
Regioselective Transfer Hydrogenative Defluorination of Polyfluoroarenes Catalyzed by Bifunctional Azairidacycle


Department of Chemical Science and Engineering, School of Materials and Chemical Technology, Tokyo Institute of Technology, 2-12-1-E4-1 O-okayama, Meguro-ku, Tokyo 152-8552, Japan
*
Author to whom correspondence should be addressed.
†
Current address: Department of Applied Chemistry, College of Life Sciences, Ritsumeikan University, 1-1-1 Noji-higashi, Kusatsu, Shiga 525-8577, Japan.
Organics 2022, 3(3), 150-160; https://doi.org/10.3390/org3030012
Submission received: 4 April 2022
/
Revised: 9 June 2022
/
Accepted: 15 June 2022
/
Published: 22 June 2022
(This article belongs to the Collection Advanced Research Papers in Organics)
Abstract
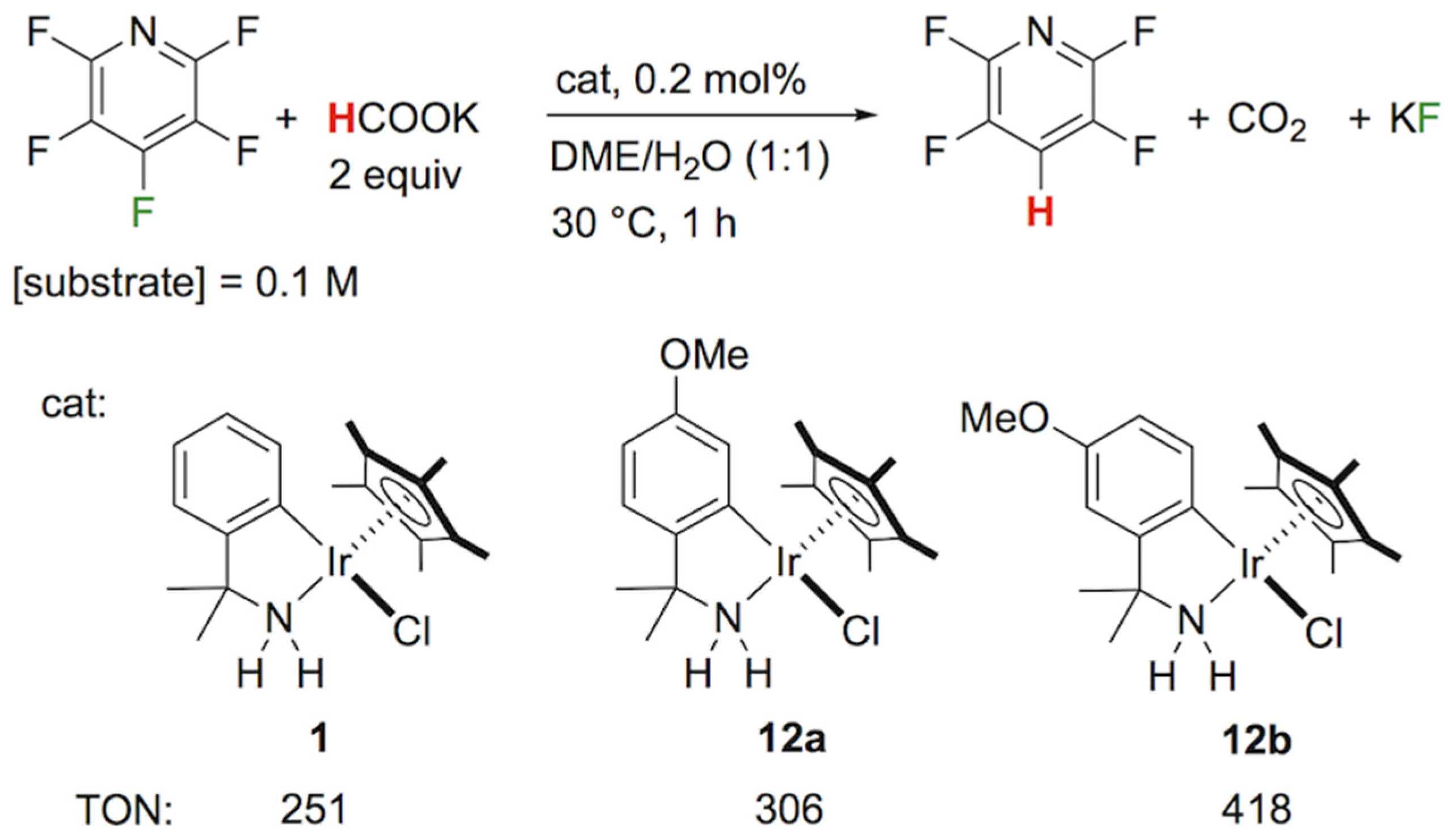
:The catalytic hydrodefluorination (HDF) with a bifunctional azairidacycle using HCOOK was examined for cyano- and chloro-substituted fluoroarenes, including penta- and tetrafluorobenzonitriles, tetrafluoroterephthalonitrile, tetrafluorophthalonitrile, 3-chloro-2,4,5,6-tetrafluoropyridine, and 4-cyano-2,3,5,6-tetrafluoropyridine. The reaction was performed in the presence of a controlled amount of HCOOK with a substrate/catalyst ratio (S/C) of 100 in a 1:1 mixture of 1,2-dimethoxyethane (DME) and H2O at an ambient temperature of 30 °C to obtain partially fluorinated compounds with satisfactory regioselectivities. The C–F bond cleavage proceeded favorably at the para position of substituents other than fluorine, which is in consonance with the nucleophilic aromatic substitution mechanism. In the HDF of tetrafluoroterephthalonitrile and 4-cyano-2,3,5,6-tetrafluoropyridine, which do not contain a fluorine atom at the para position of the cyano group, the double defluorination occurred solely at the 2- and 5-positions, as confirmed by X-ray crystallography. The HDF of 3-chloro-2,4,5,6-tetrafluoropyridine gave preference to the C–F bond cleavage over the C–Cl bond cleavage, unlike the dehalogenation pathway via electron-transfer radical anion fragmentation. In addition, new azairidacycles with an electron-donating methoxy substituent on the C–N chelating ligand were synthesized and served as a catalyst precursor (0.2 mol%) for the transfer hydrogenative defluorination of pentafluoropyridine, leading to 2,3,5,6-tetrafluoropyridine with up to a turnover number (TON) of 418.
1. Introduction
Fluorine containing organic compounds has been receiving much attention in the field of pharmaceuticals, agrochemicals, and functional materials. In general, perfluorinated aromatic compounds are practically obtained by fluorination of hydrocarbons using fluorine gas or hydrogen fluoride, whereas the synthesis of partially fluorinated compounds is still under development [1,2,3]. Aside from specific arene fluorinations using electrophilic, nucleophilic, or radical fluorine sources [4,5,6,7,8,9,10], hydrodefluorination (HDF) can be regarded as a valuable method, especially in the regioselective transformation of C–F bonds on perfluoroarenes [11,12,13]. Among the catalytic systems using transition metals, hazardous fluorophilic reductants such as hydrosilanes [14,15,16,17,18,19,20,21,22,23] and aluminum hydrides [24,25] have been widely used to facilitate the cleavage of strong C–F bonds. Hydrogenolysis of fluoroarenes has also been reported as an atom-economical HDF system [26,27,28,29,30,31,32].
