Abstract
The stunning advances in understanding the reactivity and selectivity principles of asymmetric pericyclic reactions have had a profound impact on the synthetic planning of complex natural products. Indeed, electrocyclizations, cycloadditions, and sigmatropic rearrangements enable synthetic chemists to craft highly functionalized scaffolds that would not otherwise be possible with a similar atom-, step-, and redox-economy. In this review, selected examples from the last two decades of research (2003–2020) on tandem processes combining oxa-6π electrocyclic reactions are discussed in terms of reactivity challenges, inherent reversibility, and key structural bond formation in the assembly of natural products. A particular emphasis is given to the electrocyclic ring-closures in the tandem processes featuring Knoevenagel-type condensations, Diels–Alder cycloadditions, Stille couplings, and oxidative dearomatizations. The synthetic manifolds reviewed here illustrate how oxa-6π electrocyclizations are intimately linked to the construction of complex natural product scaffolds and have inspired a number of biomimetic syntheses in the laboratory.
1. Introduction
Natural products have long held societal value as folk medicines, and even in this modern era, many FDA-approved drugs are still being derived from terpenes and alkaloids found in nature. From a fundamental stand-point, the total synthesis of natural products remains one of the most exhilarating areas of chemical research by providing a unique platform for the discovery of novel technologies, methodologies, and synthetic strategies to provide a more sustainable and reliable access to biologically active molecules. In this context, pericyclic reactions [1] presented in this special issue of Organics (e.g., electrocyclizations [2,3], cycloadditions [4], and sigmatropic rearrangements [5]) are some of the most powerful manifolds to streamline the construction of complex polycyclic natural products. This short review covers the most recent advances in oxa-6π electrocyclization and aims to complement the remarkable reviews by Trauner [6], Ding [7], and Sheikh [8] with an emphasis on complex natural product synthesis. Unlike its aza-congener [9], applications of oxa-6π electrocyclization in building the molecular complexity found in natural products has not yet been reviewed. However, natural products displaying a heterocyclic pyran-derived scaffold are quite common. Cyclic 2H-(benzo)pyrans (e.g., 2) are often not overly more stable than their opened oxatriene counterpart 1″ and are quite reactive moieties which not surprisingly undergo subsequent transformation to generate a rapid structural complexity wrapped around a highly functionalized pyran core (Scheme 1). This review was therefore primarily organized along the rapid generation of complex molecules with examples of tandem and cascade processes (Section 2.1 and Section 2.2) and rare instances of transannular oxa-6π electrocyclizations involved macrocyclic ring contraction (Section 2.3).
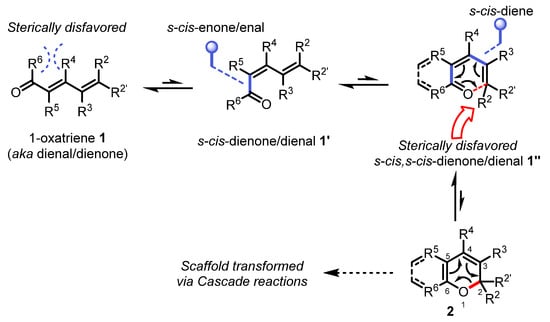
Scheme 1.
Isomerization and conformational equilibrium characteristic of the oxa-6π electrocyclic reaction reversibility in forming the 2H-(benzo)pyran scaffold.
2. Discussion
Although all carbon electrocyclic reactions (4π, 6π, 8π,and even 12π [10]) have played an important role in advancing asymmetric intermediates towards complex natural products, excellent reviews on this topic exist and will not be repeated herein [6,7,8]. 2H-pyrans and related 2H-benzopyrans are privileged scaffolds found in natural products which have attracted the interests of synthetic chemists for decades [11,12]. Interestingly, synthetic examples of these heterocycles are relatively scarce due to their innate instability as a consequence of the high reversibility of the oxa-6π electrocyclization (forward- and retro-). This electrocyclic reaction entails a key σ-bond formation between the termini of an 1-oxatriene scaffold 1 via a π-electron redistribution permitted by the principle of orbital symmetry conservation (Scheme 1). The oxa-6π electrocyclization is typically in fast equilibrium with the retro-oxa-6π due to the thermoneutrality often invoked between the opened and closed valence tautomers (isomers) of 2H-(benzo)pyrans, and the relatively low-energy barriers of the various interconversion transition states (Scheme 1) [13,14]. For example, if R5 = R6 = H, s-cis/s-trans dienal conformers 1′ and 1″ will exist in dynamic equilibrium between 1 and 2. In regards to enones 1, if R5 and R6 are forming a ring, or if R4 is a bulkier substituent than R5, the R4/R6 steric clash will effectively destabilize the s-trans conformation towards s-cis enone 1′ exclusively, thus enabling a possible equilibrium shift towards the s-cis,s-cis dienone 1″ [15,16,17]. Another steric clash occurs when R2 ≠ H or for disubstituted R2/R2′ ≠ H in terminal E-alkenes, thus forcing the s-cis,s-cis dienone π-system of 1″ out of conjugation which ultimately results in a favorable oxa-6π electrocyclic ring-closure towards the 2H-pyran isomer 2. Aside from these steric factors, the oxa-6π electrocyclic ring-closure is also favored towards 2H-(benzo)pyrans 2 by the presence of either electron-withdrawing groups at the C5 and/or C6 positions (reduced nucleophilicity of the enol ether moiety), or the stabilizing π-aromaticity of arenes (C5-C6) [18,19]. For most oxatriene substrates 1 which do not possess the stereoelectronic characteristics mentioned above, the oxa-6π electrocyclic ring-closure is often reversible unless being funneled through a cascade reaction [20,21]. As such, the total synthesis of (+)-torreyanic acid by Porco involving a oxa-6π electrocyclization/Diels−Alder cycloaddition cascade rapidly became a classic biomimetic reaction demonstrating how a 2H-pyran can be seamlessly transformed into a much more complex natural scaffold (Scheme 2).
