Abstract
DFT calculations on the photoisomerization of 2,6-dimethylpyrazine allowed us to confirm the role of benzvalene isomers in the isomerization of hexatomic heterocyclic compounds. 2,6-Dimethylpyrazine in the excited singlet states can be converted into the corresponding Dewar isomers. If the S2 state is populated, two Dewar isomers can be obtained, while the S1 state allows the formation of only one of the possible Dewar isomers. Both Dewar isomers can be converted into the benzvalene isomer, that is, the precursor of 4,5-dimethylpyrimidine, the reaction product. In fact, the benzvalene isomer can be obtained from the Dewar isomers in processes that occur without an activation energy, and it is the more stable benzvalene isomers that can be obtained from the Dewar isomers. CASSCF study indicates the presence of a conical intersection allowing the direct formation of the benzvalene isomer.
1. Introduction
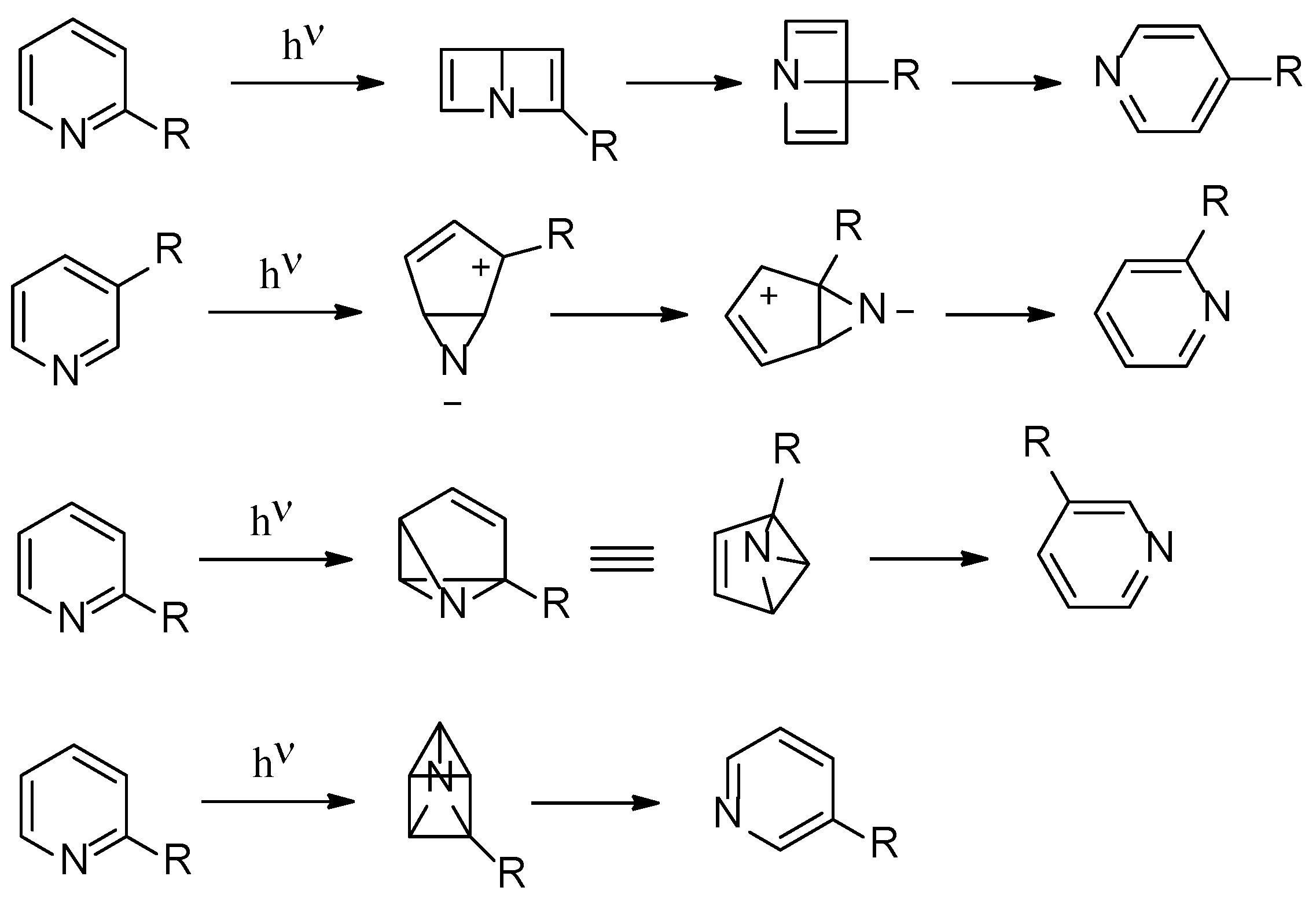
The photoisomerization of hexatomic heterocyclic compounds is an important research field, able to give, in some cases, noticeable synthetic procedures [1,2,3,4]. In order to explain the observed reactivity, several mechanistic hypotheses have been formulated. In this way, Dewar aromatic compounds, azaprefulvene intermediates, benzvalene intermediates, aziridines, and azaprismane intermediates, have been postulated in the reaction of this type of compounds. Figure 1 collects some of these mechanistic hypotheses.
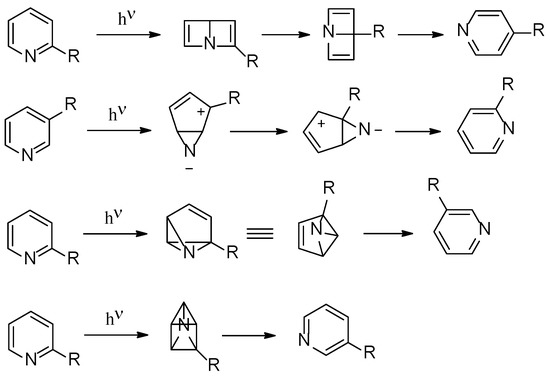
Figure 1.
Proposed photoisomerization routes for hexatomic heterocycles.
In this study, we want to present the results of DFT and CASSCF study on the photochemical isomerization of 2,6-dimethylpyrazine.
2. Materials and Methods
Gaussian09 has been used for the discussions about the computed geometries [5]. All the computations were based on the Density Functional Theory (DFT) [6] and Time-Dependent DFT (TD-DFT) [7,8] by using the B3LYP hybrid xc functional [9]. Geometry optimizations and TD-DFT results from the Gaussian09 program have been obtained at the B3LYP/6-311G+(d,p) level of approximation. Geometry optimizations were performed with default settings on geometry convergence (gradients and displacements), integration grid and electronic density (SCF) convergence. Redundant coordinates were used for the geometry optimization as produced by the Gaussian09 program. Analytical evaluation of the energy second derivative matrix w.r.t. Cartesian coordinates (Hessian matrix) at the B3LYP/6-311G+(d,p) level of approximation confirmed the nature of minima on the energy surface points associated with the optimized structures. The transition states were calculated in S0 state.
Some computations were based on the Complete Active Space Multiconfiguration Self-Consistent Field (CASSCF) [10,11,12,13,14,15,16,17,18]. Geometry optimizations from the Gaussian09 program have been obtained at the CASSCF(6,6)/6-31G(d,p) level of approximation. Geometry optimizations were performed with default settings on geometry convergence (gradients and displacements), integration grid and electronic density (SCF) convergence.