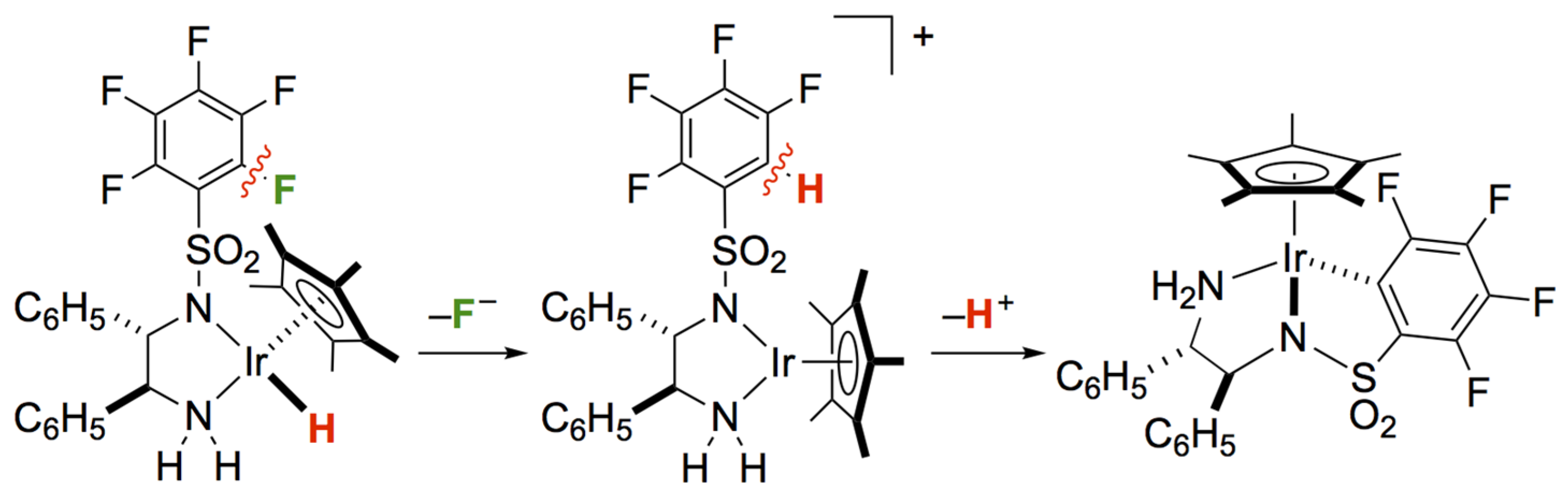
As an alternative approach to avoid unsafe procedures, we have developed the HDF of perfluoroaromatic compounds promoted by an Ir-based transfer hydrogenation catalyst 1 using 2-propanol or formate salts as mild reducing agents (Scheme 1) [33,34]. A catalytically active hydridoiridium complex possessing a σ-donating aryl-metal bond and a protic amine ligand engaged in the HDF at an ambient temperature of 30 °C, leading to an outstanding catalytic performance characterized by bifunctional amido/amine complexes. For example, pentafluoropyridine and monosubstituted pentafluorobenzenes were selectively defluorinated at the 4-position to give the corresponding HDF products in satisfactory yields. The reduction of fluorinated benzene derivatives was markedly facilitated by the introduction of electron-withdrawing substituents. These experimental results suggest a nucleophilic aromatic substitution (SNAr) mechanism involving the powerful hydridoiridium species. Separately, we also investigated the stoichiometric C–F bond cleavage reactions of aromatic fluorides, illustrated by the related iridium complexes bearing fluorinated phenylsulfonyl-1,2-diphenylethylenediamine ligands [35,36,37]. The thermolysis of the isolable hydridoiridium revealed a successive conversion into iridacycles via intramolecular HDF caused by the nucleophilic attack of the hydrido ligand (Scheme 2) [35].
In order to reinforce the SNAr mechanism [38,39] in the transfer hydrogenative defluorination catalyzed by 1, we herein investigated the advanced control of the regio- and chemoselectivity by extension of the substrates, which can provide valuable insight into the hydride transfer process. On the basis of the aromatic C–F bond cleavage by Ir-bonded nucleophilic ligands, we also modified the azairidacycle catalyst to enhance the hydridicity via the introduction of an electron-donating methoxy substituent on the C–N chelating ligand. The influence of electronic tuning on the catalytic HDF performance is elucidated.
2. Results and Discussion
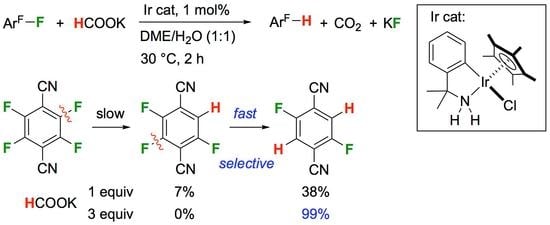
According to the transfer hydrogenation system promoted by the azairidacycle 1 [33], the HDF of selected polyfluoroarenes was performed with a substrate/catalyst molar ratio of 100 in a mixture of aqueous potassium formate and 1,2-dimethoxyethane. The results are summarized in Table 1. When pentafluorobenzonitrile (2a) was treated with 2 equiv of HCOOK at 30 °C for 1 h, the fluorine atom at the para position was substituted to give 2,3,5,6-tetrafluorobenzonitrile (3a) in a quantitative yield (Entry 1). Even with the extra amount of the hydride source, a successive defluorination hardly proceeded. The further defluorination of 3a with the elongation of the reaction time to 12 h resulted in the formation of 2,3,5-trifluorobenzonitrile (4) in a moderate yield of 20% (Entry 2). Notably, the same compound 4 was smoothly accessible via the HDF of 2,3,4,5-tetrafluorobenzonitrile (3b), which ascertained the trifluorinated structure at the 2-, 3-, and 5-positions (Entry 3). Doubling the catalytic amount to 2 mol% led to a complete conversion and selectivity within 1 h. These results indicated that the C–F bond at the para position of the substituent was rather susceptible to the reduction [40,41].
The lower reactivity of 3a than that of 3b accommodates the fact that C–F bonds at para positions are inherently unfavorable for the nucleophilic aromatic substitution mechanism. Considering the essential contribution of the negatively charged electronic structure at the para position of the carbon to which the nucleophile is introduced [42,43,44,45,46,47,48], the fluorine substituent at the para position possibly destabilizes the transition state due to the repulsion between the occupied p-orbital on the carbon and the lone electron pair on the fluorine (Scheme 3). Therefore, nucleophilic addition proceeds preferentially at the para position of substituents other than fluorine. Actually, 19F NMR monitoring experiments of the HDF using 1 mol% of 1 revealed that hexafluorobenzene (2b) was intact in the presence of 3 equiv of HCOOK at 30 °C after 24 h (Entry 4 in Table 1).
