Abstract
Stabilized arylzinc iodides, synthesized by direct insertion of zinc into the corresponding halides, were used as nucleophiles into an acylative Negishi coupling reaction to synthesize chalcones. The reaction conditions were optimized to afford optimal results on a model reaction and then applied to synthesize nine compounds. Esters, chlorides, electron-rich, electron-poor and sterically hindered substrates are well tolerated and even heteroaryl derivatives can be synthesized.
1. Introduction
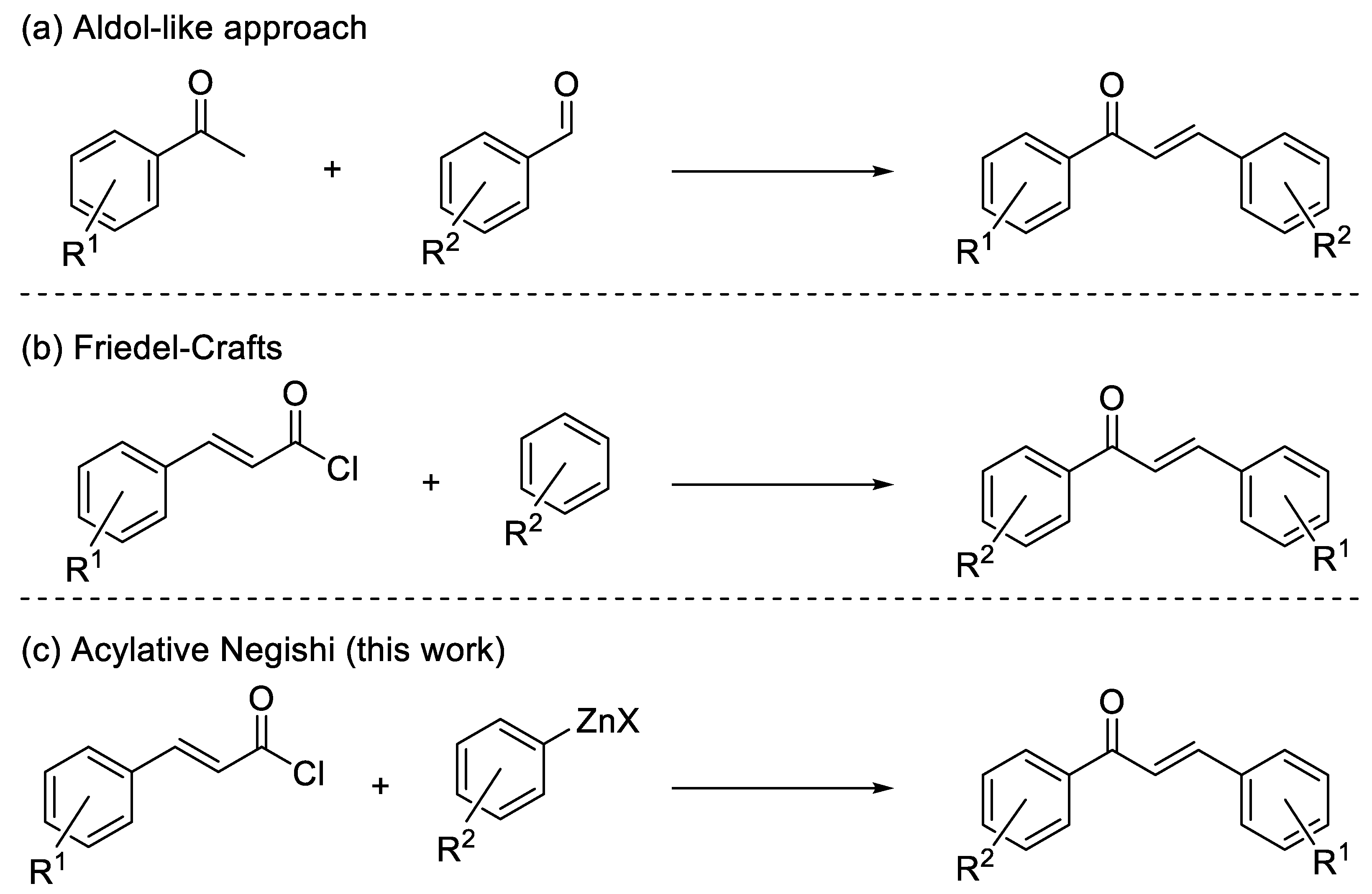
Chalcones, namely 1,3-diaryl-2-propen-1-ones, belong to the flavonoid family, being open-chain flavonoids where the two aromatic rings are conjugated to an α,β-unsaturated carbonyl system [1,2]. As with the majority of flavonoids, naturally occurring and synthetic chalcones exhibit different biological activities [2,3,4,5,6]. As a matter of fact, chalcone-containing plants, which possess beneficial biological effects, have been used for a long time in traditional medical practice [6]. However, isolation of chalcone derivatives from natural sources requires complicated procedures, so the development of efficient protocols for their synthesis has been pursued (Scheme 1) [2]. Most of the strategies to prepare chalcones make use of aldol-like reactions [2], by which the conjugated system can be built starting from an arylaldehyde and an arylmethylketone [2]. This approach usually makes use of strong bases and provides good results for simple substrates, but in the case of complex molecules, the result can be scarcely selective [2]; in addition, the synthesis of the required aldehydes and methylketones can be tricky [2]. Another approach is based on the use of the Friedel–Craft acylation, an economical option even if usually limited to electron-rich substrates [7]. Therefore, the search for alternative synthetic protocols overcoming these problems is intriguing. Among the possible alternatives, the Negishi acylative cross-coupling between acyl chlorides and organozinc halides, a well-known strategy to form ketones, attracted our attention [8]. Such a strategy can be interesting for the synthesis of chalcones, making use of cinnamoyl chlorides readily available from the corresponding carboxylic acids, which, in turn, can be synthesized in several ways, for instance by Knoevenagel–Doebner reaction. Curiously, there are no examples in the literature of such a reaction: only the synthesis of chalcone (1,3-Diphenylprop-2-en-1-one) through a Negishi coupling starting from phenylzinc chloride and a mixed anhydride of cinnamic acid was reported [9]. On the side of the nucleophile, the required arylzinc halides can be made through different paths. Among all other methods, the preparation of arylzinc is usually achieved through transmetalation, starting from a more reactive organometallic compound, or via direct insertion of zinc metal into the corresponding halide [10].