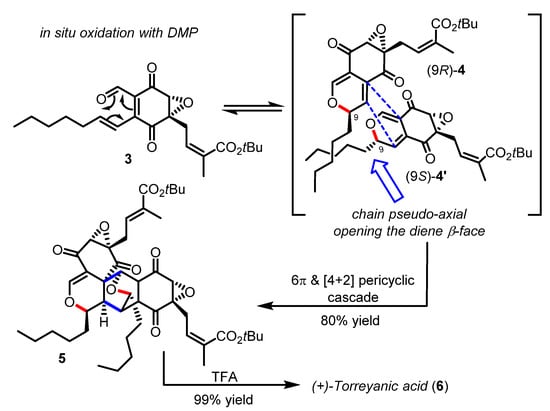
Scheme 2.
Total synthesis of (+)-torreyanic acid by Porco (2000–2003).
2.1. 6π-Electrocyclization/Diels−Alder Cycloaddition Cascade: The Domino Effect
Torreyanic acid (6) isolated from the endophytic fungus Pestalotiopsis microspore was proposed by Clardy to arise from a Diels−Alder dimerization of two 2H-pyran epimers which could have been formed in situ through the oxa-6π electrocyclization of a single epoxy-quinone precursor [22]. Indeed, Porco was able to validate this biosynthetic hypothesis, by designing a biomimetic cascade reaction that entailed the oxidation of a starting alcohol (not shown) into the enantiopure quinone epoxide 3 as a trigger for the spontaneous disrotatory oxa-6π electrocyclization and the ensuing dimerization (Scheme 2) [23,24]. A related strategy of mild oxidation as a means to produce a reactive oxatriene moiety and initiate an oxa-6π electrocyclic reaction in tandem was recently reported in a route towards the papuaforin A-C natural products [25]. The exquisite diastereoselectivity observed in the heterodimer product 5 was consistent with the theoretical energy-minima calculations from several competitive reaction pathways, reaction intermediates, and transition states computed on a model substrate at B3LYP/6-31G* level of theory. Briefly, the results suggested that both 2H-pyran epimers 4/4′ could be easily accessible (similar potential energy) and in rapid equilibrium (ΔΔG‡ ~5.0 Kcal/mol), while the syn-pyran (9S)-4′ in which the pentyl side-chain adopts an axial orientation possess a thus more sterically accessible diene (anti face to the epoxide moiety) for the Diels−Alder cycloaddition to occur. It was therefore clearly demonstrated that the Diels−Alder dimerization stereoselectivity obtained in the heterodimer 5 was fully controlled by the pair of 2H-pyran epimers. These results were further confirmed by Hayashi during the synthesis of related epoxyquinol A-C natural products [26]. A similar platform involving a tandem oxa-6π electrocyclization/Diels−Alder cycloaddition has found interesting applications in the synthesis of highly functionalized “natural product-like” molecules [20].
2.2. Tandem Sequences Geared towards Oxa-6π Electrocyclization Reactivity
Another highly effective strategy to introduce 2H-pyran structures within the skeleton of complex natural products requires transformations that install an appropriate oxatriene moiety to achieve oxa-6π electrocyclization in a tandem fashion. The most productive tactic for the formation of various 2H-(benzo)pyrans certainly entails the combination of Knoevenagel condensations with oxa-6π electrocyclizations, although the more recent approaches via Stille cross-coupling and oxidative dearomatization will also be discussed in this section. Indeed, the formal [3 + 3] cycloaddition strategy consisting of Knoevenagel-type condensation followed by an oxa-6π electrocyclic ring closure (often reversible) enables two σ bonds and one stereocenter to be installed simultaneously [27]. This methodology has enjoyed many applications by Hsung and others in the synthesis of complex natural products (Scheme 3) [28].
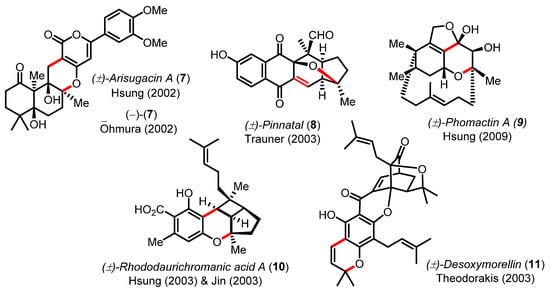
Scheme 3.
Examples of structural bond formation from natural products synthesized by oxa-6π electrocyclizations in tandem Knoevenagel condensations or Claisen rearrangement.
Although this cascade reaction has been pivotal in the synthesis of numerous natural products (e.g., arisugacin A (7) [29,30,31], pinnatal (8) [32], phomactin A (9) [33,34], rhododaurichromanic acid A (10) [35,36], or desoxymorellin (11) [37]), no real improvements in terms of catalysis (piperidine, piperidinium, or ethylene diamine diacetate (EDDA)) have been recently proposed except for the practical and efficient reaction in water by Lee [38] and the DABCO-catalyzed electrocyclization of dienyl α-diketones by Frontier [39]. This cascade reaction was also exploited by Hirama in the synthesis the marine steroidal alkaloids, cortistatins A and J (Scheme 4) [40,41]. The one-step union of cyclohexane-1,3-dione 12 with the advanced CD-ring building block 13 was mediated by piperidine to irreversibly deliver the tetracyclic product 14 in 87% yield and high diastereoselectivity (~5:1 dr). In this effort, Yamashita and Hirama had to meticulously optimized the Knoevenagel/oxa-6π electrocyclization cascade to form the pivotal B ring common to all the cortistatin natural products and avoid the formation of undesired structural isomers [41].