3. Results and Discussion
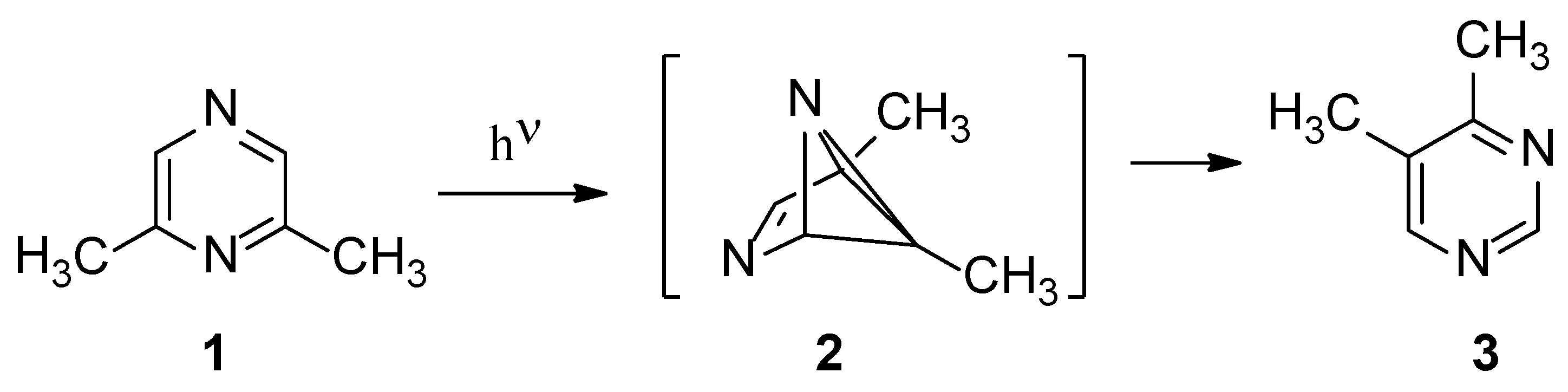
The vapor phase isomerization of 2,6-dimethylpyrazine (1) has been described several years ago, showing that the only detectable product was 4,5-dimethylpyrimidine (3) (Scheme 1) [19]. To explain the observed behavior, a benzvalene intermediate had been postulated (Scheme 1). In fact, the possible isomerization of the corresponding Dewar isomers cannot be used to justify the observed reaction product (Scheme 2). Some years ago, a study appeared where, on the basis of a CASSCF study, the direct conversion of 1 into 3 without the presence of any intermediate, has been proposed [20,21].
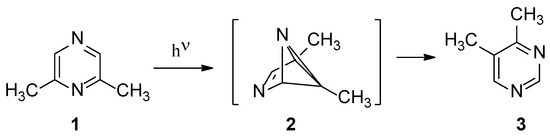
Scheme 1.
Photoisomerization of 2,6-dimethylpyrazine.
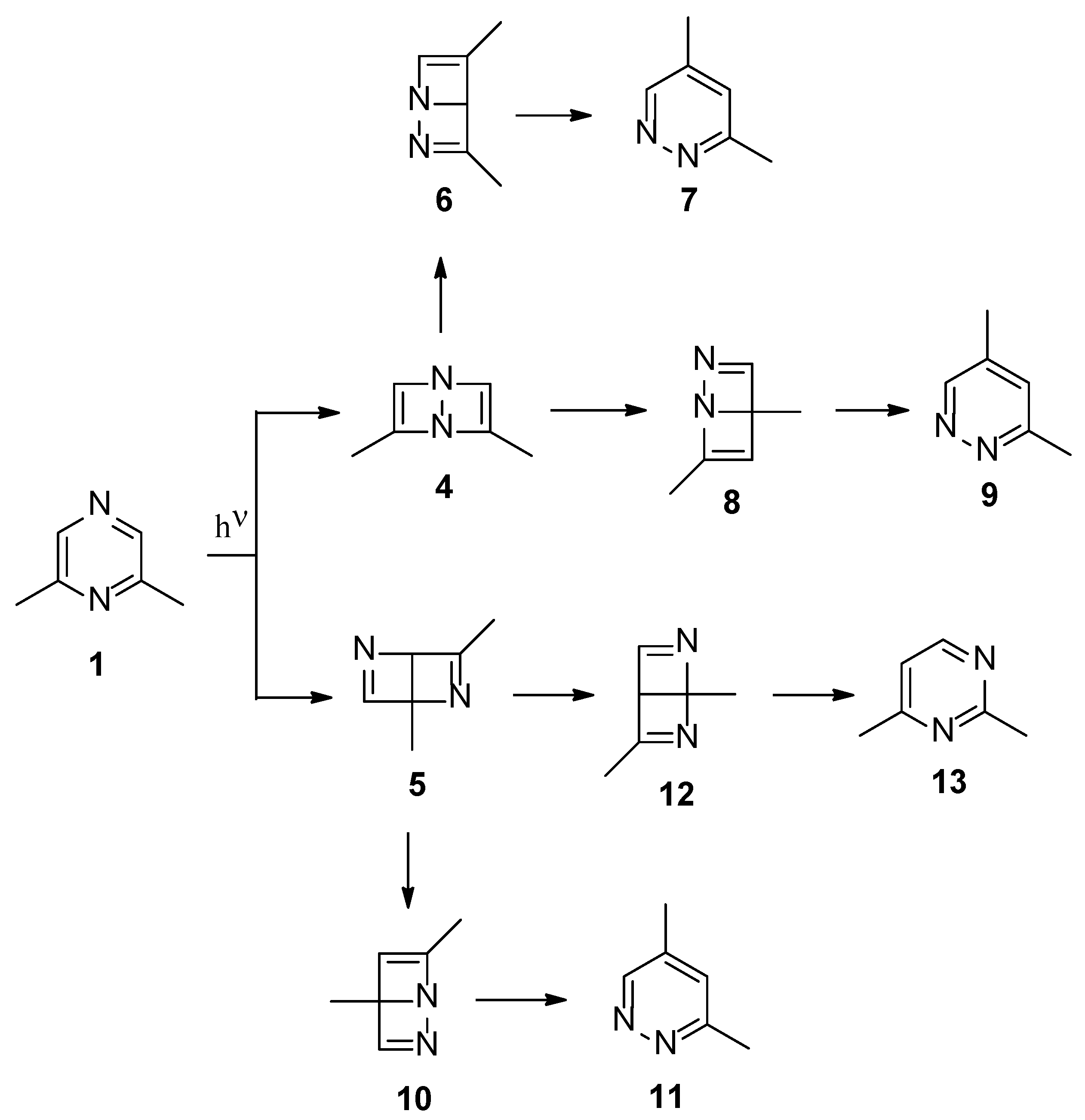
Scheme 2.
Possible isomerization reactions of the Dewar 2,6-dimethylpyrazine isomers.
