
Synthesis of Indole-Based Derivatives Containing Ammonium Salts, Diamines and Aminoureas for Organocatalysis

 ,
,  , and
, and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
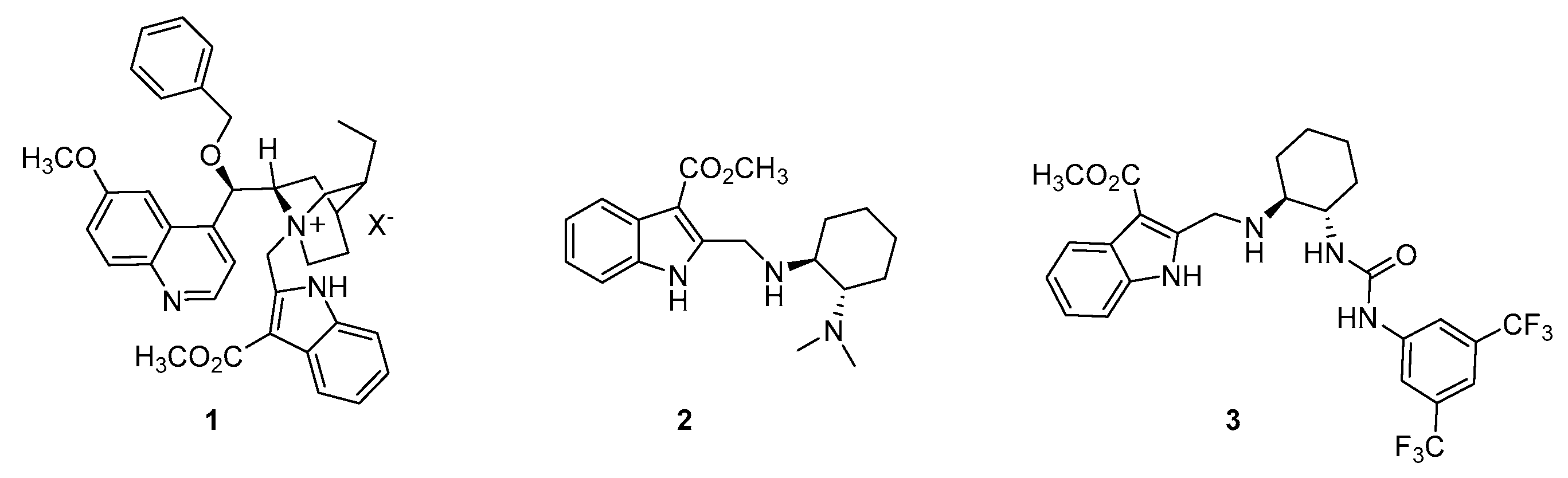
2. Results and Discussion
3. Materials and Methods
3.1. General Experiment
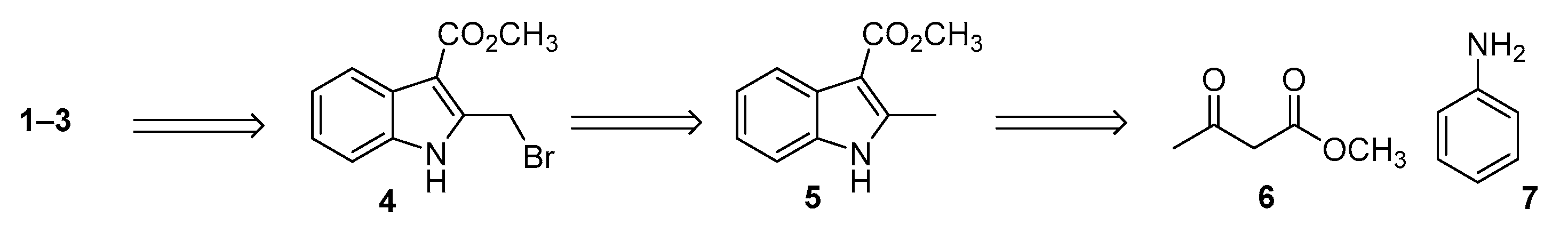
3.2. Synthetic Protocols for the Preparation of Key Synthon 4
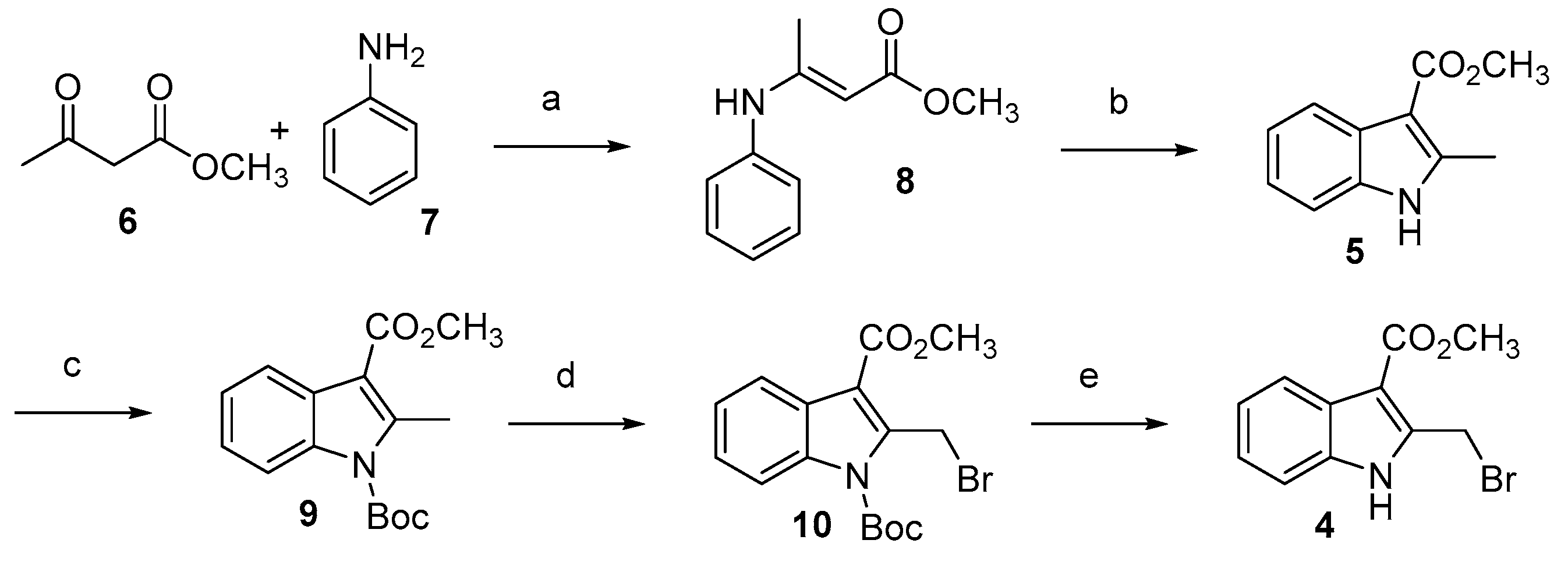
3.2.1. Synthesis of Methyl (E)-3-(Phenylimino)butanoate 8
3.2.2. Synthesis of Methyl 2-Methilindole-3-carboxylate 5
3.2.3. Synthesis of 1-(Tert-butyl) 3-Methyl 2-Methyl-1H-indole-1,3-dicarboxylate 9
3.2.4. Synthesis of 1-(Tert-butyl) 3-Methyl 2-(Bromomethyl)-1H-indole-1,3-dicarboxylate 10
3.2.5. Synthesis of Methyl 2-(Bromomethyl)-1H-indole-3-carboxylate 4
3.3. Synthetic Protocols for the Preparation of Quaternary Ammonium Salt 1
3.3.1. Synthesis of O-Benzyl Hydroquinine 12
3.3.2. Synthesis of (1S,2S)-2-((R)-(Benzyloxy)(6-methoxyquinolin-4-yl)methyl)-5-ethyl-1-((3-(methoxycarbonyl)-1H-indol-2-yl)methyl)quinuclidin-1-ium Bromide 1
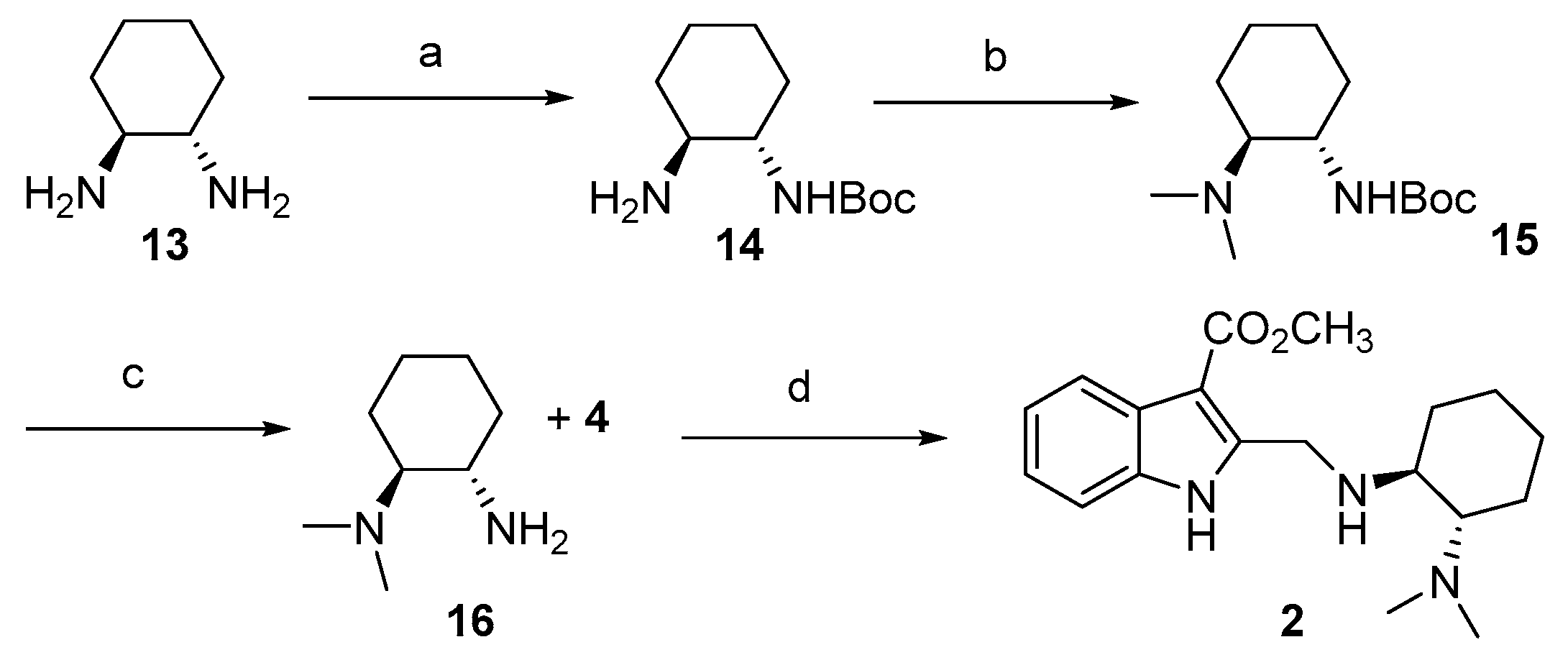
3.4. Synthetic Protocols for the Preparation of Indolediamine 2
3.4.1. Synthesis of (1S,2S)-Trans-N-Boc-1,2-cyclohexanediamine 14
3.4.2. Synthesis of Tert-butyl ((1S,2S)-2-(Dimethylamino)cyclohexyl)carbamate 15
3.4.3. Synthesis of (1S,2S)-N1,N1-Dimethylcyclohexane-1,2-diamine 16
3.4.4. Synthesis of Methyl 2-((((1S,2S)-2-(Dimethylamino)cyclohexyl)amino)methyl)-1H-indole-3-carboxylate 2
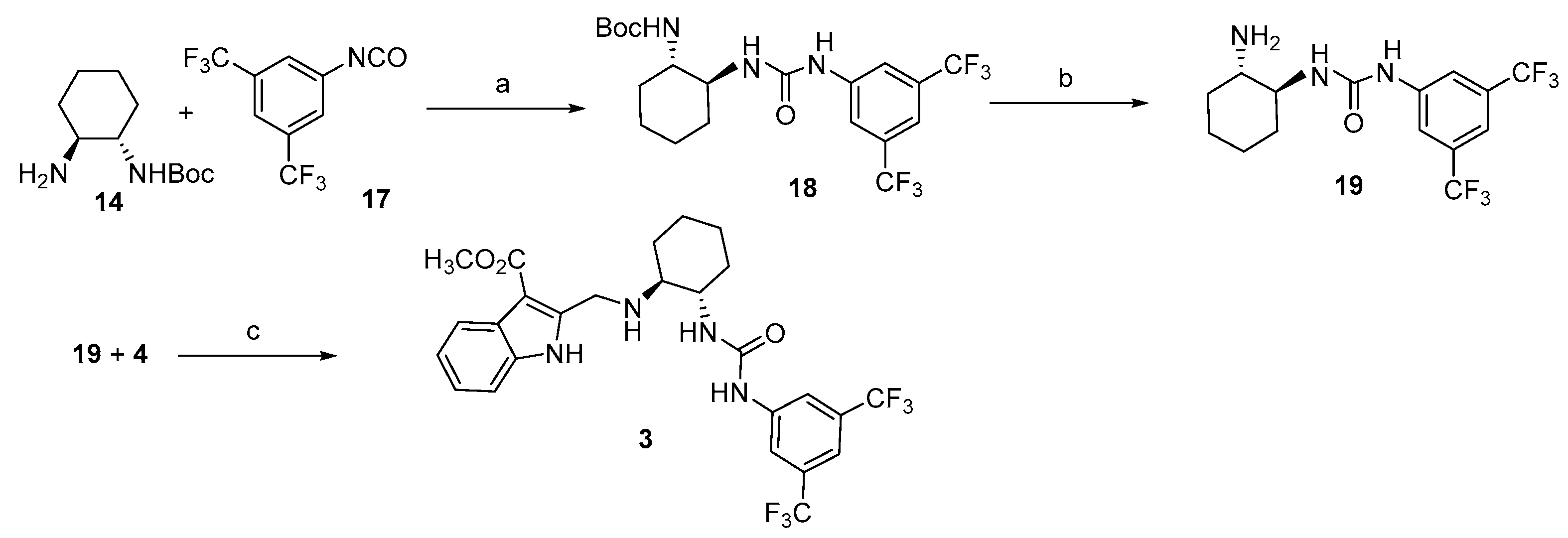
3.5. Synthetic Protocols for the Preparation of Indoleaminourea 3