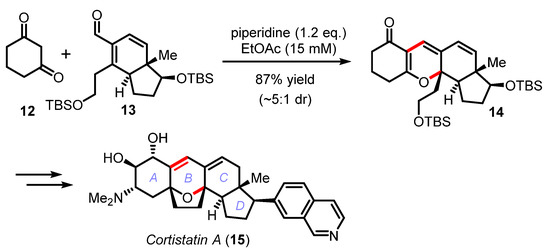
Scheme 4.
Total synthesis of cortistatin A by Yamashita and Hirama (2008–2011).
Along these lines, Quinn [42] and more recently Heretsch and Christmann [43] have exploited an organocatalyzed Knoevenagel/oxa-6π electrocyclization cascade towards the assembly of pyranopyridone-based natural products [44]. In their streamlined synthesis of yaequinolones J1/J2 (17a-b) in five steps, Heretsch and Christmann reported their efforts in optimizing the 2H-benzopyran forming step catalyzed by EDDA in toluene to prepare substrate 16 in 49% yield (Scheme 5A) [43]. As shown in Scheme 5B, the pyranopyridone scaffold can also be applied directly to the synthesis of 2H-pyran 18 through a Knoevenagel/oxa-6π electrocyclization sequence catalyzed by ytterbium triflate, thus affording a genuinely straightforward synthesis of melicodenine C (19) [42]. The same year, Zografos and coworkers reported a more systematic study to access pyranopyridones via a Knoevenagel/oxa-6π electrocyclization process for the synthesis of structurally diverse natural-product-like pyridines [45].
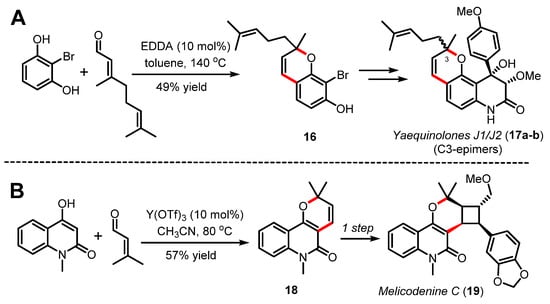
Scheme 5.
(A) Total synthesis of yaequinolones J1/J2 (17a–b) by Heretsch and Christmann (2020) and (B) total synthesis of melicodenine C (19) by Quinn (2012).
Another exciting example was reported recently by Newhouse with the ten-step synthesis of a limonoid alkaloid (+)-granatumine A (24) (Scheme 6). In this synthesis, Newhouse and his group fully exploited the reversibility of the oxa-6π electrocyclization to cleverly transform the central 2H-pyran heterocycle of 23 into the highly substituted pyridine ring of the natural product 24. The Knoevenagel/oxa-6π electrocyclization route between the cyclohexane 1,3-dione substrate 20 and the α,β—unsaturated aldehyde 21—once again catalyzed by EDDA [46] delivered the pivotal pentacyclic fused scaffold 23 in 47% yield with high diastereo- and regioselectivity. Through computational DFT (density functional theory) investigations and the experimental interconversion between pyran scaffolds, the authors demonstrated that the 2H-pyran C1-regioisomer 23 was indeed thermodynamically (ΔΔG° of ~2.8 Kcal/mol) and kinetically preferred and that the electrocyclization proceeded with an exquisite disrotatory torquoselectivity.
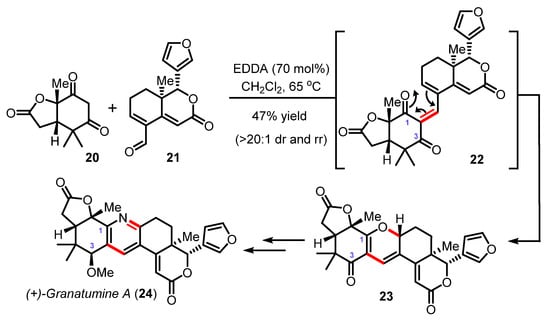
Scheme 6.
Total synthesis of (+)-granatumine A (24) by Newhouse (2019).
It is important to note that even if significant progress has been made in understanding the general diastereoselectivity outcome of the thermal oxa-6π electrocyclization (π6s disrotatory), the development of catalytic enantioselective methods towards enantioenriched 2H-pyrans is lagging far behind [47]. This statement is largely exemplified by comparing the synthetic strategies developed separately by both the Ito [48,49] and Gong/Yang [50] groups to deliver the asymmetric syntheses of several quignardone meroterpenoids (Scheme 7). The Ito group exploited the reactivity of a chiral synthetic C2-symetrical 1,3-diketone 25 in the oxa-6π electrocyclization towards the tricyclic pyran 26 in order to control facial selectivity at late-stage hydrogenation and deliver (−)-guinardone I (27) in an enantioslective manner. On the other hand, Gong and Yang exploited the chirality of (D)-quinic acid available from the chiral pool to secure the two stereocenters of 1,3-diketone 28 in a relatively long sequence of sixteen steps. As shown in Scheme 7B, the cascade reaction developed to construct pyran 29 was mediated by piperidine and acid similarly to the report by Ito.
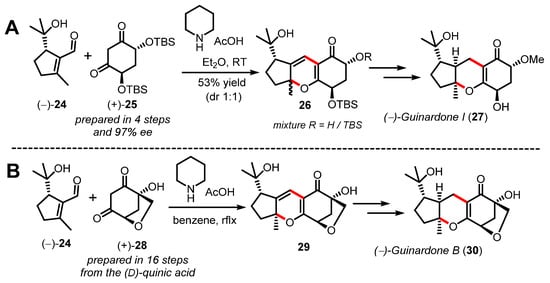
Scheme 7.
Total syntheses of guinardones (27/30), (A) by Ito (2019) and (B) by Gong and Yang (2020).
