, 622–631. [Google Scholar]
- Dwyer, A.A.; Raivio, T.; Pitteloud, N. MANAGEMENT OF ENDOCRINE DISEASE: Reversible hypogonadotropic hypogonadism. Eur. J. Endocrinol. 2016, 174, R267–R274. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.G.; Ahn, J.-W.; Kurth, I.; Ullmann, R.; Kim, H.-T.; Kulharya, A.; Ha, K.-S.; Itokawa, Y.; Meliciani, I.; Wenzel, W.; et al. WDR11, a WD protein that interacts with trascription factor EMX1, is mutated in idiopathic hypogonadotropic hypogonadism and Kallmann Syndrome. Am. J. Hum. Genet. 2010, 87, 465–479. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Osborn, D.P.; Lee, J.; Araki, M.; Araki, K.; Mohun, T.; Känsäkoski, J.; Brandstack, N.; Miralles, F.; Kim, C.; et al. WDR11-mediated Hedgehog signalling defects underlie a new ciliopathy related to Kallmann syndrome. EMBO Rep. 2018, 19, 269–289. [Google Scholar] [CrossRef] [PubMed]
- Yamada, R.; Yamakita, N.; Yasuda, K.; Imai, A. Adult-onset reversible idiopathic hypogonadotropic hypogonadism in male adult carryng a WDR11 missense mutation. BMJ Case Rep. 2022, 15, e250444. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Kim, Y.; Ataliotis, P.; Kim, H.G.; Kim, D.W.; Bennett, D.C.; Brown, N.A.; Layman, L.C.; Kim, S.H. Coordination of canonical and noncanonical Hedgehog signalling pathways mediated by WDR11 during primordial germ cell development. Sci. Rep. 2023, 13, 12309. [Google Scholar] [CrossRef] [PubMed]
- Bernard, G.; Chouery, E.; Putorti, M.L.; Tétreault, M.; Takanohashi, A.; Carosso, G.; Clément, I.; Boespflug-Tanguy, O.; Rodriguez, D.; Delague, V.; et al. Mutations of POLR3A encoding a catalytic subunit of RNA polymerase POL III cause a recessive hypomyelinating leukodystrophy. Am. J. Hum. Genet. 2011, 89, 415–423. [Google Scholar] [CrossRef] [PubMed]
- Tétreault, M.; Choquet, K.; Orcesi, S.; Tonduti, D.; Balottin, U.; Teichmann, M.; Fribourg, S.; Schiffmann, R.; Brais, B.; Vanderver, A.; et al. Recessive Mutations in POLR3B, Encoding the Second Largest Subunit of Pol III, Cause a Rare Hypomyelinating Leukodystrophy. Am. J. Hum. Genet. 2011, 89, 652–655. [Google Scholar] [CrossRef] [PubMed]
- Matthis, S.; Christoph, K.; Tobias, B.H.; Ludger, S. Ataxia meets chorioretinal dystrophy and hypogonadism: Boucher-Neuhäuser syndrome due to PNPLA6 mutations. J. Neurol. Neurosurg. Psychiatry A 2014, 86, 580–581. [Google Scholar] [CrossRef]
- Seminara, S.B.; Acierno, J.S., Jr.; Abdulwahid, N.A.; Crowley, W.F., Jr.; Margolin, D.H. Hypogonadotropic hypogonadism and cerebellar ataxia: Detailed phenotypic characterization of a large, extended kindred. J. Clin. Endocrinol. Metab. 2002, 87, 1607–1612. [Google Scholar] [CrossRef] [PubMed]
- Margolin, D.H.; Kousi, M.; Chan, Y.M.; Lim, E.T.; Schmahmann, J.D.; Hadjivassiliou, M.; Hall, J.E.; Adam, I.; Dwyer, A.; Plummer, L.; et al. Ataxia, dementia, and hypogonadotropism caused by disordered ubiquitination. N. Engl. J. Med. 2013, 368, 1992–2003. [Google Scholar] [CrossRef] [PubMed]
- De Roux, N.; Carel, J.C.; Léger, J. Congenital Hypogonadotropic Hypogonadism: A Trait Shared by Several Complex Neurodevelopmental Disorders. Endocr. Dev. 2016, 29, 72–86. [Google Scholar] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| Hormone | Value | Reference Range |
|---|---|---|
| LH | 0.8 mUI/mL | 1.9–12.5 mUI/mL |
| FSH | 0.9 mUI/mL | 2.5–10.2 mUI/mL |
| Oestradiol | 10 pg/mL | 19.5–144.2 pg/mL |
| Progesterone | 0.05 ng/mL | 0.30–1.20 ng/mL |
| AMH | 1.02 ng/mL | 1.22–15.8 ng/mL |
| TSH | 1.10 µIU/mL | 0.55–4.78 µIU/mL |
| fT3 | 3.78 pg/mL | 2.3–4.2 pg/mL |
| fT4 | 1.42 ng/dL | 0.70–1.76 ng/dL |
| ACTH | 11.6 pg/mL | <47 pg/mL |
| Cortisol | 12.38 µg/dL | 4.3–22.4 µg/dL |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chiarello, P.; Seminara, G.; Bossio, S.; Rocca, V.; Colao, E.; Iuliano, R.; Aversa, A. Adult-Onset Case of Female Idiopathic Hypogonadotropic Hypogonadism and Ataxia: Genetic Background. Endocrines 2024, 5, 334-340. https://doi.org/10.3390/endocrines5030024
Chiarello P, Seminara G, Bossio S, Rocca V, Colao E, Iuliano R, Aversa A. Adult-Onset Case of Female Idiopathic Hypogonadotropic Hypogonadism and Ataxia: Genetic Background. Endocrines. 2024; 5(3):334-340. https://doi.org/10.3390/endocrines5030024
Chicago/Turabian StyleChiarello, Paola, Giuseppe Seminara, Sabrina Bossio, Valentina Rocca, Emma Colao, Rodolfo Iuliano, and Antonio Aversa. 2024. "Adult-Onset Case of Female Idiopathic Hypogonadotropic Hypogonadism and Ataxia: Genetic Background" Endocrines 5, no. 3: 334-340. https://doi.org/10.3390/endocrines5030024
APA StyleChiarello, P., Seminara, G., Bossio, S., Rocca, V., Colao, E., Iuliano, R., & Aversa, A. (2024). Adult-Onset Case of Female Idiopathic Hypogonadotropic Hypogonadism and Ataxia: Genetic Background. Endocrines, 5(3), 334-340. https://doi.org/10.3390/endocrines5030024