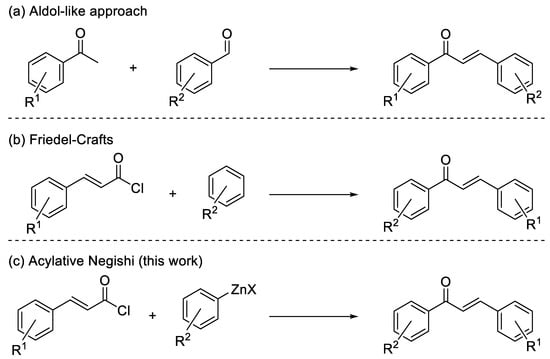
Scheme 1.
Synthesis of chalcones.
The latter strategy shows several advantages, because it is more selective, cheap, and green, as no other metal has to be used except zinc, and there is no need for other more reactive, and less selective, reactants [11,12,13]. However, the use of organozinc halides prepared by direct insertion, especially those obtained from iodides, is known to be problematic for acylative cross-couplings, because Zn species catalyze side reactions of the acyl chloride with the ethereal solvent (Scheme 2) [14].
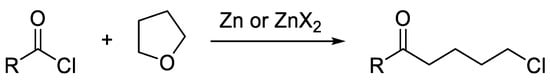
Scheme 2.
Side-reaction catalyzed by zinc and/or zinc salts.
Another issue concerns the presence of electron-rich aromatic rings in most of the natural and bioactive chalcones, because the preparation of electron-rich arylzinc reagents by direct insertion can be challenging to achieve in reasonable reaction times [11,12,13].
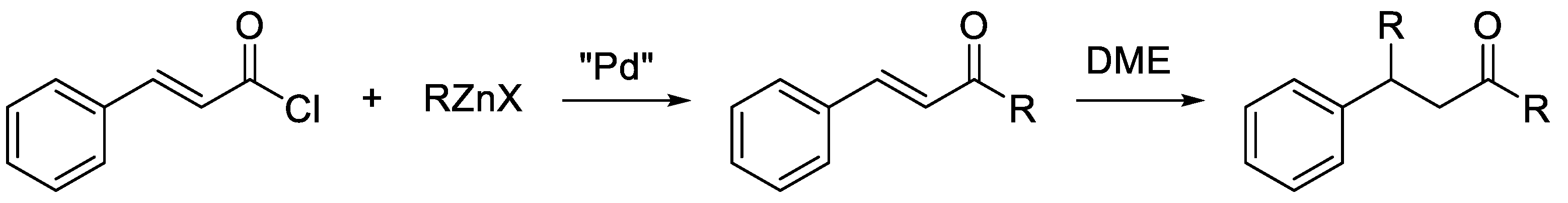
In addition, depending on the organozinc formulation, chalcones can be reactive towards the organozinc halide itself, affording the conjugated addition product of the nucleophile to the beta-carbon (Scheme 3) [15]. This aspect can complicate the tuning of the reaction conditions, as the side-reaction of the cross-coupling product with the organozinc halide must be avoided.
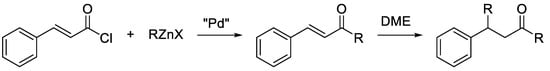
Scheme 3.
Possible side-reaction of chalcones with organozinc halides.
Problems related both to the preparation of electron-rich organozinc halides and side-reactions of the organometallic species can be overcome using a recently developed mild and efficient protocol for the preparation of organozinc iodides by silver catalyzed zinc insertion into aryl iodides in the presence of N,N,N,N-tetramethylethylenediamine (TMEDA), which allowed us to quickly obtain arylzinc iodides, also endowed with electron-rich substituent groups [13]. These organometallic reagents were successfully used in Negishi cross-coupling reactions and showed to be unreactive towards the conjugate addition [13,15].
These considerations, together with our interest in the synthesis of natural products [16,17,18], and our previous results in organozinc halides chemistry [13,15,18,19], prompted us to address our efforts in developing an affordable protocol for the synthesis of chalcones through acylative Negishi cross-coupling, using arylzinc halides prepared by direct insertion [13,15,16,17,18,19].