From all the examples discussed above, it can be concluded that EDDA and piperidine are the most common catalysts used to achieve the thermal Knoevenagel condensation/oxa-6π electrocyclization tandem processes. As a more modern synthetic alternative to the Knoevenagel condensation, Theodorakis evaluated the aromatic Claisen rearrangement in the synthesis of desoxymorellin (±)-(11) [37]. This strategy was further elegantly exploited by George in a concise and divergent biomimetic synthesis of naphterpin and marinone meroterpenoid natural products [51].
An alternative tandem reaction for the synthesis of highly functionalized 2H-pyran embedded in large and complex scaffolds was developed by the Stoltz group during their total synthesis campaign of the caged diterpenoid (−)-saudin (35) [52,53,54]. The overall synthetic strategy is shown in Scheme 8. After evaluating a number of palladium-catalyzed cross-couplings, Stoltz and coworkers were able to develop optimum conditions for a Stille coupling. Both combinations of vinylstannane 31a-b and vinyl iodide 32a-b were successfully investigated under Cu(I)-accelerated Stille conditions to provide a novel route to a variety of 2H-pyrans through a tandem of Stille coupling and the oxa-6π electrocyclization reaction of intermediates 33. During this methodological development, the authors noted that the second combination between 31b/32b was more sensitive to oxidation, which could be overcome by providing rigorous inert conditions in a glove box to generate the angular and highly substituted 2H-pyrans 34 in good yields. This tandem process was later evaluated by Mindt and successfully applied to the synthesis of benzo- and naphtopyrans in a single step from readily available bromoquinone and vinyl stannane substrates [55].
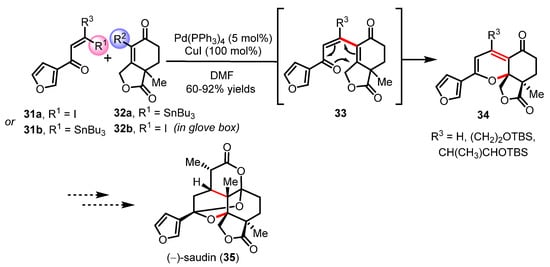
Scheme 8.
Synthetic route towards the diterpenoid (−)-saudin (35) by Stoltz (2005–2007).
In 2020, an oxidative dearomatization for the generation of ortho-quinone methide with exquisite chemoselectivity has been reported to enable a series of tandem reactions such as the oxa-6π electrocyclization [56]. This novel and efficient synthetic strategy will certainly pave the way towards a more modern approach to complex 2H-pyran scaffolds (Scheme 9). In brief, the transition-metal-free conditions reported in this study enabled the direct oxidation of phenols (e.g., 36) at their benzylic positions using hypoiodite catalysis to trigger a dearomatization process forming ortho-quinone methide 37. As shown in Scheme 9, the highly reactive π-system of o-quinone methide 37 formed by oxidative dearomatization presumably undergoes a rapid s-cis/s-trans conformational equilibrium between 37 and 38 to enable the ensuing oxa-6π electrocyclization to take place and yield the expected 2H-benzopyran products 39 through a favorable rearomatization. This study reported by Ishihara will certainly find many novel applications in the realm of natural product synthesis given the potential of reactivity of transient ortho-quinone methides for tandem reactions. To complete this overview of tandem reactions, we should also note the unique method recently reported by Bonesi and coworkers via a photo-induced Fries rearrangement/oxa-6π electrocyclization manifold for the synthesis of chromanones [57].
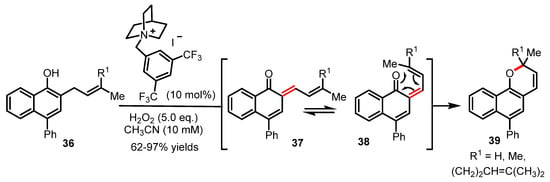
Scheme 9.
Novel synthetic route towards 2H-benzopyrans 39 by Ishihara (2020).
2.3. Strained Cyclic Oxatriene Involved in “Transannular” Oxa-6π Electrocyclization
It is important to note that unlike the all the cases of intramolecular electrocyclizations shown in Section 2.1 and Section 2.2, the examples of oxa-6π electrocyclization presented below occur within either medium- or large-size macrocycles, hence their classification as “transannular” reactions. To our knowledge, the first example of transannular oxa-6π electrocyclization was reported by Hsung and coworkers in 2003 [58] during their efforts to build the structurally unprecedented ABD-tricyclic ring system of (±)-phomactin A (9) (Scheme 10A) [33,34]. In this route, a number of steps were required to install the citral-like side chain onto the cyclohexane 1,3-dione of enal 40 designed by Hsung as the starting material for the pivotal cascade reaction. As planned, substrate 40 was subjected to iminium-based activation to construct the 14-membered ring macrocycle 42 via a Knoevenagel-type condensation under high dilution conditions. The ensuing oxa-6π electrocyclization took place in tandem to deliver product 43 bearing the contracted 12-membered D-ring (22% yield) along with an undesired structural isomer (major product) formed around a smaller and more favorable 10-membered ring macrocycle. As shown in Scheme 10B, Trauner reported in 2005 a similar approach for the synthesis of the unusual ansa-terpenoid smenochromene D (±)-(47) [59]. This macrocyclic chromene natural product (aka likonide B) was initiated by first installing a long farnesyl-type side chain onto an hydroquinone to obtain the fully functionalized building block 44. In this study, Trauner and coworkers exploited a Nagata-type reaction mediated by phenylboronic acid to obtain the ortho-hydroxyalkylation product 45, which upon the effect of temperature further spontaneously proceeded to elimination via dearomatization to form the 18-membered ring macrocyclic intermediate 46. The ensuing rearomatization into the 2H-benzopyran heterocycle is likely a strong driving force to the final transannular oxa-6π electrocyclization occurring through the ring contraction of 46 into the 16-membered ring of the natural product (±)-(47).