3.5.1. Synthesis of 1-((1S,2S)-2-Aminocyclohexyl)-3-(3,5-bis(trifluoromethyl)phenyl)urea 19
3.5.2. Synthesis of Methyl 2-((((1S,2S)-2-(3-(3,5-Bis(trifluoromethyl)phenyl)ureido)cyclohexyl)amino)methyl)-1H-indole-3-carboxylate 3
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Nagendra, K.K.; Neha, K.; Pankaj, A.; Naresh, K.; Chung, H.K.; Akhilesh, K.V.; Eun, H.C. Biomedical Importance of Indoles. Molecules 2013, 6, 6620–6662. [Google Scholar]
- Archana, K.; Rajesh, K.S. Medicinal chemistry of indole derivatives: Current to future therapeutic perspectives. Bioorg. Chem. 2019, 89, 103021–103036. [Google Scholar]
- James, P.N.; Snyder, H.R. Indole-3-aldehyde. Org. Synth. 1959, 39, 30–32. [Google Scholar]
- Heaney, H.; Ley, S.V. 1-Benzylindole. Org. Synth. 1974, 54, 54–58. [Google Scholar]
- Bergman, J.; Venemalm, L. Efficient synthesis of 2-chloro-,2-bromo-, and 2-iodoindole. J. Org. Chem. 1992, 57, 2495–2497. [Google Scholar] [CrossRef]
- Katritzky, A.R.; Li, J.; Stevens, C. Facile synthesis of 2-substituted indoles and indolo[3,2-b] carbazoles from 2-(benzotriazol-1-ylmethyl) indole. J. Org. Chem. 1995, 60, 3401–3404. [Google Scholar] [CrossRef]
- Ximenes, V.F.; Campa, A.; Catalani, L.H. The oxidation of indole derivatives catalysed by horseradish peroxidase is highly chemiluminescent. Arch. Biochem. Biophys. 2001, 387, 173–179. [Google Scholar]
- Lynch, S.M.; Bur, S.K.; Padwa, A. Intramolecular amidofuran cycloadditions across an indole pi-bond: An efficient approach to the aspidosperma and strychnos ABCE core. Org. Lett. 2002, 4, 4643–4645. [Google Scholar] [CrossRef]