2. Results and Discussion
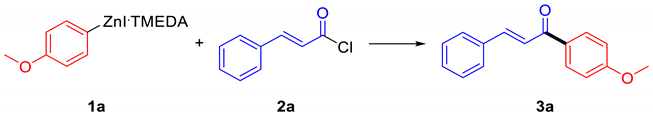
For the initial tuning of the reaction conditions, we chose to use our protocol for preparing the required organozinc halides, which proved to be affordable and fast for producing electron-rich arylzinc halides [13]. We selected the cross-coupling between 4-methoxyphenylzinc iodide (1a) and cinnamoyl chloride (2a) as the model reaction (Table 1). The first trial (Table 1, entry 1) was performed in tetrahydrofuran (THF) as the solvent, a common choice with this kind of organometallics, and we tried to use the inexpensive air-stable pre-catalyst PdCl2(PPh3)2; unfortunately, in these conditions, only a low 19% isolated yield was obtained, the more notable byproduct being the result of the reaction between the acyl chloride and the solvent (Scheme 2). So, we tried to change the reaction solvent into 1,2-dimethoxyethane (DME). DME is still an ethereal solvent, but it is not reported in the literature to give this kind of side reactions [14]. A great improvement with respect to the same reaction performed in THF was observed, the yield rising up to 46% (Table 1, entry 2).

Table 1.
Optimization of the model reaction.
Unfortunately, increasing the temperature from 50 °C to 70 °C did not improve the yield furtherly; on the contrary, we experienced lower yields working at higher temperatures, probably due to side-reactions, leading to a complex mixture of high molecular weight compounds (Table 1, entry 2 vs. entry 3). The use of diphenylphosphinoferrocene derivative Pd(dppf)Cl2 as the pre-catalyst, another air-stable compound with a bidentate phosphine, known to be a good choice in several cross-coupling reactions; temperatures at both 50 °C (Table 1, entry 4) and 70 °C (Table 1, entry 5) gave only a little improvement in isolated yields, not sufficient yet for synthetic purposes. Using PdCl2(PCy3)2, a pre-catalyst endowed with the more basic tricyclohexylphosphine ligand, afforded the cross-coupling product only in 22% yield; this result is probably attributable to a less efficient reductive elimination step in the catalytic cycle, due to the excessive basicity of the palladium ligated to the alkyl phosphine [20]. At this point, we moved our attention to Buchwald biarylic ligands SPhos and XPhos, which proved to give excellent results in other kind of Negishi cross-couplings [21,22], using palladium acetate as the source of palladium and a 1:1 stoichiometric ratio between the metal and the phosphines. While with SPhos (Table 1, entry 7), the yield was 45%, the result using XPhos (Table 1, entry 8) was even worse, as the product was obtained in only 25% yield. One of the possible problems, common to all the above-mentioned trials (Table 1, entry 1–8), was supposed to lie in the use of an oxidated source of palladium. Indeed, the catalytic cycle involves a palladium (0) catalyst, and it is generally assumed that all the pre-catalysts, in order to start their activity, must be in situ reduced by one of the other reagents [23]. Therefore, we tried to use a palladium (0) source, namely tris(dibenzylideneacetone)dipalladium (0), and SPhos as the ligand: to our delight, these conditions (Table 1, entry 9) resulted in a great improvement of the isolated yield that raised up to 90%.