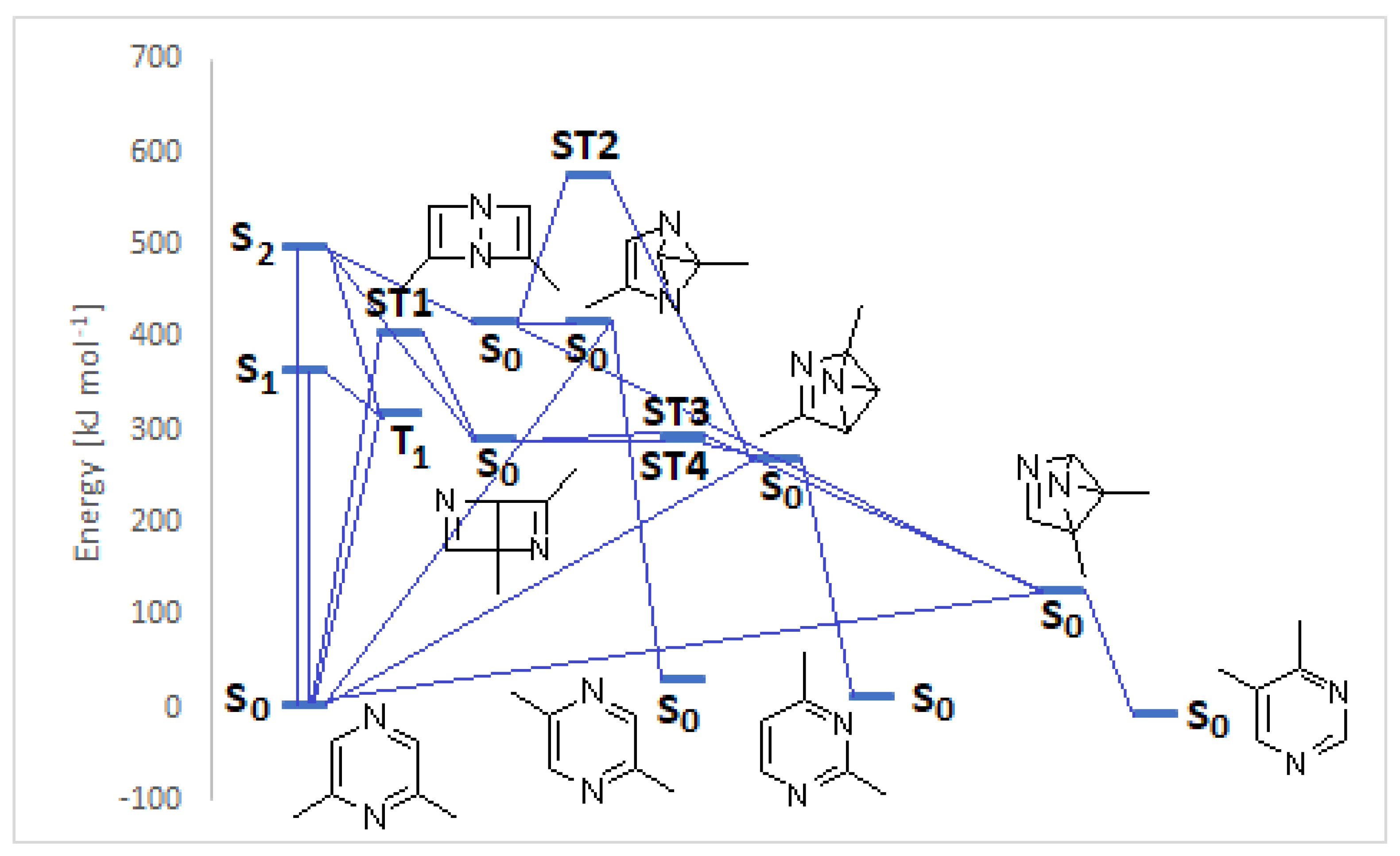
We performed both DFT and CASSCF calculations on the above-described reaction, in order to explain the observed reactivity (Table 1). 2,6-Dimethylpyrazine shows in methanol UV absorptions at λ = 208 (592 kJ mol−1) and 277 nm (432 kJ mol−1) [22]. TD-DFT calculations at B3LYP/6-31G+(d,p) level of theory on Gaussian 09 gave an absorption in the gas phase at λ = 242 nm (495 kJ mol−1), due to a NHOMO-LUMO π→π* transition, and a n→π* transition (HOMO-LUMO transition) at 329 nm (363 kJ mol−1). Calculations at CASSCF(6,6)/6-31G(d,p) level of theory on Gaussian 09 gave an absorption in the gas phase at λ = 183 nm (654 kJ mol−1), and at 217 nm (553 kJ mol−1). The possible evolution of the singlet excited states calculated at DFT level of theory has been described in Figure 2.

Table 1.
Total energy of the optimized structures.
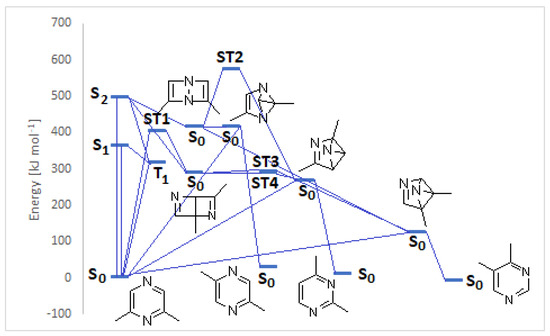
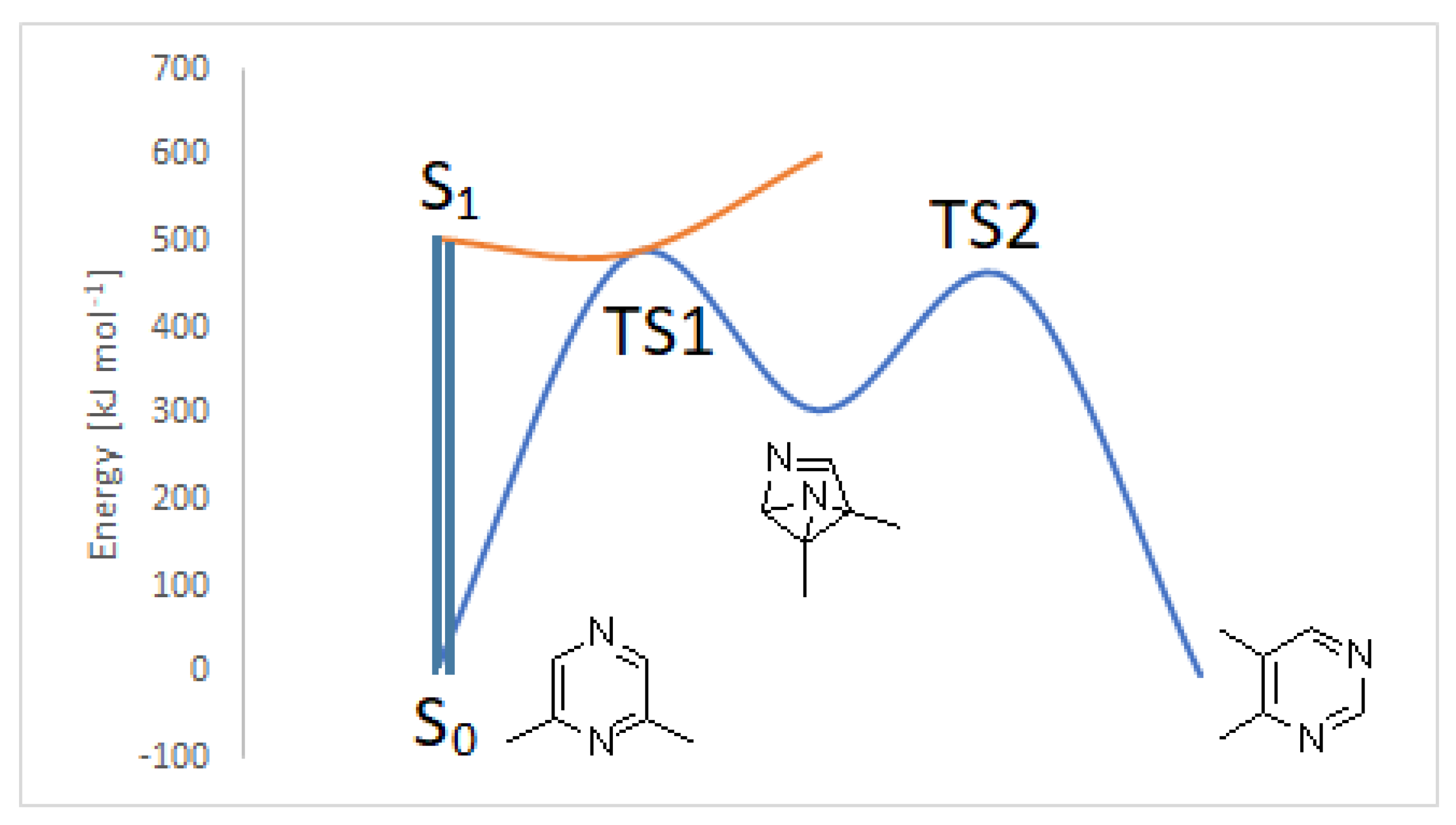
Figure 2.