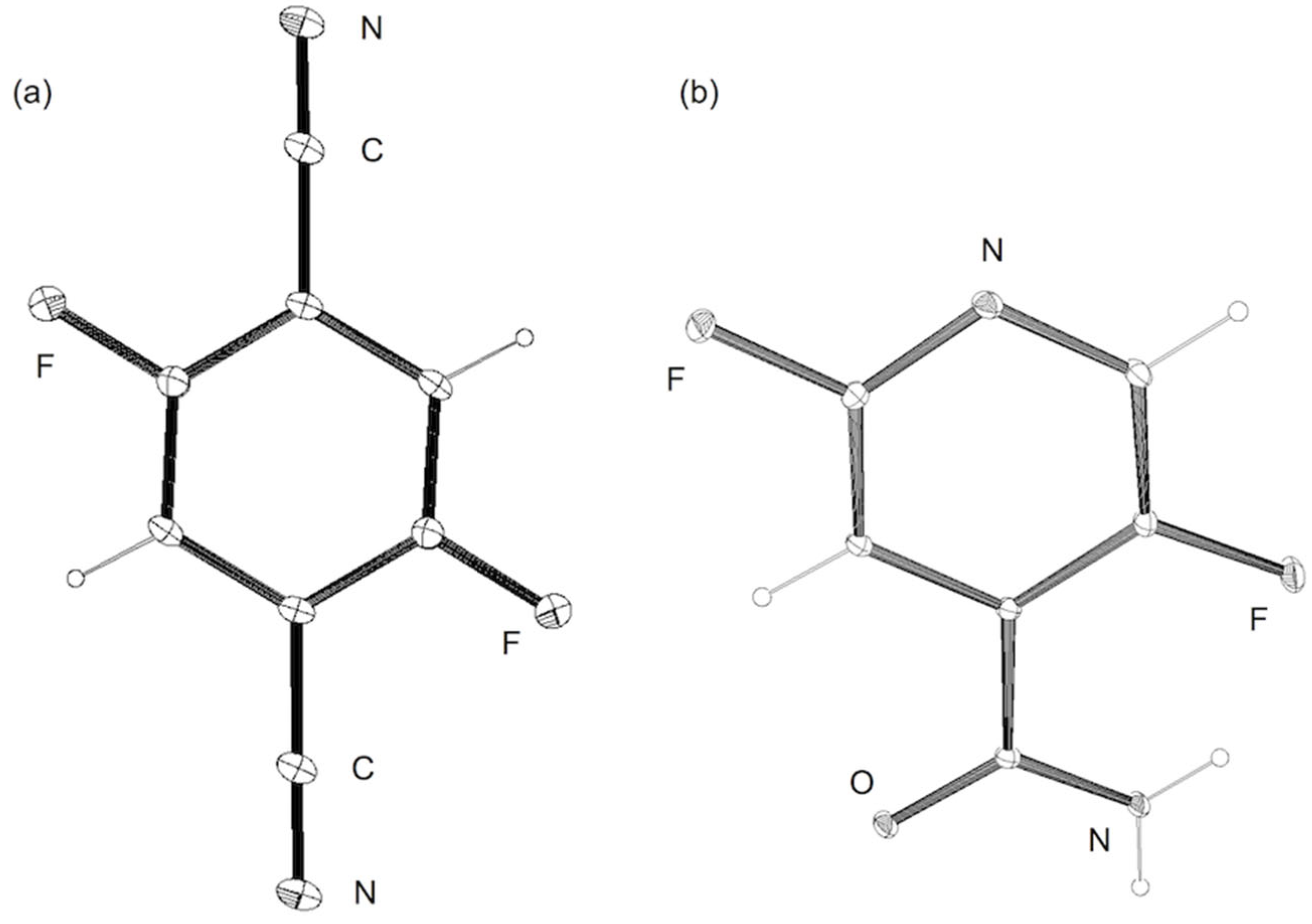
In the reaction of tetrafluoroterephthalonitrile (5a), which does not contain a fluorine atom at the para position of the cyano group, a monodefluorinated product (2,3,5-trifluoroterephthalonitrile; 6a) was hardly obtained by the reduction with an equimolar amount of HCOOK (Entry 5). Alternatively, 2,5-difluoroterephthalonitrile (7a) was formed in a 38% yield as a result of the double defluorination. By using 3 equiv of HCOOK, 5a was almost quantitatively converted into 7a as a sole product after 2 h (Entry 6). The 19F NMR analysis displayed a decay of the signal due to 5a at −128 ppm, accompanied by the appearance of signals at −109 ppm and −119 ppm due to 7a and potassium fluoride (see Figure S1 in the Supplementary Materials). The regiochemistry of the product was finally determined by X-ray crystallography, as shown in Figure 1 (left; a). These results indicated the comparatively smooth second defluorination at the para position of the initially incoming hydrogen atom.
In contrast, the regioisomeric tetrafluorophthalonitrile (5b) can be reduced stepwise to give mono- and di-defluorinated products, as investigated in our previous work [33]. When 5b was reduced with 1 equiv of HCOOK, the monosubstituted product, 3,4,6-trifluorophthalonitrile (6b), was successfully obtained in an 88% isolated yield (Entry 7). The HDF using 3 equiv of HCOOK afforded the corresponding consecutive transfer hydrogenation product (7b), in which the para positions of the two cyano groups were specifically defluorinated (Entry 8). The preferential para selectivity in the HDF of substituted perfluorobenzenes was substantiated by these experiments. The double-defluorinated product 7b is the precursor of polyfluorinated phthalocyaninato complexes, which have been synthesized via six steps from 2,5-difluorobenzoyl chloride [49]. This selective HDF, enabling the synthesis of partially fluorinated compounds directly from perfluoroarenes, simplifies the conventional methods.
A reliable regioselectivity was also observed in the HDF of fluorinated pyridine derivatives [50]. In the HDF of 3-chloro-2,4,5,6-tetrafluoropyridine (8a) under standard conditions, the carbon–fluorine bond at the 4-position was specifically cleaved to yield 3-chloro-2,5,6-trifluoropyridine (9a) quantitatively without the erosion of the chlorine substituent (Entry 9). The monodefluorination in preference to dechlorination is a peculiar feature of the nucleophilic aromatic substitution mechanism [51], in contrast with the trend of aromatic hydrodehalogenation involving electron-transfer radical anion fragmentation, in which the heavier halogen favors undergoing fragmentation [52]. The reaction was also compatible with 4-cyano-2,3,5,6-tetrafluoropyridine (8b) possessing a non-fluorine substituent at the 4-position, leading to the formation of 4-cyano-2,5-difluoropyridine (9b) in a 98% yield via double defluorination with 2 equiv of HCOOK (Entry 10). As with the HDF of 5a and 5b, the high electron deficiency of the substrates allows for the consecutive substitution. This product was converted to the carboxamide derivative (10) by hydration in the presence of a copper catalyst (Scheme 4) [53], and the regiochemistry was determined by X-ray crystallographic analysis, as shown in Figure 1 (right).