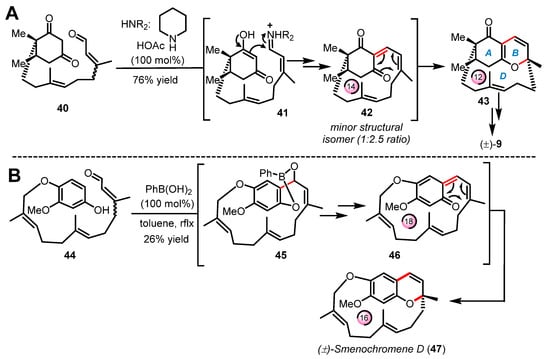
Scheme 10.
Pivotal transannular oxa-6π electrocyclization in the syntheses of (A) (±)-phomactin A (9) by Hsung (2003–2009) and (B) (±)-smenochromene D (47) by Trauner (2005).
Finally, the most recent example was reported by West and Roche during the synthesis of briareolate ester B (50) shown in Scheme 11 [60]. Given the collective isolation of briareolate esters L (48), G (49), and B (50) from the octocoral Briarium asbestinum, it was naturally proposed that these briarane cembranoids could be biosynthetically related through a mechanism of isomerization (48 with 49) and oxa-6π electrocyclization (49 to 50). Surprisingly, the naturally occurring 2H-pyran scaffold of 50 presents an anti-Bredt bridgehead alkene suggestive of a highly strained oxo-bridged macrocycle [61]. As expected, the strained (E,Z) 1,3-diene system of briareolate ester L (48) isomerized rapidly into a more conjugated (Z,Z)-system under UVA-photoirradiation to produce the reactive but observable intermediate briareolate ester G (49) [62]. After about ten minutes of photoirradiation, the isomerization reached a steady-state and a photoinduced transannular oxa-6π electrocyclization took place in tandem to afford the highly functionalized briareolate ester B (50) in 60% yield. The synthetic cascade reaction validated the biosynthetic proposal hinging on the spectacular 10- to 8-membered ring contraction of 49 to 50. In the same study, a unique photochromic switch was also developed via a rapid retro-6π electrocyclization of 50 under UVC irradiation to produce a coexisting mixture of isomers 48 and 49. Similar photochromic switches are typically valuable tools in material chemistry and the new recent study by Pei and Zhou on a 1,8-dioxatetraene model will certainly find multiple applications in the realm of photochromic films [63].
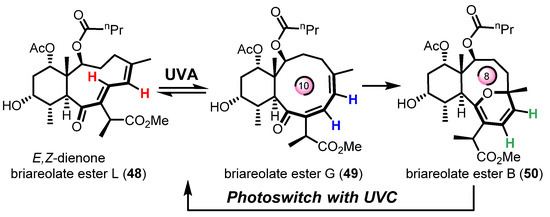
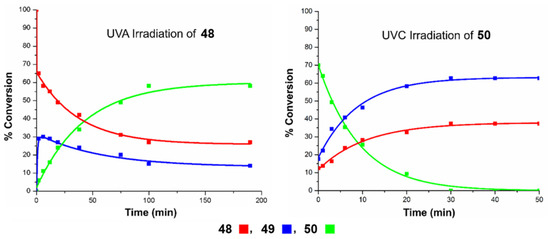
Scheme 11.
Transannular oxa-6π electrocyclization for the biomimetic interconversion of briareolate ester L (48) to B (50) controlled by a selective UVA/UVC photoswitch. Figure adapted with permission from the authors. Copyright 2017 American Chemical Society.
3. Outlook
This review summarized the most recent advances in oxa-6π electrocyclic reactions inspired by unique natural products and their biosynthetic pathways. Given the typical reversibility of 6π-electrocyclic ring closures, a systematic understanding of the stereoelectronic effects stabilizing 2H-(benzo)pyrans is critical. I hope to have demonstrated that tandem and cascade reactions combined with oxa-6π electrocyclizations play a prevalent role in constructing strategic bonds while transforming the transient 2H-(benzo)pyran rings into more stable and complex scaffolds of natural products. Significant methodological advances have been made in controlling the stereochemical outcome of oxa-6π electrocyclizations, although the lack of catalytic enantioselective variants of this reaction is evident. By using oxa-6π electrocyclization in tandem reactions, we should be able to further engineer efficient and atom-economic routes towards numerous intricate molecular structures and better understand the stereoelectronic factors enabling regio- and conformational selectivity and catalysis in these processes. I hope this review will stir further interest in these manifolds to explore novel opportunities in catalytic tandem reactions for the construction of complex (polycyclic)heterocycles.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
I would like to thank Lyndon M. West. from Florida Atlantic University for the continuous opportunity to collaborate and study biosynthetic pathways that have inspired the development of biomimetic reactions with all the talented students from his group.
Conflicts of Interest
The author declares no conflict of interest.