- Chaudhuri, A.; Haldar, S.; Sun, S.; Koeppe II, R.E.; Chattopadhyay, A. Importance of indole NH hydrogen bonding in the organization and dynamics of gramicidin channels. Biochim. Biophys. Acta 2013, 1838, 419–428. [Google Scholar] [CrossRef]
- Chen, T.; Foo, T.J.Y.; Yeung, Y.Y. Indole-Catalyzed Bromolactonization in Lipophilic Solvent: A Solid− Liquid Phase Transfer Approach. ACS Catal. 2015, 5, 4751–4755. [Google Scholar] [CrossRef]
- Shi, Y.; Ke, Z.; Yeung, Y.Y. Environmentally benign indole-catalyzed position- selective halogenation of thioarenes and other aromatic. Green Chem. 2018, 20, 4448–4452. [Google Scholar] [CrossRef]
- Wong, J.; Ke, Z.; Yeung, Y.Y. Lipophilic indole mediated chemoselective a-monobromination of 1,3-dicarbonyl compounds. Tetrahedron Lett. 2020, 61, 151772–151775. [Google Scholar] [CrossRef]
- Shi, Y.; Wong, J.; Ke, Z.; Yeung, Y.Y. Lipophilic Indole-Catalyzed Intermolecular Bromoesterification of Olefins in Nonpolar Media. J. Org. Chem. 2019, 84, 4017–4024. [Google Scholar] [CrossRef] [PubMed]
- Bencivenni, G.; Saraiva Rosa, N.; Grieco, P.; Gillick-Healy, M.W.; Kelly, B.G.; Twamley, B.; Adamo, M.F.A. Quaternary Ammonium Salts Interact with Enolates and Sulfonates via Formation of Multiple +N-C-H Hydrogen Bonding Interactions. Catalysts 2022, 12, 803. [Google Scholar] [CrossRef]
- Bencivenni, G.; Salazar Illera, D.; Moccia, M.; Houk, K.N.; Izzo, J.A.; Novacek, J.; Grieco, P.; Vetticatt, M.J.; Waser, M.; Adamo, M.F.A. Study of Ground State Interactions of Enantiopure Chiral Quaternary Ammonium Salts and Amides, Nitroalkanes, Nitroalkenes, Esters, Heterocycles, Ketones and Fluoroamides. Chem. Eur. J. 2021, 27, 11354–11366. [Google Scholar] [CrossRef]