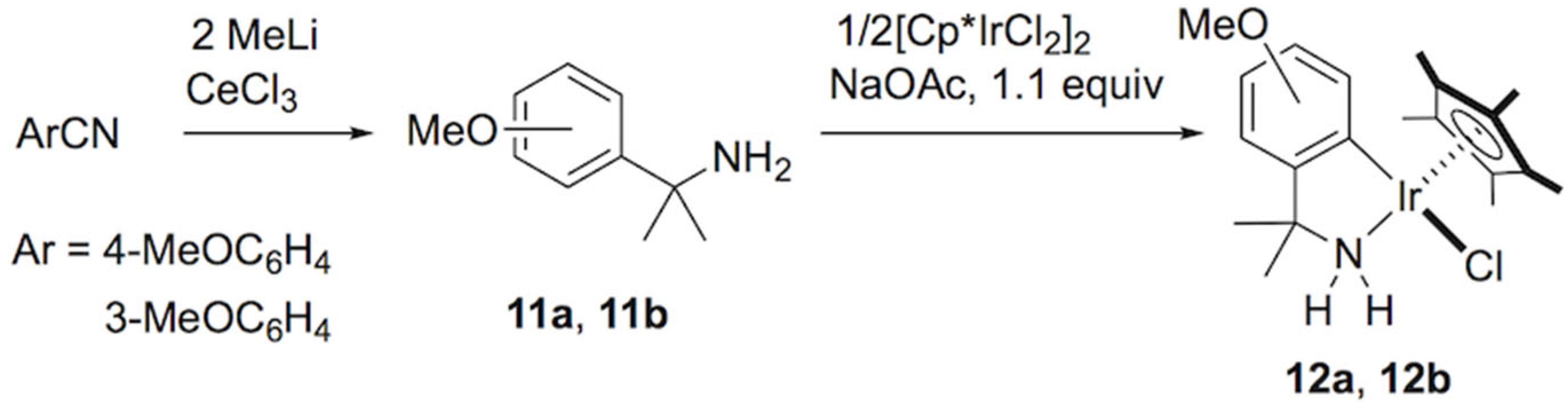
Considering the important role of interim hydridoiridium species in the C–F bond cleavage, more nucleophilic iridium systems were used to facilitate the catalytic HDF. For the modified iridacycles, we examined the coordination of new C–N chelate ligands with electron-donating substituents (Scheme 5) and investigated their catalytic activity for the HDF. The cumylamine derivatives 11a and 11b were synthesized by a treatment of methylcerium with benzonitrile derivatives having a methoxy group at the para or meta position in THF [54]. In a similar manner to our original synthetic procedure for azairidacycles [55,56], orthometalation of 11a was performed by mixing [Cp*IrCl2]2 with sodium acetate (1 equiv/Ir) in acetonitrile at 60 °C for 10 h. After recrystallization, the expected chlorido complex (12a) was obtained as yellow crystals in a 73% yield (Scheme 5). For the transformation of 3-methoxy substituted analog 11b, the metalation took place mainly at the 6-position, while a small amount of a sterically congested isomer was found in the crude products. After purification by chromatographic techniques, the major product 12b was isolated.
With the new azairidacycles, we further explored their catalytic activities for the transfer hydrogenative defluorination. The reaction of pentafluoropyridine with HCOOK (2 equiv) using the catalyst precursor 1 with a substrate/iridium ratio of 500 under otherwise standard conditions proceeded to give 2,3,5,6-tetrafluoropyridine with a turnover number of 251 after 1 h (Scheme 6). When 12a was used as the catalyst, a slightly higher TON of 306 was obtained. By switching the catalyst with 12b, a further enhancement of the catalytic activity was observed with a TON of 418. The complexes with a methoxy group on the aryl ligand showed an enhanced catalytic activity, possibly due to the electron-donating methoxy group improving the nucleophilicity of the catalytically active hydrido intermediate.
In keeping with the attractive catalytic performance of 12b, the double defluorination of 5b was performed on a 1.1 g scale for 4 h with a substrate/catalyst ratio of 180. After purification by column chromatography, the desired product 7b was successfully isolated in a 90% yield (0.81 g).
3. Materials and Methods
3.1. General Information
All manipulations of oxygen- and moisture-sensitive materials were performed under a purified argon atmosphere using standard Schlenk techniques. Solvents were purchased from Kanto Chemical Co., Inc. (Tokyo, Japan) and dried by refluxing over sodium benzophenone ketyl (THF, 1,4-dioxane, and diethyl ether), P2O5 (CH3CN and CH2Cl2), or CaH2 (2-propanol, pentane), and distilled under argon before use. Ethyl acetate and chloroform-d1 was used as delivered. Dichloromethane-d2 was degassed by three freeze-pump-thaw cycles and purified by trap-to-trap distillation after being dried with P2O5. The fluoroarene substrates were purchased from Kanto Chemical Co., Ltd. (Tokyo, Japan), Sigma-Aldrich Co. LLC. (St. Louis, MO, USA), Tokyo Chemical Industry Co., Ltd. (Tokyo, Japan), and FUJIFILM Wako Pure Chemical Corporation (Osaka, Japan), degassed, and stored under argon atmosphere. The other reagents were purchased from Sigma-Aldrich Co. LLC. (St. Louis, MO, USA), Tokyo Chemical Industry Co., Ltd. (Tokyo, Japan), and Nacalai Tesque Inc. (Kyoto, Japan), and used as delivered. The Ir catalyst 1 was prepared according to the procedures described in the literature [55]. 1H (399.8 MHz), 19F (376.2 MHz), and 13C{1H} (100.5 MHz) NMR spectra were recorded on a JNM-ECX400 spectrometer (JEOL Ltd., Tokyo, Japan) at around 25 °C. The NMR chemical shifts were referenced to an external tetramethylsilane signal (0.0 ppm) by using the signals of residual proton impurities in the deuterated solvents for 1H and 13C{1H} NMR, and referenced to an external CF3CO2H signal (−76.5 ppm) for 19F NMR. Recyclable preparative high-performance liquid chromatography was performed on an LC-9225 NEXT system (Japan Analytical Industry Co., Ltd., Tokyo, Japan) equipped with JAIGEL-1H and -2H columns using CHCl3 as an eluent at a flow rate of 14 mL min−1. Elemental analyses were carried out using a PE2400 Series II CHN analyzer (PerkinElmer, Inc., Waltham, MA, USA).
3.2. Representative Procedures of HDF of Fluoroarenes Using Potassium Formate under the Conditions of S/C = 100 at 30 °C
A mixture of the catalyst 1 (2.5 mg, 5.0 × 10−3 mmol), potassium formate (84.8 mg, 1.01 mmol), and pentafluorobenzonitrile (96.5 mg, 0.50 mmol) in 1,2-dimethoxyethane (2.5 mL) and water (2.5 mL) was stirred at 30 °C for 1 h. The product yield was determined by 19F NMR spectroscopy using a solution of trifluoromethylbenzene (0.260 M) in 1,2-dimethoxyethane (1 mL) as an internal standard. After the HDF reaction, the compound was extracted by Et2O (10 mL × 2) and was dried over MgSO4. The catalysts were removed by filtration through a pad of Florisil. The sample after the evaporation was purified by silica gel column chromatography using hexane/ethyl acetate (with a ratio of 100/5–100/8) as the eluent.
3.3. Catalytic Hydration of 9b
Copper(II) acetate (4.0 mg, 0.02 mmol), N,N-diethylhydroxylamine (292 mg, 3.27 mmol), and water were placed in a 20 mL reactor, and then 9b (0.5 mmol) was added while stirring. After stirring the mixture at 35 °C for 4 h, the solvent was removed in vacuo, and the crude product was purified over a short pad of silica gel, eluted with CH2Cl2 and methanol (95:5). Colorless crystals of 10 suited for X-ray crystallography were obtained by recrystallization from CH2Cl2 and hexane.
3.4. Synthesis of 11a and 11b
Anhydrous CeCl3 (0.84 g, 3.4 mmol) was stirred in dry THF (10 mL) at room temperature under Ar atmosphere for 2 h. The solution was cooled at −78 °C, and methyllithium (2.2 mL, 3.4 mmol, 1.6 M in diethyl ether) was added dropwise. After stirring the resulting mixture for 30 min at −78 °C, a solution of 4-methoxybenzonitrile (0.15 g, 1.1 mmol) in dry THF (5 mL) was added. The crude reaction mixture was stirred at room temperature overnight, quenched by adding 1.5 mL of concentrated ammonium chloride, and allowed to stir for 1 h at room temperature. The organic layer was filtered, dried over anhydrous Na2SO4, and concentrated in vacuo. The crude product of 11a [57] was used for the next reaction without further purification.
Compound 11b was obtained from 3-methoxybenzonitrile analogously to 11a as a yellowish oil [58].