Photochemical isomerization of 2,6-dimethylpyrazine.
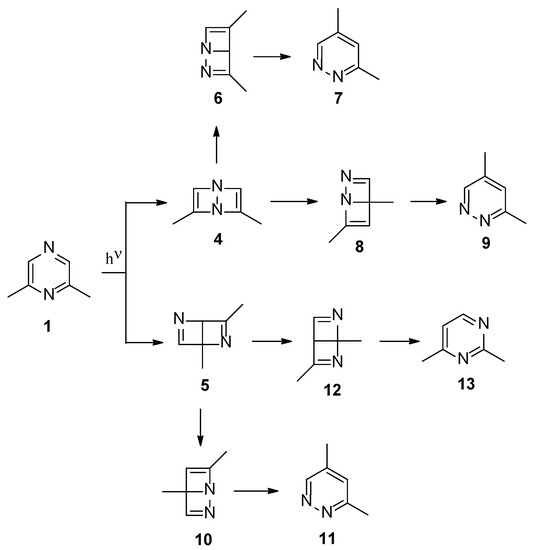
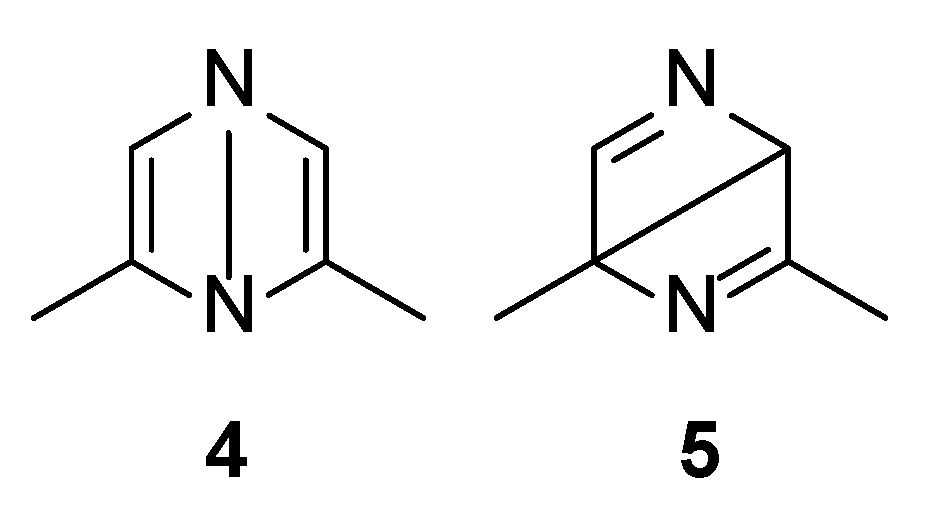
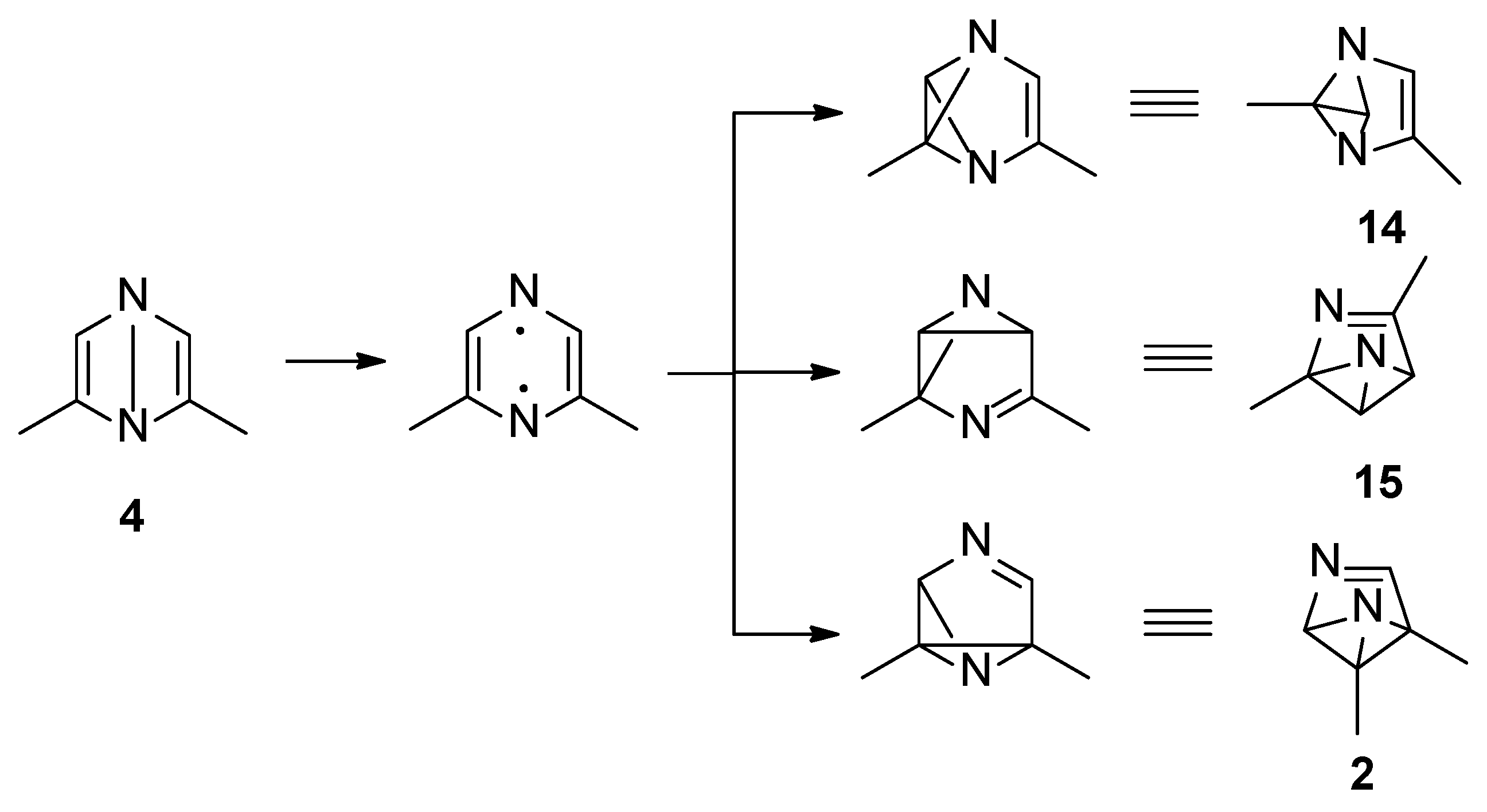
Both S1 and S2 states can evolve to give the corresponding triplet state; however, that does not play an important role in the reactivity of the pyrazine derivative. The S2 state can be converted into the corresponding Dewar isomer 4 and 5 (Figure 3).