Encouraged by this result, we tried another palladium (0) pre-catalyst, the more classical and less expensive palladium tetrakis triphenylphosphine, which provided slightly better results (Table 1, entry 10).
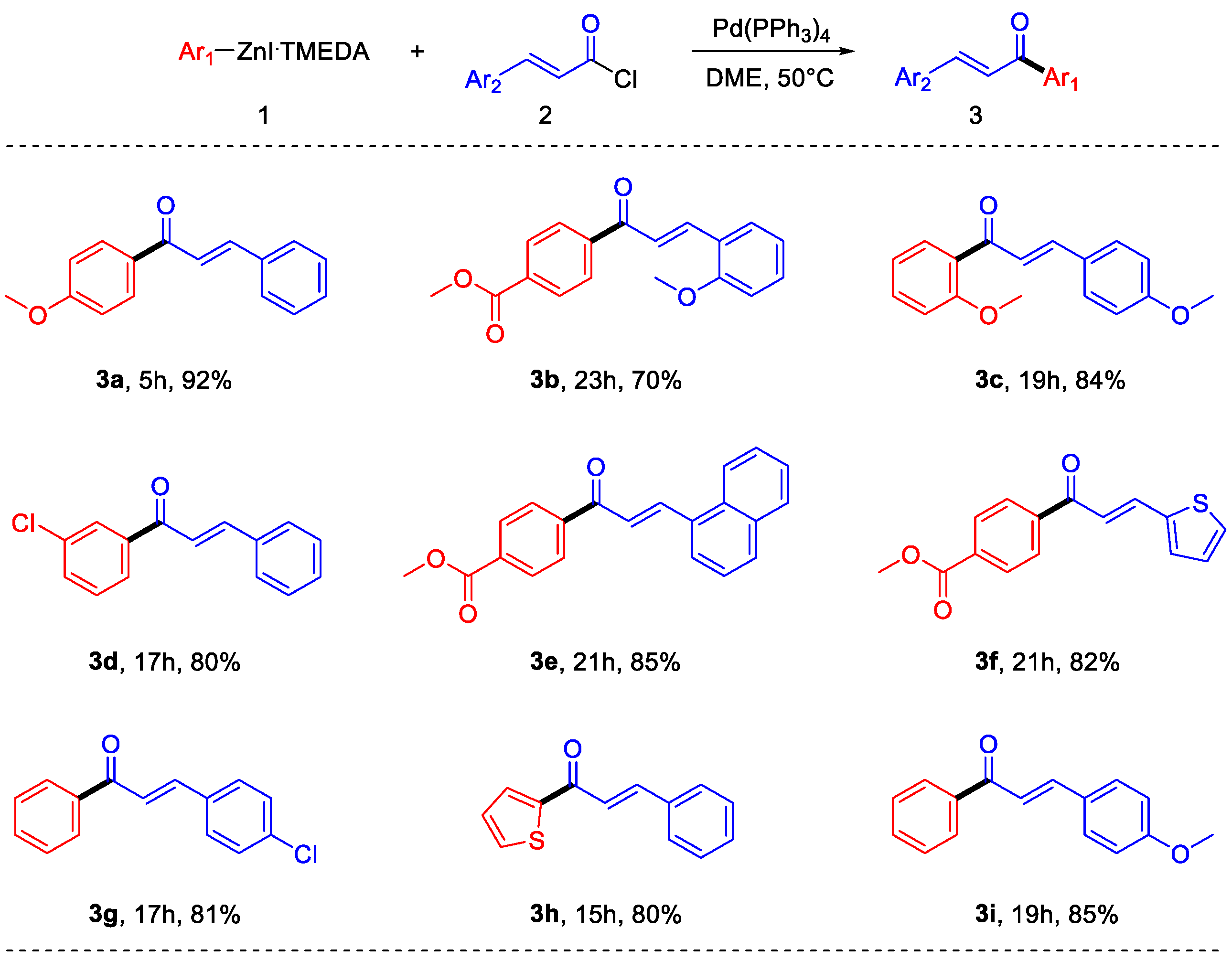
With the optimized reaction conditions, the applicability and robustness of the protocol was proved, using different organozinc iodides as well as different cinnamoyl chlorides. As shown in Scheme 4, several substituents on both the reaction partners are well-tolerated. Electron-rich, as well as electron-poor arylzinc halides can be employed and different substituted cinnamoyl chlorides have been used. Moreover, the reaction does not suffer steric hindrance on the nucleophile, and even heteroaryl derivatives can be synthesized. It is important to note that some of the obtained products (3b, 3e, 3f) have functional groups that are not compatible with polarized organometallic compounds such as Grignard or organolithium reagents; therefore, their synthesis using arylzinc halides prepared by transmetalation from these kinds of reactants is not feasible without the use of very low temperatures [24]. It is also important to note that the direct synthesis of electron-rich arylzinc halides, such as the ones required to synthesize 3a and 3c, requires very short times if compared with other literature methods [12].
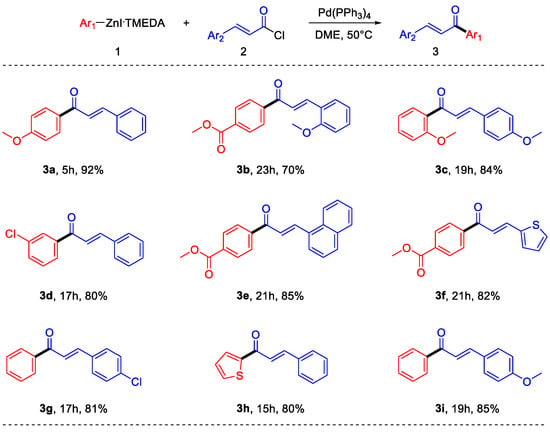
Scheme 4.
Scope of the reaction.
3. Conclusions
In conclusion, the synthesis of chalcones through a path involving an acylative Negishi coupling has been investigated and optimized. The key point of this strategy is the preparation of the organozinc by direct insertion which is reflected in a superior functional-group tolerance and an improved intrinsic greenness. Different functional groups as well as heteroaryl rings are well-tolerated. The effectiveness of the method has been proven by synthesizing nine substrates endowed with different synthetic patterns. The products were esters, chlorides, heteroaryl, electron-rich, electron-poor and sterically hindered compounds.
4. Experimental Section
4.1. General Information
Proton (1H NMR) and carbon (13C NMR) nuclear magnetic resonance spectra were recorded at 400 MHz and 100 MHz, or at 500 MHz and 125 MHz, respectively. The chemical shifts are given in parts per million (ppm) on the delta (δ) scale. GC-FID analyses were performed on GC instrument with a Split/Splitless injector and an FID detector. Analytical TLCs were performed on precoated silica gel ALUGRAM Xtra G/UV254 plates. Purifications were performed by flash chromatography on silica gel (40–63 µm). All reactions were performed in flame dried glassware under argon atmosphere. Ethereal solvents were dried twice over molecular sieves and distilled before the use. TMEDA was refluxed with CaH2 and distilled before the use. Thionyl chloride was distilled before the use. Zinc was flame dried under high vacuum before the use. Cinnamic acids were prepared according to reported procedures [25]. Acyl chlorides were prepared according to reported procedures [26]. All other solid reagents were dried under vacuum before the use.