3.5. Synthesis of 12a
To a mixture of [Cp*IrCl2]2 (614.4 mg, 0.77 mmol) and NaOAc (128.6 mg, 1.57 mmol) in CH3CN (15 mL), 11a (254.7 mg, 1.54 mmol) was added. Then, the reaction mixture was stirred at 60 °C for 10 h. The solvent was removed under reduced pressure. The resulting reaction mixture in CH2Cl2 (10 mL) was washed with degassed water several times, and the organic layer was dried over Na2SO4. After removing insoluble materials by filtration, the evaporation of the filtrate to dryness gave a yellow powder. Further purification by recrystallization from CH2Cl2 and ether gave orange crystals of 12a in a 73% yield (591 mg, 1.12 mmol). 1H NMR (CDCl3, r.t., δ/ppm): δ 1.21 (s, 3H), 1.65 (s, 3H), 1.72 (s, 15H), 3.75 (br, 1H), 3.78 (s, 3H), 4.45 (br, 1H), 6.42 (dd, 1H; J = 2.6 and 8.4 Hz), 6.71 (d, 1H; J = 8.2 Hz), 7.06 (d, 1H; J = 2.7 Hz). 13C{1H} NMR (CDCl3, r.t., δ/ppm): 9.3, 31.0, 31.9, 54.9, 65.8, 86.6, 107.5, 121.2, 121.6, 147.0, 154.8, 158.0. Anal. Calcd for [C20H29ONClIr]: C, 45.57; H, 5.55; N, 2.66; Found: C, 45.96; H, 5.40; N, 3.15%.
3.6. Synthesis of 12b
To a mixture of [Cp*IrCl2]2 (1.003 g, 1.26 mmol) and NaOAc (273.2 mg, 3.34 mmol) in CH3CN (50 mL), 11b (427.3 mg, 2.59 mmol) was added. Then, the reaction mixture was stirred at 60 °C overnight. The solvent was removed under reduced pressure. The resulting reaction mixture in CH2Cl2 (10 mL) was washed with degassed water several times, and the organic layer was dried over Na2SO4. After removing insoluble materials by filtration, the evaporation of the filtrate to dryness gave a yellow powder. Recrystallization from CH2Cl2 and ether and subsequent purification by a preparative gel permeation chromatography gave an orange powder. Further purification by recrystallization from CH2Cl2 and ether gave orange crystals of 12b in a 12% yield (158 mg, 0.30 mmol). 1H NMR (CD2Cl2, r.t., δ/ppm): δ 1.18 (br, 3H), 1.62 (br, 3H), 1.68 (s, 15H), 3.71 (s, 3H), 3.85 (br, 1H), 4.36 (br, 1H), 6.39 (d, 1H; J = 2.8 Hz), 6.59 (dd, 1H; J = 2.8 and 8.2 Hz), 7.26 (d, 1H; J = 8.3 Hz). 13C{1H} NMR (CD2Cl2, r.t., δ/ppm): 9.1, 30.6, 31.5, 55.1, 66.2, 86.4, 108.1, 112.5, 136.3, 143.2, 155.4, 156.2. Anal. Calcd for [C20H29ONClIr]: C, 45.57; H, 5.55; N, 2.66; Found: C, 45.42; H, 5.34; N, 2.63%.
3.7. Scale-Up Experiment in the Synthesis of 7b
A mixture of the catalyst 12b (16.5 mg, 3.1 × 10−2 mmol), potassium formate (1.46 g, 17.2 mmol), and tetrafluorophthalonitrile (1.10 mg, 5.5 mmol) in 1,2-dimethoxyethane (25 mL) and water (25 mL) was stirred at 30 °C for 4 h. The mixture was extracted with Et2O (20 mL), dried over MgSO4, and evaporated under reduced pressure. The residue was purified by silica gel column chromatography using hexane/ethyl acetate as the eluent to afford 7b (0.81 g, 4.94 mmol, 90% yield) as a white solid.
3.8. X-ray Crystal Structure Determination
Diffraction experiments were performed on a Rigaku Saturn CCD area detector (Rigaku Corporation, Tokyo, Japan) using graphite-monochromated Mo-Kα radiation (λ = 0.71075 Å) under a nitrogen stream at 193 or 113 K (for 7a and 10, respectively). Single crystals suitable for X-ray analyses were mounted on glass fibers. The crystal-to-detector distance was 45.0 mm. Data were collected to a maximum 2θ value of 55.0°. A total of 720 oscillation images were collected. A sweep of the data was carried out by using ω scans from −110.0 to 70.0° in 0.5° steps at χ = 45.0° and ϕ = 0.0°. A second sweep was performed by using ω scans from −110.0 to 70.0° in 0.5˚ steps at χ = 45.0° and ϕ = 90.0°. Intensity data were collected for Lorentz-polarization effects and absorption. Details of the crystal and data collection parameters for the compounds 7a and 10 are summarized in Table S1. The structure solution and refinements were performed with the CrystalStructure program package [59]. The heavy atom positions were determined by a direct program method (SIR92) [60], and the remaining non-hydrogen atoms were found by subsequent Fourier syntheses and were refined by full-matrix least-squares techniques against F2 using the SHELXL-2014/7 program [61]. The hydrogen atoms were placed at calculated positions and were refined with a riding model.
4. Conclusions
In summary, this work expands on polyfluoroarene substrates for transfer hydrogenative defluorination catalyzed by the bifunctional iridium catalyst 1. Based on the highly regioselective control in HDF, nucleophilic aromatic substitution is responsible for the C–F bond cleavage. A marked acceleration caused by the introduction of a methoxy group into the azairidacycle structure corroborates the fact that the highly σ-donating nature of the iridium catalyst contributes to enhancing the hydridicity. These results will be of assistance in the design of new reduction catalyst systems with a metal/NH bifunctionality. In addition to practical reaction procedures that avoid hazardous reducing agents, the transfer hydrogenation system is characterized by an outstanding catalytic performance under mild conditions and thus offers further innovation opportunities for synthesizing a range of partially fluorinated aromatic compounds.
Supplementary Materials
The following are available online at https://www.mdpi.com/article/10.3390/org3030012/s1. Table S1: Crystallographic data for 7a and 10; Figure S1: HDF of 5a Monitored by 19F NMR Spectroscopy; Figures S2–S8: copies of 1H and 13C{1H} NMR spectra of 10, 12a, and 12b.
Author Contributions
Conceptualization, Y.K.; methodology, A.M. and Y.K.; validation, S.K. and Y.K.; formal analysis, A.M. and Y.K.; investigation, A.M.; data curation, A.M. and Y.K.; writing, A.M., S. K. and Y.K.; project administration, S.K. and Y.K.; funding acquisition, A.M., S.K. and Y.K.; supervision, S.K. and Y.K. All authors have read and agreed to the published version of the manuscript.
Funding
This study was funded in part by the Japan Society of Promotion of Science (JSPS) for a Research Fellowship for Young Scientists (No. 17J09484) and by a Sasakawa Scientific Research Grant from the Japan Science Society (No. 2018-3015).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented in this study are available in the Supplementary Materials.