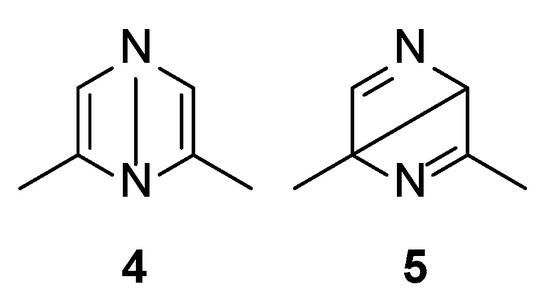
Figure 3.
Dewar isomers in the photoisomerization of 2,6-dimethylpyrazine.
The Dewar isomer 4 can be interconverted into the starting material in a step without a transition state, showing that this isomer has a very short lifetime. On the contrary, the conversion of the Dewar isomer 5, which can be obtained from S2 but also from S1 excited singlet state, into 2,6-dimethylpyrazine necessitates overcoming a transition state with ΔE = 105 kJ mol−1 (ST1, Figure 2).
The Dewar isomer 4 could be converted in a step without a transition state into the benzvalene intermediate 14 that showed the same energy as 4 (Figure 2 and Scheme 3). Furthermore, the Dewar isomer 4 can be converted into the benzvalene isomer 15 (Figure 2 and Scheme 3), but this step showed a transition state of 159 kJ mol−1 (ST2, Figure 2). Finally, 4 can be converted, without a transition state into the benzvalene isomer 2 (Figure 2 and Scheme 3). Considering the energy level of all the possible benzvalene isomers, the formation of 2 is clearly favored.
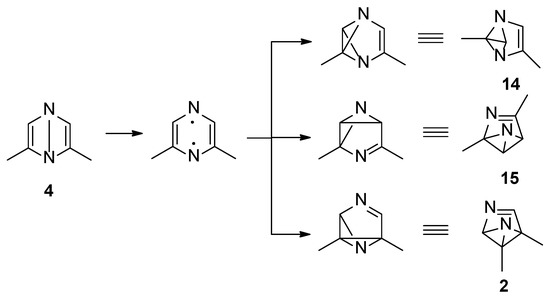
Scheme 3.
Benzvalene isomers from 4.
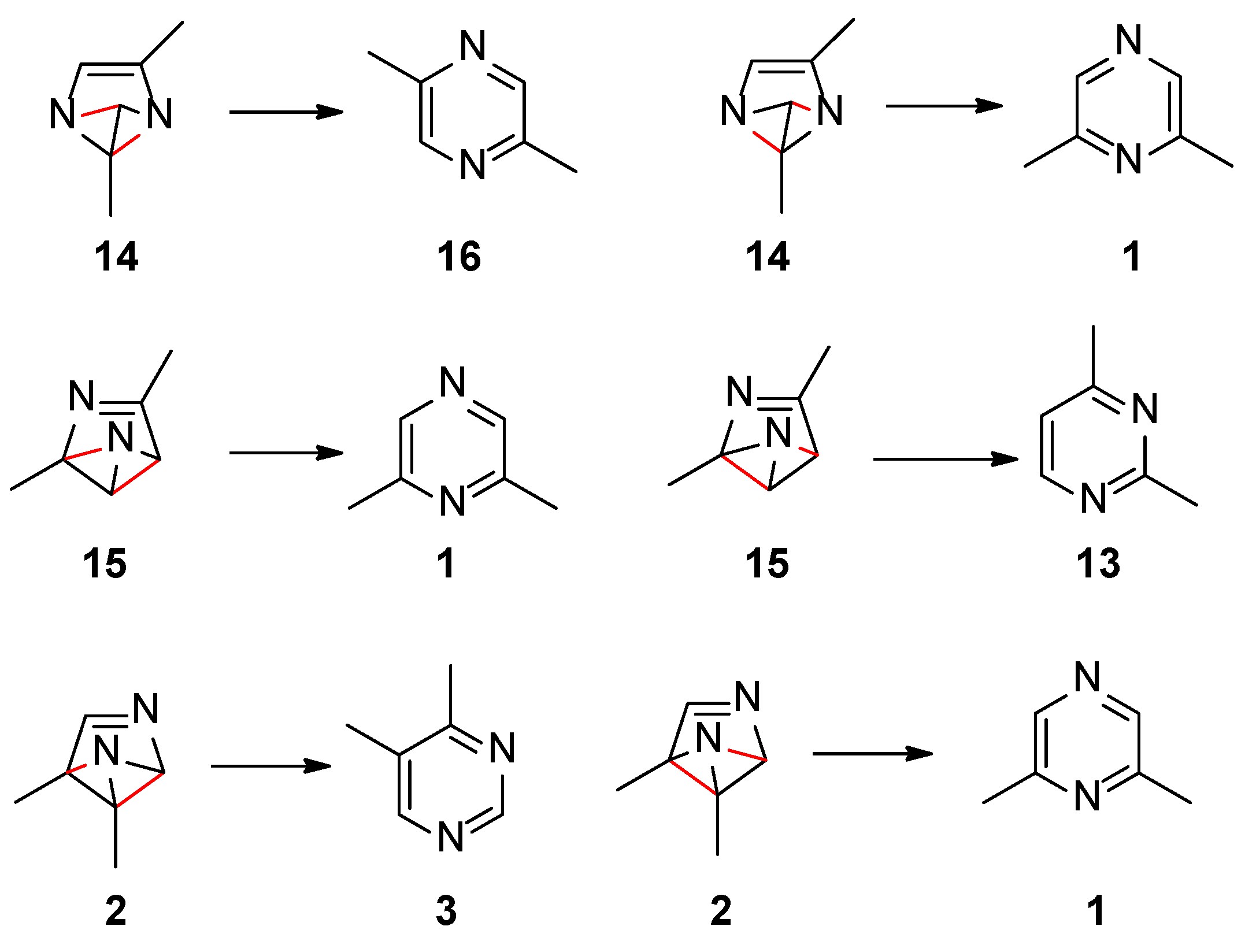
Scheme 4 collects the possible rearomatization reactions of the benzvalene intermediates 2, 14, and 15. In red the broken bonds are evidenced. The aromatization reactions occurred without a transition state. Benzvalene 14 can give 2,5-dimethylpyrazine (16), while 15 can be transformed into 2,4-dimethylpyrimidine (13). All these compounds were not found in the reaction mixture. Only benzvalene 2, the isomer showing the lowest energy, can be converted into 4,5-dimethylpyrimidine (3), the observed reaction product. This scheme is in agreement with the observed reactivity.