References
- Wiest, O.; Houk, K.N. Density Functional Theory IV, 1st ed.; Series: Topics in Current Chemistry; Springer: Berlin/Heidelberg, Germany, 1996; pp. 1–24. [Google Scholar]
- Marvell, E.N. Thermal Electrocyclic Reactions; Academic Press: New York, NY, USA, 1980; Volume 43. [Google Scholar]
- Ansari, F.L.; Qureshi, R.; Qureshi, M.L. Electrocyclic Reactions; Wiley-VCH: Weinheim, Germany, 1999. [Google Scholar]
- Nicolaou, K.C.; Snyder, S.A.; Montagnon, T.; Vassilikogiannakis, G. The Diels–Alder Reaction in Total Synthesis. Angew. Chem. Int. Ed. 2002, 41, 1668–1698. [Google Scholar] [CrossRef]
- Jones, A.C.; May, J.A.; Sarpong, R.; Stoltz, B.M. Toward a Symphony of Reactivity: Cascades Involving Catalysis and Sigmatropic Rearrangements. Angew. Chem. Int. Ed. 2014, 53, 2556–2591. [Google Scholar] [CrossRef] [PubMed]
- Beaudry, C.M.; Malerich, A.J.P.; Trauner, D. Biosynthetic and Biomimetic Electrocyclizations. Chem. Rev. 2005, 105, 4757–4778. [Google Scholar] [CrossRef] [PubMed]
- Bian, M.; Li, L.; Ding, H. Recent Advances on the Application of Electrocyclic Reactions in Complex Natural Product Synthe-sis. Synthesis 2017, 49, 4383–4413. [Google Scholar] [CrossRef]
- Sheikh, N.S. 4π electrocyclisation in domino processes: Contemporary trends and synthetic applications towards natural products. Org. Biomol. Chem. 2015, 13, 10774–10796. [Google Scholar] [CrossRef]
- Vargas, D.F.; Larghi, E.L.; Kaufman, T.S. The 6π-azaelectrocyclization of azatrienes. Synthetic applications in natural products, bioactive heterocycles, and related fields. Nat. Prod. Rep. 2018, 36, 354–401. [Google Scholar] [CrossRef] [PubMed]
- Derewacz, D.K.; Covington, B.; McLean, J.A.; Bachmann, B.O. Mapping Microbial Response Metabolomes for Induced Natural Product Discovery. ACS Chem. Biol. 2015, 10, 1998–2006. [Google Scholar] [CrossRef] [PubMed]
- Nicolaou, K.C.; Pfefferkorn, J.A.; Roecker, A.J.; Cao, G.-Q.; Barluenga, S.; Mitchell, H.J. Natural Product-like Combinatorial Libraries Based on Privileged Structures. 1. General Principles and Solid-Phase Synthesis of Benzopyrans. J. Am. Chem. Soc. 2000, 122, 9939–9953. [Google Scholar] [CrossRef]
- Tejedor, D.; Delgado-Hernández, S.; Diana-Rivero, R.; Díaz-Díaz, A.; García-Tellado, F. Recent Advances in the Synthesis of 2H-Pyrans. Molecules 2019, 24, 2904. [Google Scholar] [CrossRef]
- Bishop, L.M.; Winkler, M.; Houk, K.N.; Bergman, R.G.; Trauner, D. Mechanistic Investigations of the Acid-Catalyzed Cyclization of a Vinyl ortho-Quinone Methide. Chem. Eur. J. 2008, 14, 5405–5408. [Google Scholar] [CrossRef]
- Krasnaya, Z.A. Dienone ⇄ 2H-pyran valence isomerization. Chem. Heterocycl. Comp. 1999, 35, 1255–1271. [Google Scholar] [CrossRef]
- Tsuda, T.; Kiyoi, T.; Miyane, T.; Saegusa, T. Nickel(0)-catalyzed reaction of diynes with aldehydes. J. Am. Chem. Soc. 1988, 110, 8570–8572. [Google Scholar] [CrossRef]
- Lillya, C.P.; Kluge, A.F. Molecular spectra and conformations of conjugated dienones. J. Org. Chem. 1971, 36, 1977–1988. [Google Scholar] [CrossRef]
- Gosink, T.A. Valence isomers. Substituent effects on the equilibrium between 2H-pyrans and cis-dienones. J. Org. Chem. 1974, 39, 1942–1944. [Google Scholar] [CrossRef]
- Peng, W.; Hirabaru, T.; Kawafuchi, H.; Inokuchi, T. Substituent-Controlled Electrocyclization of 2,4-Dienones: Synthesis of 2,3,6-Trisubstituted 2H-Pyran-5-carboxylates and Their Transformations. Eur. J. Org. Chem. 2011, 2011, 5469–5474. [Google Scholar] [CrossRef]
- Menz, H.; Kirsch, S.F. Synthesis of Stable 2H-Pyran-5-carboxylates via a Catalyzed Propargyl-Claisen Rearrangement/Oxa-6π Electrocyclization Strategy. Org. Lett. 2006, 8, 4795–4797. [Google Scholar] [CrossRef]
- Tejedor, D.; Delgado-Hernández, S.; Peyrac, J.; González-Platas, J.; García-Tellado, F. Integrative Pericyclic Cascade: An Atom Economic, Multi C−C Bond-Forming Strategy for the Construction of Molecular Complexity. Chem. A Eur. J. 2017, 23, 10048–10052. [Google Scholar] [CrossRef] [PubMed]
- Bankura, A.; Naskar, S.; Chowdhury, S.R.; Maity, R.; Mishra, S.; Das, I. C 3 -Thioester/-Ester Substituted Linear Dienones: A Pluripotent Molecular Platform for Diversification via Cascade Pericyclic Reactions. Adv. Synth. Catal. 2020, 362, 3604–3612. [Google Scholar] [CrossRef]
- Lee, J.C.; Strobel, G.A.; Lobkovsky, E.; Clardy, J. Torreyanic Acid: A Selectively Cytotoxic Quinone Dimer from the Endophytic Fungus Pestalotiopsis microspora. J. Org. Chem. 1996, 61, 3232–3233. [Google Scholar] [CrossRef]