- Salazar Illera, D.; Pacifico, R.; Adamo, M.F.A. Asymmetric Phase Transfer Catalysed Michael Addition of γ-Butenolide and N-Boc-Pyrrolidone to 4-Nitro-5-styrylisoxazoles. Catalysts 2022, 12, 634. [Google Scholar] [CrossRef]
- Destro, D.; Bottinelli, C.; Ferrari, L.; Albanese, D.C.A.; Bencivenni, G.; Gillick Healy, M.; Kelly, B.; Adamo, M.F.A. Enantioselective Synthesis of 3, 4-Dihydropyran-2-ones via Phase-Transfer-Catalyzed Addition–Cyclization of Acetylacetone to Cinnamic Thioesters. J. Org. Chem. 2020, 85, 5183–5192. [Google Scholar] [CrossRef]
- Del Fiandra, C.; Piras, L.; Fini, F.; Disetti, P.; Moccia, M.; Adamo, M.F.A. Phase transfer catalyzed enantioselective cyclopropanation of 4-nitro-5-styrylisoxazoles. Chem. Commun. 2012, 48, 3863–3865. [Google Scholar]
- Baschieri, A.; Bernardi, L.; Ricci, A.; Suresh, S.; Adamo, M.F.A. Catalytic Asymmetric Conjugate Addition of Nitroalkanes to 4-Nitro-5-styrylisoxazoles. Angew. Chem. Int. Ed. 2009, 48, 9342–9345. [Google Scholar] [CrossRef]
- Pineda, A.; Carr, J.; Rodriguez-Padron, D.; Lazaro Ronco, N.; Fox, K.; Gonzalez-Arellano, C.; Gillick-Healy, M.W.; Kelly, B.G.; Adamo, M.F.A.; Luque, R. A continuous flow approach for the desulfurative bromination of sulfides. Sustain. Chem. Pharm. 2024, 38, 101490–101495. [Google Scholar] [CrossRef]
- Canestrani, D.; Cioffi, C.; Biancofiore, I.; Lancianesi, S.; Ghisu, L.; Reuther, M.; O´Brien, J.; Adamo, M.F.A.; Ibrahim, H. Sulphide as a leaving group: Highly stereoselective bromination of alkyl phenyl sulphides. Chem. Sci. 2019, 10, 9042–9050. [Google Scholar] [CrossRef] [PubMed]