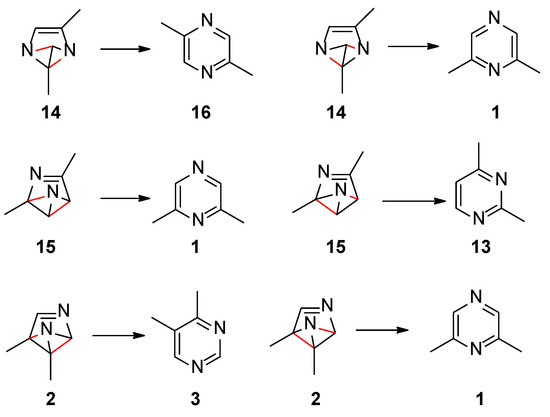
Scheme 4.
Rearomatization processes of benzvalene isomers.
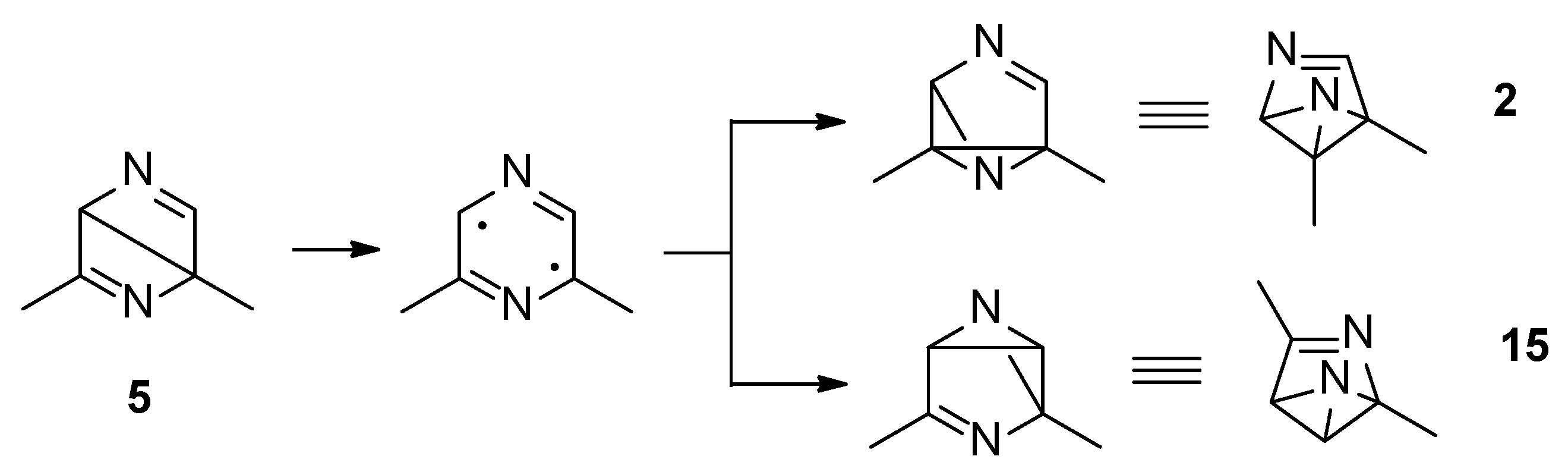
Both the excited singlet states of 2,6-dimethylpyrazine (1) can allow the formation of the Dewar isomer 5. This isomer can return to the starting material, but this step has an activation energy of 105 kJ mol−1 (ST1, Figure 2). The Dewar isomer 5 can be a precursor of several benzvalene isomers (Scheme 5). In particular, benzvalene isomer 2 and 15 can be obtained.
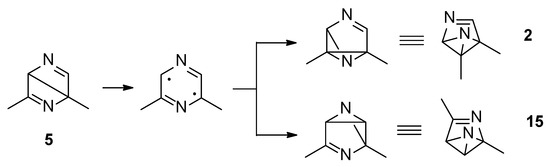
Scheme 5.
Benzvalene isomers from 5.
The formation of 2 and 15 from 5 does not require high transition state. The conversion of 5 into 15 showed a transition energy of 1 kJ mol−1, while that of 5 into 2 has a transition state at 2 kJ mol−1 (Figure 4). This is an early transition state with a structure similar to the starting material. Additionally, in this case, the formation of 2 is favored considering that it is the most stable compound. The low value of the transition states energy could be in agreement with the transposition nature of the reaction. Furthermore, we consider that two sp2 carbon atoms in a cyclobutene ring disappeared, and also the other cyclobutene ring was not present in the products.
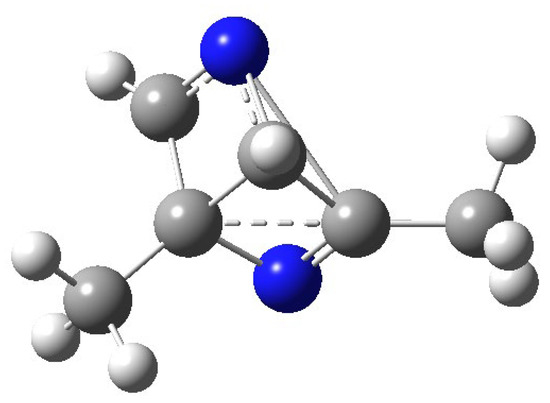
Figure 4.
Transition state of the conversion of 5 into 2.
On the other hand, the CASSCF study offers a completely different possible evolution of the first excited singlet state. The results we obtained are summarized in Figure 5.
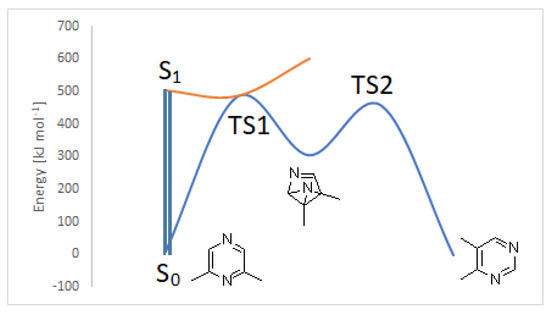
Figure 5.
CASSCF evolution of the singlet excited state of 2,6-dimethylpyrazine.