4.2. General Procedure for the Synthesis of the Arylzinc Halides
In a typical procedure, zinc powder (490 mg, 7.5 mmol) was flame dried under vacuum in a round-bottomed flask equipped with a reflux condenser and a magnetic stirrer; silver acetate (8.4 mg, 0.05 mmol) was then added under argon and the mixture dried again under vacuum; the flask was refilled with argon and anhydrous DME (5 mL) and chlorotrimethylsilane (15 µL, 0.075 mmol) were added. The mixture was stirred and heated with a hot-gun for 5 min. After cooling, anhydrous TMEDA (750 µL, 5 mmol) and the aromatic iodide (5 mmol) were added.
The mixture was heated at the reflux and stirred with TLC check on hydrolyzed aliquot until full conversion.
4.3. General Procedure for the Synthesis of Chalcones
In a typical procedure, Pd(PPh3)4 (38 mg, 0.033 mmol, 1%) was dried under vacuum in a round-bottomed flask equipped with magnetic stirrer; the flask was refilled with argon and an acyl halide (3.3 mmol) solution in anhydrous DME (3.3 mL) and the organozinc halide solution [13] (5.8 mL, 5 mmol) were added. The mixture was heated at 50 °C and stirred for the time indicated for each trial (Scheme 2) before being quenched with NH4Cl and extracted with EtOAc (3 × 10 mL). Flash chromatography purification on silica gel with hexane/ethyl acetate mixtures afforded the pure compounds.
4.3.1. (E)-1-(4-Methoxyphenyl)-3-phenylprop-2-en-1-one (3a)
It was prepared according to the general procedure from cinnamoyl chloride and 4-(methoxy)phenylzinc iodide: 723 mg (3.04 mmol, 92%) of white solid were obtained after flash chromatography (SiO2, Hex:EtOAc 9:1) [27] 1H NMR (500 MHz, CDCl3) δ 8.08–8.01 (m, 2H), 7.80 (d, J = 15.6 Hz, 1H), 7.68–7.60 (m, 2H), 7.55 (d, J = 15.7 Hz, 1H), 7.45–7.36 (m, 3H), 7.01–6.93 (m, 2H), 3.86 (s, 3H). 13C NMR (125 MHz, CDCl3) δ 188.8, 163.5, 144.0, 135.2, 131.2, 130.9, 130.5, 129.0, 128.5, 121.9, 114.0, 55.6.
4.3.2. Methyl (E)-4-(3-(2-Methoxyphenyl)acryloyl)benzoate (3b)
It was prepared according to the general procedure from (E)-3-(2-methoxyphenyl)acryloyl chloride and (4-(methoxycarbonyl)phenyl)zinc iodide: 684 mg (2.31 mmol, 70%) of pale yellow solid were obtained after flash chromatography (SiO2, Hex:EtOAc 8:2) [28]. 1H NMR (400 MHz, CDCl3) δ 8.22–7.98 (m, 5H), 7.69–7.54 (m, 2H), 7.45–7.36 (m, 1H), 7.04–6.90 (m, 2H), 3.96 (s, 3H), 3.92 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 190.8, 166.4, 158.9, 142.1, 141.4, 133.3, 132.1, 129.8, 129.4, 128.4, 123.6, 122.7, 120.8, 111.3, 55.6, 52.4.
4.3.3. (E)-1-(2-Methoxyphenyl)-3-(4-methoxyphenyl)prop-2-en-1-one (3c)
It was prepared according to the general procedure from (E)-3-(4-methoxyphenyl)acryloyl chloride and 2-(methoxy)phenylzinc iodide: 744 mg (2.77 mmol, 84%) of pale yellow oil were obtained after flash chromatography (SiO2, Hex:EtOAc 8:2) [29]. 1H NMR (400 MHz, CDCl3) δ: 7.58-7.52 (m, 3H), 7.45 (t, 1H, J = 7.8 Hz), 7.04-6.97 (m, 2H), 6.90 (d, 2H, J = 8.4 Hz), 3.88 (s, 3H), 3.83 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 193.2, 161.6, 158.0, 143.4, 132.6, 130.2, 130.1, 129.7, 127.9, 125.0, 120.7, 114.4, 111.7, 55.8, 55.4.
4.3.4. (E)-1-(3-Chlorophenyl)-3-phenylprop-2-en-1-one (3d)
It was prepared according to the general procedure from cinnamoyl chloride and 3-chlorophenylzinc iodide: 641 mg (2.64 mmol, 80%) of pale yellow solid were obtained after flash chromatography (SiO2, Hex) [30]. 1H NMR (400 MHz, CDCl3) δ 7.99 (t, J = 1.9 Hz, 1H), 7.89 (dt, J = 7.7, 1.4 Hz, 1H), 7.83 (d, J = 15.6 Hz, 1H), 7.69–7.61 (m, 2H), 7.56 (ddd, J = 8.0, 2.2, 1.1 Hz, 1H), 7.50–7.39 (m, 5H). 13C NMR (100 MHz, CDCl3) δ 189.0, 145.7, 139.8, 134.9, 134.6, 132.7, 130.9, 130.0, 129.1, 128.6, 128.6, 126.6, 121.4.