- Alletto, F.; Adamo, M.F.A. Enantiospecific on-water bromination: A mild and efficient protocol for the preparation of alkyl bromides. Green Chem. 2020, 22, 8692–8698. [Google Scholar] [CrossRef]
- Canestrari, D.; Lancianesi, S.; Badiola, E.; Strinna, C.; Ibrahim, H.; Adamo, M.F.A. Desulfurative chlorination of alkyl phenyl sulfides. Org. Lett. 2017, 19, 918–921. [Google Scholar] [PubMed]
- Würtz, S.; Rakshit, S.; Neumann, J.J.; Dröge, T.; Glorius, F. Palladium-Catalyzed Oxidative Cyclization of N-Aryl Enamines: From Anilines to Indoles. Angew. Chem. Int. Ed. 2008, 38, 7230–7233. [Google Scholar]
- Patil, S.A.; Patil, R.; Miller, D.D. Microwave-assisted synthesis of medicinally relevant indoles. Curr. Med. Chem. 2011, 4, 615–637. [Google Scholar] [CrossRef] [PubMed]
- Nigam, V.; Singh, S.; Kasana, S.; Kumar, S.; Das Kurmi, B.; Das Gupta, G.; Patel, P. Revolutionizing Indole Synthesis: A Microwave-Powered Approach. Chem. Select 2024, 9, e202402171. [Google Scholar] [CrossRef]
- Huang, Y.; Yang, Y.; Song, H.; Liu, Y.; Wang, Q. Synthesis of Structurally Diverse 2,3-Fused Indoles via Microwave-Assisted AgSbF6-Catalysed Intramolecular Difunctionalization of o-Alkynylanilines. Sci. Rep. 2015, 5, 13516. [Google Scholar] [CrossRef]
- Bellavita, R.; Casertano, M.; Grasso, N.; Gillick-Healy, M.W.; Kelly, B.G.; Adamo, M.F.A.; Menna, M.; Merlino, F.; Grieco, P. Microwave-Assisted synthesis of 2-methyl-1H-indole-3-carboxylate derivatives via Pd-catalysed heterocyclisation. Symmetry 2022, 3, 435–445. [Google Scholar]