Acknowledgments
We are grateful for the support from Sumitomo Foundation and Mizuho Foundation for the Promotion of Sciences.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Senaweera, S.M.; Singh, A.; Weaver, J.D. Photocatalytic Hydrodefluorination: Facile Access to Partially, Fluorinated Aromatics. J. Am. Chem. Soc. 2014, 136, 3002–3005. [Google Scholar] [CrossRef] [PubMed]
- Weaver, J.; Senaweera, S. C−F activation and functionalization of perfluoro- and polyfluoroarenes. Tetrahedron 2014, 70, 7413–7428. [Google Scholar] [CrossRef]
- Amii, H.; Uneyama, K. C−F Bond Activation in Organic Synthesis. Chem. Rev. 2009, 109, 2119–2183. [Google Scholar] [CrossRef]
- Liang, T.; Neumann, C.N.; Ritter, T. Introduction of Fluorine and Fluorine-Containing Functional Groups. Angew. Chem. Int. Ed. 2013, 52, 8214–8264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campbell, M.G.; Ritter, T. Modern Carbon-Fluorine Bond Forming Reactions for Aryl Fluoride Synthesis. Chem. Rev. 2015, 115, 612–633. [Google Scholar] [CrossRef] [PubMed]
- Champagne, P.A.; Desroches, J.; Hamel, J.-D.; Vandamme, M.; Paquin, J.-F. Monofluorination of Organic Compounds: 10 Years of Innovation. Chem. Rev. 2015, 115, 9073–9174. [Google Scholar] [CrossRef]
- Sather, A.C.; Buchwald, S.L. The Evolution of Pd0/PdII-Catalyzed Aromatic Fluorination. Acc. Chem. Res. 2016, 49, 2146–2157. [Google Scholar] [CrossRef]
- Szpera, R.; Moseley, D.F.J.; Smith, L.B.; Sterling, A.J.; Gouverneur, V. The Fluorinayion of C–H Bonds: Developments and Perspectives. Angew. Chem. Int. Ed. 2019, 58, 14824–14848. [Google Scholar] [CrossRef]
- See, Y.Y.; Morales-Colón, M.T.; Bland, D.C.; Sanford, M.S. Development of SNAr Nucleophilic Fluorination: A Fruitful Academia-Industry Collaboration. Acc. Chem. Res. 2020, 53, 2372–2383. [Google Scholar] [CrossRef]
- Rozatian, N.; Hodgson, D.R.W. Reactivities of electrophilic N–F fluorinating reagents. Chem. Commun. 2021, 57, 683–712. [Google Scholar] [CrossRef]
- Kuehnel, M.F.; Lentz, D.; Braun, T. Synthesis of Fluorinated Building Blocks by Transition-Metal-Mediated Hydrodefluorination Reactions. Angew. Chem. Int. Ed. 2013, 52, 3328–3348. [Google Scholar] [CrossRef] [PubMed]
- Whittlesey, M.K.; Peris, E. Catalytic Hydrodefluorination with Late Transition Metal Complexes. ACS Catal. 2014, 4, 3152–3159. [Google Scholar] [CrossRef]
- Hu, J.-Y.; Zhang, J.-L. Hydrodefluorination Reactions Catalyzed by Transition-Metal Complexes. In Organometallic Fluorine Chemistry; Braun, T., Hughes, R.P., Eds.; Topics in Organometallic Chemistry; Springer: Cham, Switzerland, 2015; Volume 52, pp. 143–196. [Google Scholar]
- Aizenberg, M.; Milstein, D. Catalytic Activation of Carbon-Fluorine Bonds by a Soluble Transition Metal Complex. Science 1994, 265, 359–361. [Google Scholar] [CrossRef] [PubMed]
- Vela, J.; Smith, J.M.; Yu, Y.; Ketterer, N.A.; Flaschenriem, C.J.; Lachicotte, R.J.; Holland, P.L. Synthesis and Reactivity of Low-Coordinate Iron(II) Fluoride Complexes and Their Use in the Catalytic Hydroefluorination of Fluorocarbons. J. Am. Chem. Soc. 2005, 127, 7857–7870. [Google Scholar] [CrossRef]
- Reade, S.P.; Mahon, M.F.; Whittlesey, M.K. Catalytic Hydrodefluorination of Aromatic Fluorocarbons by Ruthenium N-Heterocyclic Carbene Complexes. J. Am. Chem. Soc. 2009, 131, 1847–1861. [Google Scholar] [CrossRef] [PubMed]
- Beltrán, T.F.; Feliz, M.; Llusar, R.; Mata, J.A.; Safont, V.S. Mechanism of the Catalytic Hydrodefluorination of Pentafluoropyridine by Group Six Triangular Cluster Hydrides Containing Phosphines: A Combined Experimental and Theoretical Study. Organometallics 2011, 30, 290–297. [Google Scholar] [CrossRef]
- Fischer, P.; Götz, K.; Eichhorn, A.; Radius, U. Decisive Steps of the Hydrodefluorination of Fluoroaromatics Using [Ni(NHC)2]. Organometallics 2012, 31, 1374–1383. [Google Scholar] [CrossRef]
- Lv, H.; Zhan, J.-H.; Cai, Y.-B.; Yu, Y.; Wang, B.; Zhang, J.-L. π–π Interaction Assisted Hydrodefluorination of Perfluoroarenes by Gold Hydride: A Case of Synergistic Effect on C–F Bond Activation. J. Am. Chem. Soc. 2012, 134, 16216–16227. [Google Scholar] [CrossRef]
- Chen, Z.; He, C.-Y.; Yin, Z.; Chen, L.; He, Y.; Zhang, X. Palladium-Catalyzed Ortho-Selective C–F Activation of Polyfluoroarenes with Triethylsilane: A Facile Access to Partially Fluorinated Aromatics. Angew. Chem. Int. Ed. 2013, 52, 5813–5817. [Google Scholar] [CrossRef]
- Podolan, G.; Lentz, D.; Reissig, H.-U. Selective Catalytic Hydrdefluorination as a Key Step for the Synthesis of Hitherto Inaccessible Aminopyridine Derivatives. Angew. Chem. Int. Ed. 2013, 52, 9491–9494. [Google Scholar] [CrossRef]
- Arévalo, A.; Tlahuext-Aca, A.; Flores-Alamo, M.; García, J.J. On the Catalytic Hydrodefluorination of Fluoroaromatics Using Nickel Complexes: The True Role of the Phosphine. J. Am. Chem. Soc. 2014, 136, 4634–4639. [Google Scholar] [CrossRef] [PubMed]
- Hein, N.M.; Pick, F.S.; Fryzuk, M.D. Synthesis and Reactivity of a Low-Coordinate Iron(II) Hydride Complex: Applications in Catalytic Hydrodefluorination. Inorg. Chem. 2017, 56, 14513–14523. [Google Scholar] [CrossRef] [PubMed]
- Yow, S.; Gates, S.J.; White, A.J.P.; Crimmin, M.R. Zirconocene Dichloride Catalyzed Hydrodefluorination of Csp3−F bonds. Angew. Chem. Int. Ed. 2012, 51, 12559–12563. [Google Scholar] [CrossRef] [PubMed]
- Ekkert, O.; Strudley, S.D.A.; Rozenfeld, A.; White, A.J.P.; Crimmin, M.R. Rhodium Catalyzed, Carbon−Hydrogen Bond Directed Hydrodefluorination of Fluoroarenes. Organometallics 2014, 33, 7027–7030. [Google Scholar] [CrossRef]
- Aizenberg, M.; Milstein, D. Homogeneous Rhodium Complex-Catalyzed Hydrogenolysis of C–F Bonds. J. Am. Chem. Soc. 1995, 117, 8674–8675. [Google Scholar] [CrossRef]