4.3.5. Methyl (E)-4-(3-(Naphthalen-1-yl)acryloyl)benzoate (3e)
It was prepared according to the general procedure from (E)-3-(naphthalen-1-yl)acryloyl chloride and (4-(methoxycarbonyl)phenyl)zinc iodide: 887 mg (2.81 mmol, 85%) of pale yellow solid were obtained after flash chromatography (SiO2, Hex:EtOAc 8:2) [28]. 1H NMR (400 MHz, CDCl3) δ 8.69 (d, J = 15.5 Hz, 1H), 8.24 (dd, J = 8.4, 1.2 Hz, 1H), 8.21–8.09 (m, 4H), 7.98–7.86 (m, 3H), 7.66–7.49 (m, 4H), 3.97 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 189.9, 166.4, 142.7, 141.7, 133.9, 133.7, 132.1, 131.9, 131.3, 130.0, 128.9, 128.6, 127.2, 126.5, 125.5, 125.3, 124.4, 123.5, 52.6.
4.3.6. Methyl (E)-4-(3-(Thiophen-2-yl)acryloyl)benzoate (3f)
It was prepared according to the general procedure from (E)-3-(thiophen-2-yl)acryloyl chloride and (4-(methoxycarbonyl)phenyl)zinc iodide: 737 mg (2.71 mmol, 82%) of pale yellow solid were obtained after flash chromatography (SiO2, Hex:EtOAc 8:2) [31]. 1H NMR (400 MHz, CDCl3) δ 8.18–8.01 (m, 4H), 7.96 (dt, J = 15.3, 0.8 Hz, 1H), 7.45 (dt, J = 5.0, 1.0 Hz, 1H), 7.40–7.36 (m, 1H), 7.30 (d, J = 15.3 Hz, 1H), 7.10 (dd, J = 5.1, 3.6 Hz, 1H), 3.96 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 189.5, 166.4, 141.7, 140.2, 138.2, 133.6, 132.7, 129.9, 129.4, 128.6, 128.4, 120.5, 52.6.
4.3.7. (E)-3-(4-Chlorophenyl)-1-phenylprop-2-en-1-one (3g)
It was prepared according to the general procedure from (E)-3-(4-chloro)acryloyl chloride and phenylzinc iodide: 649 mg (2.67 mmol, 81%) of pale yellow oil were obtained after flash chromatography (SiO2, Hex:EtOAc 8:2) [32]. 1H NMR (500 MHz, CDCl3) δ 8.06–7.98 (m, 2H), 7.76 (d, J = 15.7 Hz, 1H), 7.63–7.55 (m, 3H), 7.55–7.47 (m, 3H), 7.43–7.36 (m, 2H). 13C NMR (125 MHz, CDCl3) δ 190.3, 143.4, 138.1, 136.5, 133.5, 133.1, 129.7, 129.4, 128.8, 128.6, 122.5.
4.3.8. (E)-3-Phenyl-1-(thiophen-2-yl)prop-2-en-1-one (3h)
It was prepared according to the general procedure from cinnamoyl chloride and thiophen-2-ylzinc iodide: 566 mg (2.64 mmol, 80%) of pale yellow solid were obtained after flash chromatography (SiO2, Hex). 1H NMR (400 MHz, CDCl3) δ 7.88–7.80 (m, 2H), 7.68–7.59 (m, 3H), 7.45–7.37 (m, 4H), 7.16 (dd, J = 4.9, 3.8 Hz, 1H) [7]. 13C NMR (100 MHz, CDCl3) δ 182.1, 145.7, 144.2, 134.8, 134.1, 132.0, 130.7, 129.1, 128.6, 128.4, 121.7.
4.3.9. (E)-3-(4-Methoxyphenyl)-1-phenylprop-2-en-1-one (3i)
It was prepared according to the general procedure from (E)-3-(4-methoxyphenyl)acryloyl chloride and phenylzinc iodide: 668 mg (2.81 mmol, 85%) of pale yellow oil were obtained after flash chromatography (SiO2, Hex:EtOAc 8:2) [33]. 1H NMR (400 MHz, CDCl3) δ 8.06–7.95 (m, 2H), 7.79 (d, J = 15.7 Hz, 1H), 7.63–7.54 (m, 3H), 7.53–7.46 (m, 2H), 7.42 (d, J = 15.6 Hz, 1H), 6.97–6.89 (m, 2H), 3.84 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 190.7, 161.8, 144.8, 138.6, 132.7, 130.4, 128.7, 128.5, 127.7, 119.9, 114.5, 55.5.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/org3020006/s1: 1H-NMR and 13C-NMR spectra of synthetized compounds.
Author Contributions
Conceptualization, G.C.; methodology, G.C.; investigation, G.C. and M.P.; writing—original draft preparation, G.C.; writing—review and editing, A.I., G.A. and G.C.; supervision, A.I. and G.C.; project administration, A.I. and G.A.; funding acquisition, A.I. and G.A. All authors have read and agreed to the published version of the manuscript.