- Kohler, M.C.; Yost, M.J.; Garnsey, M.R.; Coltart, D.M. Direct Carbon−Carbon Bond Formation via Soft Enolization: A Biomimetic Asymmetric Mannich Reaction of Phenylacetate Thioesters. Org. Lett. 2010, 15, 3376–3379. [Google Scholar]
- Bennani, Y.L.; Hanessian, S. trans-1,2-Diaminocyclohexane Derivatives as Chiral Reagents, Scaffolds, and Ligands for Catalysis: Applications in Asymmetric Synthesis and Molecular Recognition. Chem. Rev. 1997, 97, 3161–3195. [Google Scholar]
- Kopyt, M.; Glowacki, M.P. Trans-1,2-Diaminocyclohexane and Its Derivatives in Asymmetric Organocatalysis. In Chiral Building Blocks in Asymmetric Synthesis: Synthesis and Applications; Wiley VCH, GmbH: Weinheim, Germany, 2022; ISBN 9783527349463/9783527834204. [Google Scholar]
- Lee, D.W.; Ha, H.J.; Lee, W.K. Selective Mono-BOC Protection of Diamines. Synth. Commun. 2007, 37, 737–742. [Google Scholar]
- Imperatore, C.; Valadan, M.; Tartaglione, L.; Persico, M.; Ramunno, A.; Menna, M.; Casertano, M.; Dell’Aversano, C.; Singh, M.; d’Aulisio Garigliota, M.L.; et al. Exploring the Photodynamic Properties of Two Antiproliferative Benzodiazopyrrole Derivatives. Int. J. Mol. Sci. 2020, 21, 1246. [Google Scholar] [CrossRef] [PubMed]
- Casertano, M.; Genovese, M.; Piazza, L.; Balestri, F.; Del Corso, A.; Vito, A.; Paoli, P.; Santi, A.; Imperatore, C.; Menna, M. Identifying Human PTP1B Enzyme Inhibitors from Marine Natural Products: Perspectives for Developing of Novel Insulin-Mimetic Drugs. Pharmaceuticals 2022, 15, 325. [Google Scholar] [CrossRef]