- Li, C.; Lobkovsky, E.; Porco, J.A. Total Synthesis of (±)-Torreyanic Acid. J. Am. Chem. Soc. 2000, 122, 10484–10485. [Google Scholar] [CrossRef]
- Li, C.; Johnson, R.P.; Porco, J.A. Total Synthesis of the Quinone Epoxide Dimer (+)-Torreyanic Acid: Application of a Biomimetic Oxidation/Electrocyclization/Diels−Alder Dimerization Cascade1. J. Am. Chem. Soc. 2003, 125, 5095–5106. [Google Scholar] [CrossRef] [PubMed]
- Bellavance, G.; Barriault, L. Modular Total Syntheses of Hyperforin, Papuaforins A, B, and C via Gold(I)-Catalyzed Carbocy-clization. J. Org. Chem. 2018, 83, 7215–7230. [Google Scholar] [CrossRef]
- Shoji, M.; Imai, H.; Mukaida, M.; Sakai, K.; Kakeya, H.; Osada, H.; Hayashi, Y. Total Synthesis of Epoxyquinols A, B, and C and Epoxytwinol A and the Reactivity of a 2H-Pyran Derivative as the Diene Component in the Diels−Alder Reaction. J. Org. Chem. 2005, 70, 79–91. [Google Scholar] [CrossRef]
- Shen, H.C.; Wang, J.; Cole, K.P.; McLaughlin, M.J.; Morgan, C.D.; Douglas, C.J.; Hsung, R.P.; Coverdale, H.A.; Gerasyuto, A.I.; Hahn, J.M.; et al. A Formal [3 + 3] Cycloaddi-tion Reaction. Improved Reactivity Using α,β-Unsaturated Iminium Salts and Evidence for Reversibility of 6π-Electron Elec-trocyclic Ring Closure of 1-Oxatrienes. J. Org. Chem. 2003, 68, 1729–1735. [Google Scholar] [CrossRef] [PubMed]
- Richard, P.H.; Aleksey, V.K.; Sydorenko, N. A Formal [3 + 3] Cycloaddition Approach to Natural-Product Synthesis. Eur. J. Org. Chem. 2005, 2005, 23–44. [Google Scholar]
- Sunazuka, T.; Handa, M.; Nagai, K.; Shirahata, T.; Harigaya, Y.; Otoguro, K.; Kuwajima, I.; Omura, S. The First Total Synthesis of (±)-Arisugacin A, a Potent, Orally Bioavailable Inhibitor of Acetylcholinesterase. Org. Lett. 2002, 4, 367–369. [Google Scholar] [CrossRef]
- Zehnder, L.R.; Hsung, R.P.; Wang, J.; Golding, G.M. A Concise Stereoselective Route to the Pentacyclic Frameworks of Arisugacin A and Territrem B. Angew. Chem. Int. Ed. 2000, 39, 3876–3879. [Google Scholar] [CrossRef]
- Cole, K.P.; Hsung, R.P. The first enantioselective total synthesis of (−)-arisugacin A. Tetrahedron Lett. 2002, 43, 8791–8793. [Google Scholar] [CrossRef]
- Malerich, J.P.; Trauner, D. Biomimetic Synthesis of (±)-Pinnatal and (±)-Sterekunthal A. J. Am. Chem. Soc. 2003, 125, 9554–9555. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Cole, K.P.; Buchanan, G.S.; Li, G.; Hsung, R.P. Total Synthesis of Phomactin A. Org. Lett. 2009, 11, 1591–1594. [Google Scholar] [CrossRef] [PubMed]
- Buchanan, G.S.; Cole, K.P.; Tang, Y.; Hsung, R.P. Total Synthesis of (±)-Phomactin A. Lessons Learned from Respecting a Challenging Structural Topology. J. Org. Chem. 2011, 76, 7027–7039. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.; Mei, Y.; Du, Y.; Jin, Z. Total Synthesis of the Highly Potent Anti-HIV Natural Product Daurichromenic Acid along with Its Two Chromane Derivatives, Rhododaurichromanic Acids A and B. Org. Lett. 2003, 5, 4481–4484. [Google Scholar] [CrossRef] [PubMed]
- Kurdyumov, A.V.; Hsung, R.P.; Ihlen, K.; Wang, J. Formal [3 + 3] Cycloaddition Approach to Chromenes and Chromanes. Concise Total Syntheses of (±)-Rhododaurichromanic Acids A and B and Methyl (±)-Daurichromenic Ester. Org. Lett. 2003, 5, 3935–3938. [Google Scholar] [CrossRef]
- Tisdale, E.J.; Slobodov, I.; Theodorakis, E.A. Biomimetic total synthesis of forbesione and desoxymorellin utilizing a tandem Claisen/Diels–Alder/Claisen rearrangement. Org. Biomol. Chem. 2003, 1, 4418–4422. [Google Scholar] [CrossRef] [PubMed]
- Jung, E.J.; Park, B.H.; Lee, Y.R. Environmentally benign, one-pot synthesis of pyrans by domino Knoevenagel/6π-electrocyclization in water and application to natural products. Green Chem. 2010, 12, 2003–2011. [Google Scholar] [CrossRef]
- Jacob, S.D.; Brooks, J.L.; Frontier, A.J. No Acid Required: 4π and 6π Electrocyclization Reactions of Dienyl Diketones for the Synthesis of Cyclopentenones and 2H-Pyrans. J. Org. Chem. 2014, 79, 10296–10302. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, S.; Iso, K.; Hirama, M. A Concise Synthesis of the Pentacyclic Framework of Cortistatins. Org. Lett. 2008, 10, 3413–3415. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, S.; Iso, K.; Kitajima, K.; Himuro, M.; Hirama, M. Total Synthesis of Cortistatins A and J. J. Org. Chem. 2011, 76, 2408–2425. [Google Scholar] [CrossRef] [PubMed]
- Holla, H.; Jenkins, I.; Neve, J.E.; Pouwer, R.H.; Pham, N.; Teague, S.J.; Quinn, R. Synthesis of melicodenines C, D and E. Tetrahedron Lett. 2012, 53, 7101–7103. [Google Scholar] [CrossRef]