Funding
This research was supported by the University of Pisa.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented in this study are available in the article and supporting information.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Bukhari, S.N.A.; Jasamai, M.; Jantan, I.; Ahmad, W. Review of Methods and Various Catalyst Used for Chalcones Synthesis. Mini-Rev. Org. Chem. 2013, 10, 73–83. [Google Scholar] [CrossRef]
- Suwito, H.; Jumina, J.; Mustofa, M.; Kristanti, A.N.; Tri Puspaningsih, N.N. Chalcones: Synthesis, Structure Diversity and Pharmacological Aspects. J. Chem. Pharm. Res. 2014, 6, 1076. [Google Scholar] [CrossRef]
- Chavan, B.B.; Gadekar, A.S.; Mehta, P.P.; Vawhal, P.K.; Kolsure, A.K.; Chabukswar, A.R. Synthesis and Medicinal Significance of Chalcones—A Review. Asian J. Pharm. Biol. Res. 2016, 6, 1–7. [Google Scholar]
- Yazdan, S.K.; Sagar, D.V.; Shaik, A.B. Chemical and Biological Potentials of Chalcones: A Review. Org. Med. Chem. 2015, 1, 20–28. [Google Scholar] [CrossRef]
- Singh, P.; Anand, A.; Kumar, V. Recent Developments in Biological Activities of Chalcones: A Mini Review. Eur. J. Med. Chem. 2014, 85, 758–777. [Google Scholar] [CrossRef]
- Rozmer, Z.; Perjési, P. Naturally Occurring Chalcones and Their Biological Activities. Phytochem. Rev. 2016, 15, 87–120. [Google Scholar] [CrossRef]
- More, P.E.; Bandgar, B.P.; Kamble, V.T. Zinc Oxide as a Regioselective and Heterogeneous Catalyst for the Synthesis of Chalcones at Room Temperature. Catal. Commun. 2012, 27, 30–32. [Google Scholar] [CrossRef]
- Negishi, E.; Bagheri, V.; Chatterjee, S.; Luo, F.-T.; Miller, J.A.; Stoll, A.T. Palladium-Catalyzed Acylation of Organozincs and Other Organometallics as a Convenient Route to Ketones. Tetrahedron Lett. 1983, 24, 5181–5184. [Google Scholar] [CrossRef]
- Wang, D.; Zhang, Z. Palladium-Catalyzed Cross-Coupling Reactions of Carboxylic Anhydrides with Organozinc Reagents. Org. Lett. 2003, 5, 4645–4648. [Google Scholar] [CrossRef]
- Paul Knochel, P.J. Organozinc Reagents—A Practical Approach; Oxford University Press: Oxford, UK, 1999; ISBN 9780198501213. [Google Scholar]
- Knochel, P.; Millot, N.; Rodriguez, A.L.; Tucker, C.E. Preparation and Applications of Functionalized Organozinc Compounds. In Organic Reactions; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2001; pp. 417–759. ISBN 9783527615179. [Google Scholar]
- Krasovskiy, A.; Malakhov, V.; Gavryushin, A.; Knochel, P. Efficient Synthesis of Functionalized Organozinc Compounds by the Direct Insertion of Zinc into Organic Iodides and Bromides. Angew. Chem. Int. Ed. 2006, 45, 6040–6044. [Google Scholar] [CrossRef]
- Casotti, G.; Iuliano, A.; Carpita, A. Arylzinc Halides by Silver Catalyzed Zinc Insertion into Aryl Iodides. Eur. J. Org. Chem. 2019, 2019, 1021–1026. [Google Scholar] [CrossRef]
- Bhar, S.; Ranu, B.C. Zinc-Promoted Selective Cleavage of Ethers in Presence of Acyl Chloride. J. Org. Chem. 1995, 60, 745–747. [Google Scholar] [CrossRef]
- Casotti, G.; Ciancaleoni, G.; Lipparini, F.; Nieri, C.; Iuliano, A. Uncatalyzed Conjugate Addition of Organozinc Halides to Enones in DME: A Combined Experimental/Computational Study on the Role of the Solvent and the Reaction Mechanism. Chem. Sci. 2020, 11, 257–263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laudadio, G.; Fusini, G.; Casotti, G.; Evangelisti, C.; Angelici, G.; Carpita, A. Synthesis of Pterostilbene through Supported-Catalyst Promoted Mizoroki-Heck Reaction, and Its Transposition in Continuous Flow Reactor. J. Flow Chem. 2019, 9, 133–143. [Google Scholar] [CrossRef]
- Casotti, G.; Fusini, G.; Ferreri, M.; Pardini, L.F.; Evangelisti, C.; Angelici, G.; Carpita, A. Total Synthesis of Asparenydiol by Two Sonogashira Cross-Coupling Reactions Promoted by Supported Pd and Cu Catalysts. Synthesis 2020, 52, 1795–1803. [Google Scholar] [CrossRef] [Green Version]
- Fusini, G.; Barsanti, D.; Angelici, G.; Casotti, G.; Canale, A.; Benelli, G.; Lucchi, A.; Carpita, A. Identification and Synthesis of New Sex-Specific Components of Olive Fruit Fly (Bactrocera Oleae) Female Rectal Gland, through Original Negishi Reactions on Supported Catalysts. Tetrahedron 2018, 74, 4381–4389. [Google Scholar] [CrossRef]