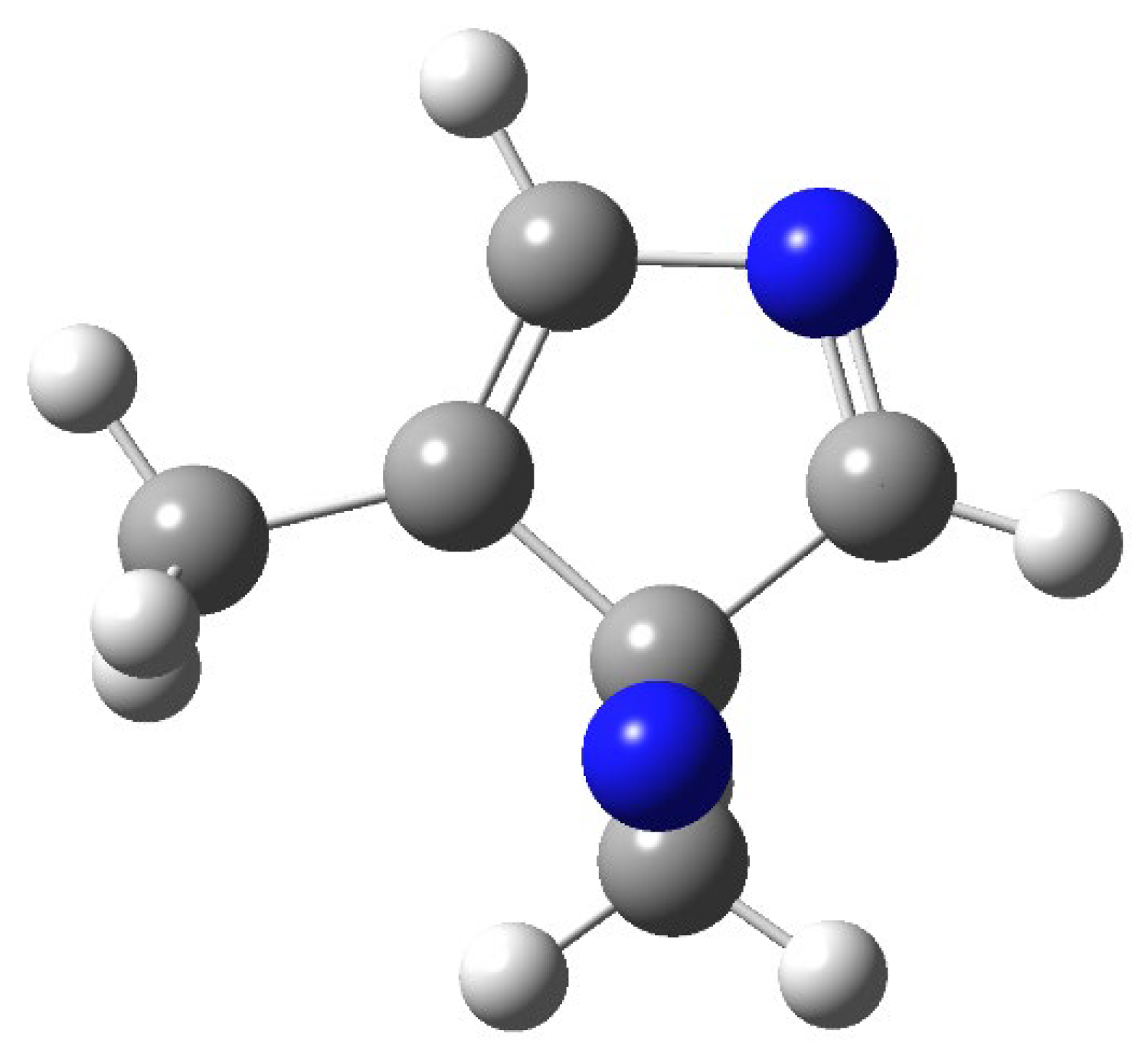
In this case, we can observe a conical intersection that allows the direct formation of 2. The benzvalene isomer 2 can be converted into the product, on the basis of the process described in Scheme 4, with a transition state (TS2) with an energy barrier of 153 kJ mol−1.
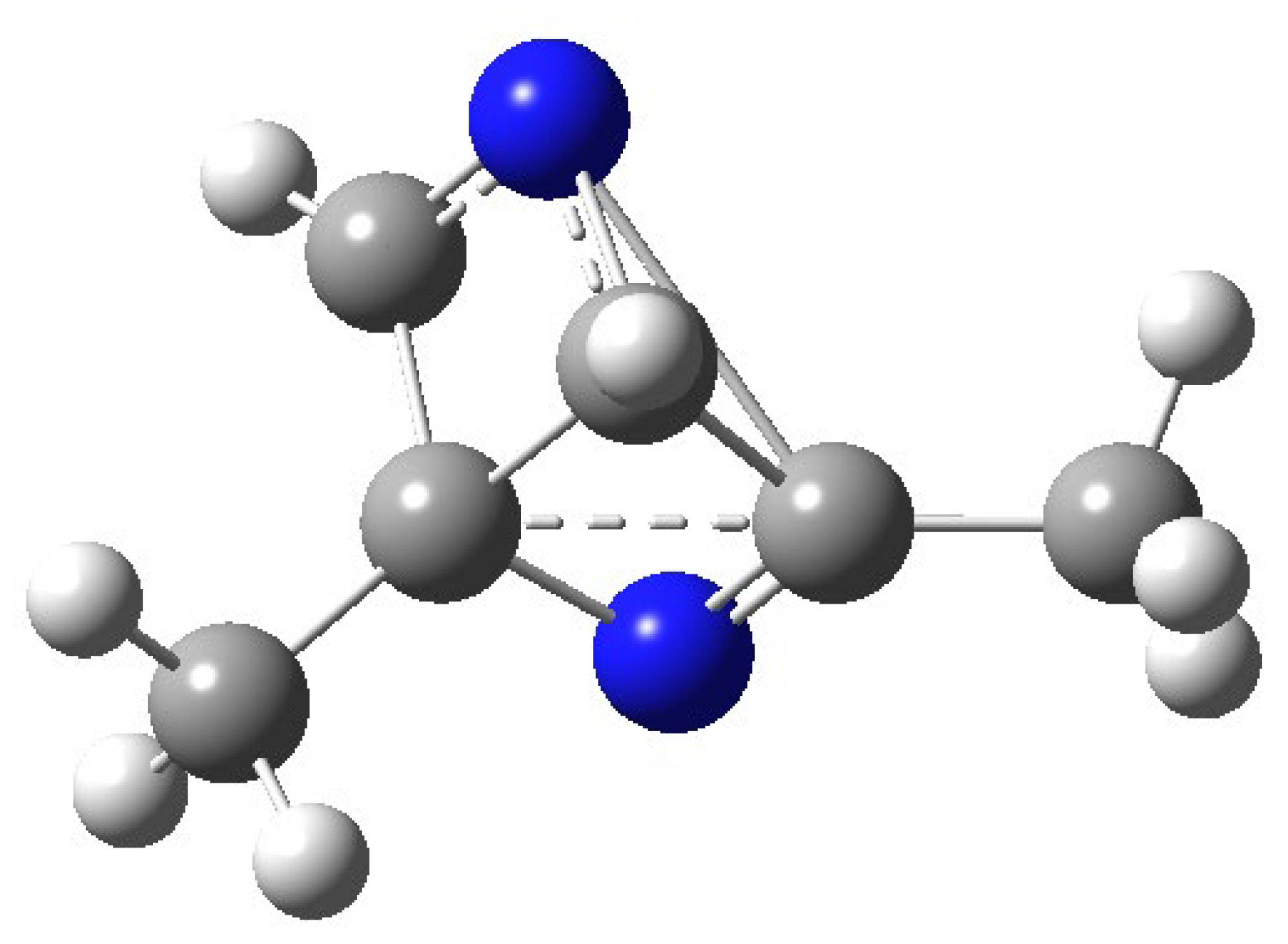
The structure of the compound at the conical intersection is reported in Figure 6.