- Amarasinghe, N.R.; Turner, P.; Todd, M.H. The First Catalytic, Enantioselective Aza-Henry Reaction of an Unactivated Cyclic Imine. Adv. Synth. Catal. 2012, 354, 2954–2958. [Google Scholar]
- Bulman Page, P.; Farah, M.; Buckley, B.; Chan, Y.; Blacker, A. Preparation of C2-Symmetric Biaryl Bisiminium Salts and Their Use as Organocatalysts for Asymmetric Epoxidation. Synlett 2015, 27, 126–130. [Google Scholar] [CrossRef]
- Berkessel, A.; Seeling, B. A simplified synthesis of Takemoto’s catalyst. Synthesis 2009, 12, 2113–2115. [Google Scholar] [CrossRef]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Casertano, M.; Kelly, B.G.; Gillick-Healy, M.W.; Grieco, P.; Adamo, M.F.A. Synthesis of Indole-Based Derivatives Containing Ammonium Salts, Diamines and Aminoureas for Organocatalysis. Organics 2025, 6, 15. https://doi.org/10.3390/org6020015
Casertano M, Kelly BG, Gillick-Healy MW, Grieco P, Adamo MFA. Synthesis of Indole-Based Derivatives Containing Ammonium Salts, Diamines and Aminoureas for Organocatalysis. Organics. 2025; 6(2):15. https://doi.org/10.3390/org6020015
Chicago/Turabian StyleCasertano, Marcello, Brian G. Kelly, Malachi W. Gillick-Healy, Paolo Grieco, and Mauro F. A. Adamo. 2025. "Synthesis of Indole-Based Derivatives Containing Ammonium Salts, Diamines and Aminoureas for Organocatalysis" Organics 6, no. 2: 15. https://doi.org/10.3390/org6020015
APA StyleCasertano, M., Kelly, B. G., Gillick-Healy, M. W., Grieco, P., & Adamo, M. F. A. (2025). Synthesis of Indole-Based Derivatives Containing Ammonium Salts, Diamines and Aminoureas for Organocatalysis. Organics, 6(2), 15. https://doi.org/10.3390/org6020015