- Edelbach, B.L.; Jones, W.D. Mechanism of Carbon–Fluorine Bond Activation by (C5Me5)Rh(PMe3)H2. J. Am. Chem. Soc. 1997, 119, 7734–7742. [Google Scholar] [CrossRef]
- Young, R.J., Jr.; Grushin, V.V. Catalytic C–F Bond Activation of Nonactivated Monofluoroarenes. Organometallics 1999, 18, 294–296. [Google Scholar] [CrossRef]
- Braun, T.; Noveski, D.; Ahijado, M.; Wehmeier, F. Hydrodefluorination of pentafluoropyridine at rhodium using dihydrogen: Detection of unusual rhodium hydrido complexes. Dalton Trans. 2007, 34, 3820–3825. [Google Scholar] [CrossRef]
- Konnick, M.M.; Bischof, S.M.; Periana, R.A.; Hashiguchi, B.G. The Hydroxide-Promoted Catalytic Hydrodefluoriation of Fluorocarbons by Ruthenium in Aqueous Media. Adv. Synth. Catal. 2013, 355, 632–636. [Google Scholar] [CrossRef]
- Nakai, H.; Jeong, K.; Matsumoto, T.; Ogo, S. Catalytic C–F Bond Hydrogenolysis of Fluoroaromatics by [(η2-C5Me5)RhI(2,2’-bipyridine)]. Organometallics 2014, 33, 4349–4352. [Google Scholar] [CrossRef]
- Moore, J.T.; Lu, C.C. Catalytic Hydrogenolysis of Aryl C–F Bonds Using a Bimetallic Rhodium–Indium Complex. J. Am. Chem. Soc. 2020, 142, 11641–11646. [Google Scholar] [CrossRef] [PubMed]
- Matsunami, A.; Kuwata, S.; Kayaki, Y. Hydrodefluorination of Fluoroarenes Using Hydrogen Transfer Catalysts with a Bifunctional Iridium/NH Moiety. ACS Catal. 2016, 6, 5181–5185. [Google Scholar] [CrossRef]
- Matsunami, A.; Kuwata, S.; Kayaki, Y. Hydrogen Evolution from Formic Acid and Hydrodefluorination of Fluoroarenes by Bifunctional Iridium Catalysts—Beyond the Transfer Hydrogenation. J. Synth. Org. Chem. Jpn. 2018, 76, 315–324. [Google Scholar] [CrossRef]
- Matsunami, A.; Kayaki, Y.; Kuwata, S.; Ikariya, T. Nucleophilic Aromatic Substitution in Hydrodefluorination Exemplified by Hydridoiridium(III) Complexes with Fluorinated Phenylsulfonyl-1,2-diphenylethylenediamine Ligands. Organometallics 2018, 37, 1958–1969. [Google Scholar] [CrossRef]
- Dub, P.A.; Wang, H.; Matsunami, A.; Gridnev, I.D.; Kuwata, S.; Ikariya, T. C−F Bond Breaking through Aromatic Nucleophilic Substitution with a Hydroxo Ligand Mediated via Water Bifunctional Activation. Bull. Chem. Soc. Jpn. 2013, 86, 557–568. [Google Scholar] [CrossRef] [Green Version]
- Dub, P.A.; Matsunami, A.; Kuwata, S.; Kayaki, Y. Cleavage of N−H Bond of Ammonia via Metal–Ligand Cooperation Enables Rational Design of a Conceptually New Noyori-Ikariya Catalyst. J. Am. Chem. Soc. 2019, 141, 2661–2677. [Google Scholar] [CrossRef]
- McKay, D.; Riddlestone, I.M.; Macgregor, S.A.; Mahon, M.F.; Whittlesey, M.K. Mechanistic Study of Ru-NHC-Catalyzed Hydrodefluorination of Fluoropyridines: The Influence of the NHC on the Regioselectivity of C–F Activation and Chemoselectivity of C–F versus C–H Bond Cleavage. ACS Catal. 2015, 5, 776–787. [Google Scholar] [CrossRef] [Green Version]
- Cybulski, M.K.; McKay, D.; Macgregor, S.A.; Mahon, M.F.; Whittlesey, M.K. Room Temperature Regioselective Catalytic Hydrodefluorination of Fluoroarenes with trans-[Ru(NHC)4H2] through a Concerted Nucleophilic Ru–H Attack Pathway. Angew. Chem. Int. Ed. 2017, 56, 1515–1519. [Google Scholar] [CrossRef] [Green Version]
- Kvíčala, J.; Beneš, M.; Paleta, O.; Král, V. Regiospecific nucleophilic substitution in 2,3,4,5,6-pentafluorobiphenyl as model compound for supramolecular systems. Theoretical study of transition states and energy profiles, evidence for tetrahedral SN2 mechanism. J. Fluorine Chem. 2010, 131, 1327–1337. [Google Scholar] [CrossRef]
- Kikushima, K.; Koyama, H.; Kodama, K.; Dohi, T. Nucleophilic Aromatic Substitution of Polyfluoroarene to Access Highly Functionalized 10-Phenylphenothiazine Derivatives. Molecules 2021, 26, 1365. [Google Scholar] [CrossRef]
- Zimmerman, H.E. Orientation in Metal Ammonia Reduction. Tetrahedron 1961, 16, 169–176. [Google Scholar] [CrossRef]
- Chambers, R.D.; Seabury, M.J.; Lyn, D.; Williams, H.; Hughes, N. Mechanisms for Reactions of Halogenated Compounds. Part 5. Orientating Effects of Fluorine Substituents on Nucleophilic Substitution in Naphthalene and Other Polycyclic Systems. J. Chem. Soc. PerkinTrans. 1988, 1, 251–254. [Google Scholar] [CrossRef]
- Baker, J.; Muir, M. The Meisenheimer model for predicting the principal site for nucleophilic substitution in aromatic perfluorocarbons—Generalization to include ring-nitrogen atoms and non-fluorine ring substituents. Can. J. Chem. 2010, 88, 588–597. [Google Scholar] [CrossRef]
- Liljenberg, M.; Brinck, T.; Herschend, B.; Rein, T.; Tomasi, S.; Svensson, M. Predicting Regioselectivity in Nucleophilic Aromatic Substitutiton. J. Org. Chem. 2012, 77, 3262–3269. [Google Scholar] [CrossRef] [PubMed]
- Liljenberg, M.; Brinck, T.; Rein, T.; Svensson, M. Utilizing the σ-complex stability for quantifying reactivity in nucleophilic substitution of aromatic fluorides. Beilstein J. Org. Chem. 2013, 9, 791–799. [Google Scholar] [CrossRef]
- Sadowsky, D.; McNeill, K.; Cramer, C.J. Thermochemical Factors Affecting Dehalogenation of Aromatics. Environ. Sci. Technol. 2013, 47, 14194–14203. [Google Scholar] [CrossRef]
- Sadowsky, D.; McNeill, K.; Cramer, C.J. Dehalogenation of Aromatics by Nucleophilic Aromatic Substitution. Environ. Sci. Technol. 2014, 48, 10904–10911. [Google Scholar] [CrossRef]
- Crucius, G.; Lyubimtsev, A.; Kramer, M.; Hanack, M.; Ziegler, T. 1,4,8,11,15,18,22,25-Octafluorophthalocyaninato Zinc (F8PcZn). Synlett 2012, 23, 2501–2503. [Google Scholar]
- Banks, R.E.; Burgess, J.E.; Cheng, W.M.; Haszeldine, R.N. Heterocyclic Polyfluoro-Compounds. Part IV. Nucleophilic Substitution in Pentafluoropyridine: The Preparation and Properties of Some 4-Substituted 2,3,5,6-Tetrafluoropyridines. J. Chem. Soc. 1965, 67, 575–581. [Google Scholar] [CrossRef]