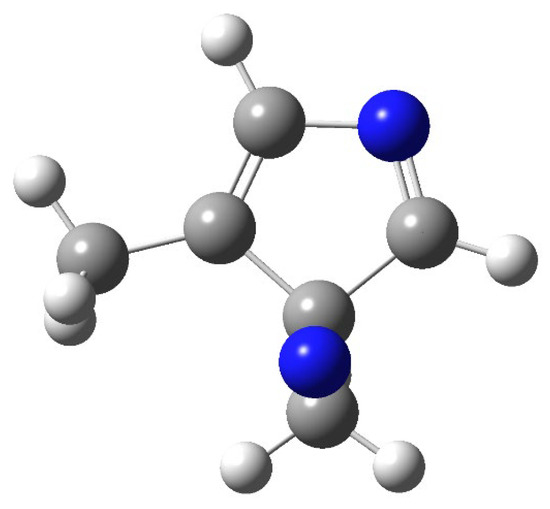
Figure 6.
Structure of 2,6-dimethylpyrazine at the conical intersection.
4. Conclusions
DFT calculations allow us to present a complete description of the reactivity of 2,6-dimethylpyrazine. This compound, in the excited singlet states, can be converted into the corresponding Dewar isomers. If the S2 state is populated, both Dewar isomers can be obtained, while S1 state allowed the formation of only the Dewar isomer 5. Both Dewar isomers can be converted into the benzvalene isomer 2, which is the precursor of 4,5-dimethylpyrimidine, the reaction product. The CASSCF study proposed a very different scenario, where a conical intersection from the S1 state allowed the direct formation of 2.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Pavlik, J.M. Photoisomerization of some nitrogen-containing hetero-aromatic compounds. In CRC Handbook of Organic Photochemistry and Photobiology, 2nd ed.; Horspool, W., Lenci, F., Eds.; CRC Press: Boca Raton, FL, USA, 2004; pp. 97.1–97.22. [Google Scholar]
- Albini, A.; Fagnoni, M. Photochemistry of N-oxides. In CRC Handbook of Organic Photochemistry and Photobiology, 2nd ed.; Horspool, W., Lenci, F., Eds.; CRC Press: Boca Raton, FL, USA, 2004; pp. 99.1–99.21. [Google Scholar]
- Mariano, P.S. A new look at pyridinium salt photochemistry. In CRC Handbook of Organic Photochemistry and Photobiology, 2nd ed.; Horspool, W., Lenci, F., Eds.; CRC Press: Boca Raton, FL, USA, 2004; pp. 100.1–100.10. [Google Scholar]
- Klán, P.; Wirz, J. Photochemistry of Organic Compounds; John Wiley & Sons Ltd.: Chichester, UK, 2009; pp. 276–279. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision A.1; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Parr, R.G.; Yang, W. Density Functional Theory of Atoms and Molecules; Oxford University Press: Oxford, UK, 1989. [Google Scholar]
- Casida, M.E. Time-dependent density-functional response theory for molecules. In Recent Advances in Density Functional Methods; Chong, D.P., Ed.; World Scientific: Singapore, 1995; Volume 1, pp. 155–192. [Google Scholar]
- Casida, M.E.; Jamorski, C.; Casida, K.C.; Salahub, D.R. Molecular excitation energies to high-lying bound states from time-dependent density-functional response theory: Characterization and correction of the time-dependent local density approximation ionization threshold. J. Chem. Phys. 1998, 108, 4439–4449. [Google Scholar] [CrossRef]
- Becke, A.D. Molecular excitation energies to high-lying bound states from time-dependent density-functional response theory: Characterization and correction of the time-dependent local density approximation ionization threshold. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef] [Green Version]
- Hegarty, D.; Robb, M.A. Application of unitary group-methods to configuration-interaction calculations. Mol. Phys. 1979, 38, 1795–1812. [Google Scholar] [CrossRef]
- Eade, R.H.A.; Robb, M.A. Direct minimization in MC SCF theory—The Quasi-Newton method. Chem. Phys. Lett. 1981, 83, 362–368. [Google Scholar] [CrossRef]
- Schlegel, H.B.; Robb, M.A. MC SCF gradient optimization of the H2CO → H2 + CO transition structure. Chem. Phys. Lett. 1982, 93, 43–46. [Google Scholar] [CrossRef]
- Bernardi, F.; Bottini, A.; McDougall, J.J.W.; Robb, M.A.; Schlegel, H.B. MCSCF gradient calculation of transition structures in organic reactions. Faraday Symp. Chem. Soc. 1984, 19, 137–147. [Google Scholar] [CrossRef]
- Frisch, M.J.; Ragazos, I.N.; Robb, M.A.; Schlegel, H.B. An Evaluation of 3 Direct MC-SCF Procedures. Chem. Phys. Lett. 1992, 189, 524–528. [Google Scholar] [CrossRef]
- Yamamoto, N.; Vreven, T.; Robb, M.A.; Frisch, M.J.; Schlegel, H.B. A Direct Derivative MC-SCF Procedure. Chem. Phys. Lett. 1996, 250, 373–378. [Google Scholar] [CrossRef]
- Siegbahn, P.E.M. A new direct CI method for large CI expansions in a small orbital space. Chem. Phys. Lett. 1984, 109, 417–423. [Google Scholar] [CrossRef]
- Robb, M.A.; Niazi, U. The Unitary Group Approach to Electronic Structure Computations. In Reports in Molecular Theory; Weinstein, H., Náray-Szabó, G., Eds.; CRC Press: Boca Raton, FL, USA, 1990; Volume 1, pp. 23–55. [Google Scholar]
- Klene, M.; Robb, M.A.; Frisch, M.J.; Celani, P. Parallel implementation of the CI-vector evaluation in full CI/CAS-SCF. J. Chem. Phys. 2000, 113, 5653–5665. [Google Scholar] [CrossRef]
- Lahmani, F.; Ivanoff, N. Photoisomerization of pyrazine and of its methylderivatives. Tetrahedron Lett. 1967, 8, 3913–3917. [Google Scholar] [CrossRef]
- Su, M.-D. CASCSF Study on the photochemical transposition reactions of pyrazines. J. Phys. Chem. A 2006, 110, 9420–9428. [Google Scholar] [CrossRef] [PubMed]
- D’Auria, M. The photochemical isomerization of hexatomic heterocyclic compounds. Curr. Org. Chem. 2021, 25, 1659–1685. [Google Scholar] [CrossRef]
- Available online: https://spectrabase.com/spectrum/JGriPcJDFzk (accessed on 10 January 2022).
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).