- Schwan, J.; Kleoff, M.; Heretsch, P.; Christmann, M. Five-Step Synthesis of Yaequinolones J1 and J2. Org. Lett. 2020, 22, 675–678. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Qiao, T.; Watanabe, K.; Tang, Y. Concise Biosynthesis of Phenylfuropyridones in Fungi. Angew. Chem. Int. Ed. 2020, 59, 19889–19893. [Google Scholar] [CrossRef]
- Fotiadou, A.D.; Zografos, A.L. Electrocyclization of Oxatrienes in the Construction of Structurally Complex Pyranopyridones. Org. Lett. 2012, 14, 5664–5667. [Google Scholar] [CrossRef]
- Wang, X.; Lee, Y.R. Efficient Synthesis of Substituted Pyranoquinolinones from 2,4-Dihydroxyquinoline: Total Synthesis of Zanthosimuline, cis-3′,4′-Dihydroxy-3′,4′-Dihydroflindersine, and Orixalone D. Synthesis 2007, 2007, 3044–3050. [Google Scholar] [CrossRef]
- Thompson, S.; Coyne, A.G.; Knipe, P.C.; Smith, M.D. Asymmetric electrocyclic reactions. Chem. Soc. Rev. 2011, 40, 4217–4231. [Google Scholar] [CrossRef]
- Kobayashi, T.; Takizawa, I.; Shinobe, A.; Kawamoto, Y.; Abe, H.; Ito, H. Asymmetric Synthesis and Structure Revision of Guignardone H and I: Development of a Chiral 1,3-Diketone Possessing C2 Symmetry. Org. Lett. 2019, 21, 3008–3012. [Google Scholar] [CrossRef]
- Kobayashi, T.; Takizawa, I.; Kawamoto, Y.; Ito, H. Sequential condensation-6π-electrocyclization reaction of a chiral 1,3-Diketone possessing C2 symmetry. Tetrahedron Lett. 2020, 61, 151897. [Google Scholar] [CrossRef]
- Yan, Z.; Zhao, C.; Gong, J.; Yang, Z. Asymmetric Total Synthesis of (−)-Guignardones A and B. Org. Lett. 2020, 22, 1644–1647. [Google Scholar] [CrossRef]
- Murray, L.A.M.; Fallon, T.; Sumby, C.J.; George, J.H. Total Synthesis of Naphterpin and Marinone Natural Products. Org. Lett. 2019, 21, 8312–8315. [Google Scholar] [CrossRef]
- Tambar, U.K.; Kano, T.; Stoltz, B.M. Progress toward the Total Synthesis of Saudin: Development of a Tandem Stille-Oxa-Electrocyclization Reaction. Org. Lett. 2005, 7, 2413–2416. [Google Scholar] [CrossRef][Green Version]
- Tambar, U.K.; Kano, T.; Zepernick, A.J.F.; Stoltz, B.M. Convergent and Diastereoselective Synthesis of the Polycyclic Pyran Core of Saudin. J. Org. Chem. 2006, 71, 8357–8364. [Google Scholar] [CrossRef] [PubMed]
- Tambar, U.K.; Kano, T.; Zepernick, J.F.; Stoltz, B.M. The development and scope of a versatile tandem Stille-oxa-electrocyclization reaction. Tetrahedron Lett. 2007, 48, 345–350. [Google Scholar] [CrossRef]
- Parker, K.A.; Mindt, T.L. Convergent synthesis of 2H-chromenes—a formal [3+3] cycloaddition by a one-pot, three-step cascade. Tetrahedron 2011, 67, 9779–9786. [Google Scholar] [CrossRef][Green Version]
- Uyanik, M.; Nishioka, K.; Kondo, R.; Ishihara, K. Chemoselective oxidative generation of ortho-quinone methides and tandem transformations. Nat. Chem. 2020, 12, 353–362. [Google Scholar] [CrossRef]
- Iguchi, D.; Erra-Balsells, R.; Bonesi, S.M. Formation of 2,2-dimethylchroman-4-ones during the photoinduced rearrangement of some aryl 3-methyl-2-butenoate esters. A mechanistic insight. Tetrahedron 2016, 72, 1903–1910. [Google Scholar] [CrossRef]
- Cole, K.P.; Hsung, R.P. Intramolecular Formal oxa-[3 + 3] Cycloaddition Approach to the ABD System of Phomactin A. Org. Lett. 2003, 35, 4843–4846. [Google Scholar] [CrossRef] [PubMed]
- Olson, B.S.; Trauner, D. Concise Synthesis of (±)-Smenochromene D (= Likonide B). Synlett 2005, 2005, 700–702. [Google Scholar] [CrossRef]
- Hall, A.J.; Roche, S.P.; West, L.M. Synthesis of Briarane Diterpenoids: Biomimetic Transannular Oxa-6π electrocyclization Induced by a UVA/UVC Photoswitch. Org. Lett. 2017, 19, 576–579. [Google Scholar] [CrossRef] [PubMed]
- Kutateladze, A.G.; Krenske, E.H.; Williams, C.M. Reassignments and Corroborations of Oxo-Bridged Natural Products Di-rected by OSE and DU8+ NMR Computation. Angew. Chem. Int. Ed. 2019, 58, 7107–7112. [Google Scholar] [CrossRef] [PubMed]
- Boon, B.A.; Green, A.G.; Liu, P.; Houk, K.N.; Merlic, C.A. Using Ring Strain to Control 4π-Electrocyclization Reactions: Torquoselectivity in Ring Closing of Medium-Ring Dienes and Ring Opening of Bicyclic Cyclobutenes. J. Org. Chem. 2017, 82, 4613–4624. [Google Scholar] [CrossRef] [PubMed]
- Si, X.; Jia, Y.; Luan, X.; Yang, L.; Pei, Y.; Zhou, W. Insight into 6π Electrocyclic Reactions of 1,8-Dioxatetraene. Angew. Chem. Int. Ed. 2019, 58, 2660–2664. [Google Scholar] [CrossRef] [PubMed]












Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).