- Schoch, T.D.; Mondal, M.; Weaver, J.D. Catalyst-Free Hydrodefluorination of Perfluoroarenes with NaBH4. Org. Lett. 2021, 23, 1588–1593. [Google Scholar] [CrossRef]
- Beregovaya, I.V.; Shchegoleva, L.N. Potential Energy Surface and dissociative cleavage of chlorobenzene radial anion. Chem. Phys. Lett. 2001, 348, 501–506. [Google Scholar] [CrossRef]
- Marcé, P.; Lynch, J.; Blacker, A.J.; Williams, J.M.J. A mild hydration of nitriles catalyzed by copper(II) acetate. Chem. Commun. 2016, 52, 1436–1438. [Google Scholar] [CrossRef] [PubMed]
- Ciganek, E.J. Tertiary Carbinamines by Addition of Organocerium Reagents to Nitriles and Ketimines. Org. Chem. 1992, 57, 4521–4527. [Google Scholar] [CrossRef]
- Arita, S.; Koike, T.; Kayaki, Y.; Ikariya, T. Synthesis and Reactivities of Cp*Ir Amide and Hydride Complexes Bearing C−N Chelate Ligands. Organometallics 2008, 27, 2795–2802. [Google Scholar] [CrossRef]
- Sato, Y.; Kawata, Y.; Yasui, S.; Kayaki, Y.; Ikariya, T. New Bifunctional Bis(azairidacycle) with Axial Chirality via Double Cyclometalation of 2,2’-Bis(aminomethyl)-1,1’-binaphthyl. Molecules 2021, 26, 1145. [Google Scholar] [CrossRef] [PubMed]
- Gallardo-Macias, R.; Kumar, P.; Jaskowski, M.; Richmann, T.; Shrestha, R.; Russo, R.; Singleton, E.; Zimmerman, M.D.; Ho, H.P.; Dartois, V.; et al. Optimization of N-benzyl-5-nitrofuran-2-carboxamide as an antitubercular agent. Bioorg. Med. Chem. Lett. 2019, 29, 601–606. [Google Scholar] [CrossRef] [PubMed]
- Jaudzems, K.; Tars, K.; Maurops, G.; Ivdra, N.; Otikovs, M.; Leitans, J.; Kanepe-Lapsa, I.; Domraceva, I.; Mutule, I.; Trapencieris, P.; et al. Plasmepsin Inhibitory Activity and Structure-Guided Optimization of a Potent Hydroxyethylamine-Based Antimalarial Hit. ACS Med. Chem. Lett. 2014, 5, 373–377. [Google Scholar] [CrossRef] [Green Version]
- Rigaku Coorporation. CrystalStructure 4.1: Crystal Structure Analysis Package; Rigaku Coorporation: Tokyo, Japan, 2015. [Google Scholar]
- Altomare, A.; Cascarano, G.; Giacovazzo, C.; Guagliardi, A.; Burla, M.; Polidori, G.; Camalli, M. SIR92—A program for automatic solution of crystal structures by direct methods. J. Appl. Cryst. 1994, 27, 435. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C 2015, 71, 3–8. [Google Scholar] [CrossRef]
Scheme 1.
Transfer hydrogenative defluorination catalyzed by azairidacycle 1.

Scheme 2.
Cyclometalation of hydridoiridium bearing N-pentafluorobenzenesulfonylated 1,2-diphenylethylenediamine via intramolecular hydrodefluorination.
Scheme 2.
Cyclometalation of hydridoiridium bearing N-pentafluorobenzenesulfonylated 1,2-diphenylethylenediamine via intramolecular hydrodefluorination.
Scheme 3.
Destabilization by vicinal electron-pair repulsion through nucleophilic attack on the para position of a fluorine atom.
Scheme 3.
Destabilization by vicinal electron-pair repulsion through nucleophilic attack on the para position of a fluorine atom.

Figure 1.
ORTEP drawings of 7a (left; (a)) and 10 (right; (b)). Thermal ellipsoids are shown at the 30% probability level.
Figure 1.
ORTEP drawings of 7a (left; (a)) and 10 (right; (b)). Thermal ellipsoids are shown at the 30% probability level.
Scheme 4.
Cu(OAc)2-catalyzed hydration of 9b.

Scheme 5.
Synthesis of new azairidacycles 12a and 12b.
Scheme 6.
Comparison of the catalytic activities of 1, 12a, and 12b for the transfer hydrogenative defluorination of pentafluoropyridine with HCOOK.
Scheme 6.
Comparison of the catalytic activities of 1, 12a, and 12b for the transfer hydrogenative defluorination of pentafluoropyridine with HCOOK.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Synthesis of partially fluorinated arenes by catalytic transfer hydrogenative defluorination with HCOOK using 1 1.
Table 1.
Synthesis of partially fluorinated arenes by catalytic transfer hydrogenative defluorination with HCOOK using 1 1.
| Entry | Substrate | HCOOK, eq | Time, h | % Yield 2 | |
|---|---|---|---|---|---|
| 1 |  2a | 2 | 1 | 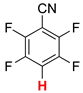 3a, 100 | |
| 2 | 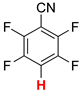 3a | 2 | 12 |  4, 20 | |
| 3 3 | 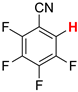 3b | 2 | 2 | 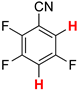 4, 100 | |
| 4 4 | C6F6 2b | 2 | 12 | N.R. | |
| 5 |  5a | 1 | 2 |  6a, 7 |  7a, 38 |
| 6 | 5a | 3 | 2 | 6a, 0 | 7a, 99 |
| 7 | 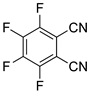 5b | 1 | 1 | 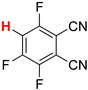 6b, 90(88) 5 | 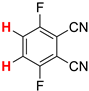 7b, 2 |
| 8 | 5b | 3 | 1 | 6b, 0 | 7b, 100(86) 5 |
| 9 |  8a | 2 | 1 | 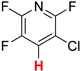 9a, 100 | |
| 10 | 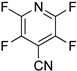 8b | 2 | 1 | 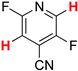 9b, 98 | |
1 Standard conditions: Substrate (0.5 mmol), 1 (5.0 × 10−3 mmol), 1,2-dimethoxyethane (2.5 mL), H2O (2.5 mL), 30 °C. 2 Determined by 19F NMR using trifluoromethylbenzene as an internal standard. 3 Catalyst loading = 2 mol%. 4 Catalyst loading = 5 mol%. 5 Isolated yield in parenthesis.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Matsunami, A.; Kuwata, S.; Kayaki, Y. Regioselective Transfer Hydrogenative Defluorination of Polyfluoroarenes Catalyzed by Bifunctional Azairidacycle. Organics 2022, 3, 150-160. https://doi.org/10.3390/org3030012
AMA Style
Matsunami A, Kuwata S, Kayaki Y. Regioselective Transfer Hydrogenative Defluorination of Polyfluoroarenes Catalyzed by Bifunctional Azairidacycle. Organics. 2022; 3(3):150-160. https://doi.org/10.3390/org3030012
Chicago/Turabian StyleMatsunami, Asuka, Shigeki Kuwata, and Yoshihito Kayaki. 2022. "Regioselective Transfer Hydrogenative Defluorination of Polyfluoroarenes Catalyzed by Bifunctional Azairidacycle" Organics 3, no. 3: 150-160. https://doi.org/10.3390/org3030012