- Casotti, G.; Rositano, V.; Iuliano, A. Enantioselective Conjugate Addition of Stabilized Arylzinc Iodide to Enones: An Improved Protocol of the Hayashi Reaction. Adv. Synth. Catal. 2021, 363, 1126–1131. [Google Scholar] [CrossRef]
- Culkin, D.A.; Hartwig, J.F. Carbon−Carbon Bond-Forming Reductive Elimination from Arylpalladium Complexes Containing Functionalized Alkyl Groups. Influence of Ligand Steric and Electronic Properties on Structure, Stability, and Reactivity. Organometallics 2004, 23, 3398–3416. [Google Scholar] [CrossRef]
- Han, C.; Buchwald, S.L. Negishi Coupling of Secondary Alkylzinc Halides with Aryl Bromides and Chlorides. J. Am. Chem Soc. 2009, 131, 7532–7533. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Oldenhuis, N.J.; Buchwald, S.L. Mild and General Conditions for Negishi Cross-Coupling Enabled by the Use of Palladacycle Precatalysts. Angew. Chem. Int. Ed. 2013, 52, 615–619. [Google Scholar] [CrossRef] [Green Version]
- D’Alterio, M.C.; Casals-Cruañas, È.; Tzouras, N.V.; Talarico, G.; Nolan, S.P.; Poater, A. Mechanistic Aspects of the Palladium-Catalyzed Suzuki-Miyaura Cross-Coupling Reaction. Chem. A Eur. J. 2021, 27, 13481–13493. [Google Scholar] [CrossRef] [PubMed]
- Tucker, C.E.; Majid, T.N.; Knochel, P. Preparation of Highly Functionalized Magnesium, Zinc, and Copper Aryl and Alkenyl Organometallics via the Corresponding Organolithiums. J. Am. Chem. Soc. 1992, 114, 3983–3985. [Google Scholar] [CrossRef]
- Qian, Y.; Zhang, H.-J.; Zhang, H.; Xu, C.; Zhao, J.; Zhu, H.-L. Synthesis, Molecular Modeling, and Biological Evaluation of Cinnamic Acid Metronidazole Ester Derivatives as Novel Anticancer Agents. Bioorganic Med. Chem. 2010, 18, 4991–4996. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Yan, W.; Zhang, C.; Zhang, Y.; Liang, M.; Chu, F.; Gong, Y.; Xu, B.; Wang, P.; Lei, H. New Synthesis Method for Sultone Derivatives: Synthesis, Crystal Structure and Biological Evaluation of S-CA. Molecules 2015, 20, 4307–4318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, X.-F.; Neumann, H.; Spannenberg, A.; Schulz, T.; Jiao, H.; Beller, M. Development of a General Palladium-Catalyzed Carbonylative Heck Reaction of Aryl Halides. J. Am. Chem. Soc. 2010, 132, 14596–14602. [Google Scholar] [CrossRef] [PubMed]
- Tripathi, S.; Kapoor, R.; Yadav, L.D.S. Visible Light Activated Radical Denitrative Benzoylation of β -Nitrostyrenes: A Photocatalytic Approach to Chalcones. Adv. Synth. Catal. 2018, 360, 1407–1413. [Google Scholar] [CrossRef]
- Stevenson, S.M.; Higgins, R.F.; Shores, M.P.; Ferreira, E.M. Chromium Photocatalysis: Accessing Structural Complements to Diels–Alder Adducts with Electron-Deficient Dienophiles. Chem. Sci. 2017, 8, 654–660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shrestha, A.; Shrestha, A.; Park, P.; Lee, E. Hydroxyl- and Halogen-containing Chalcones for the Inhibition of LPS-stimulated ROS Production in RAW 264.7 Macrophages: Design, Synthesis and Structure–Activity Relationship Study. Bull. Korean Chem. Soc. 2019, 40, 729–734. [Google Scholar] [CrossRef]
- Mekky, A.E.M.; Sanad, S.M.H.; Said, A.Y.; Elneairy, M.A.A. Synthesis, Cytotoxicity, in-Vitro Antibacterial Screening and In-Silico Study of Novel Thieno[2,3-b]Pyridines as Potential Pim-1 Inhibitors. Synth. Commun. 2020, 50, 2376–2389. [Google Scholar] [CrossRef]
- Schmink, J.R.; Holcomb, J.L.; Leadbeater, N.E. Testing the Validity of Microwave-Interfaced, in Situ Raman Spectroscopy as a Tool for Kinetic Studies. Org. Lett. 2009, 11, 365–368. [Google Scholar] [CrossRef]
- Romanelli, G.; Pasquale, G.; Sathicq, Á.; Thomas, H.; Autino, J.; Vázquez, P. Synthesis of Chalcones Catalyzed by Aminopropylated Silica Sol–Gel under Solvent-Free Conditions. J. Mol. Catal. A Chem. 2011, 340, 24–32. [Google Scholar] [CrossRef]
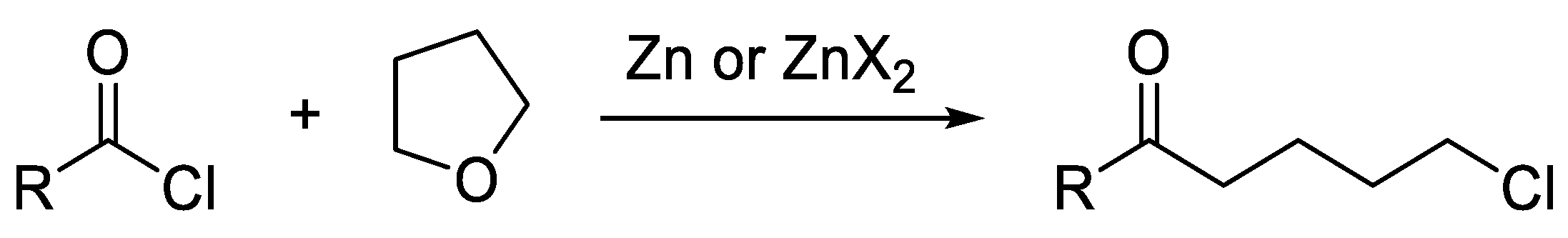
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).