Three-Dimensional Imaging of Plant Cell Wall Deconstruction Using Fluorescence Confocal Microscopy



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Experimental Design
2.1. Materials
- Acetic acid (Carlo Ebra, Barcelona, Spain, 401422)
- Citric acid (Merck, Darmstadt, Germany, 60910)
- Sodium citrate (Fluka, 71183)
- Monobasic sodium phosphate (Sigma, St. Louis, MO, USA, 71500)
- Dibasic sodium phosphate (Sigma Aldrich, St. Louis, MO, USA, S2002)
- Ethanol (VWR Prolabo, 2082132)
- Sulphuric acid (Carlo Erba, SDS, 410301)
- Glycerol (sigma ALDRICH, 9012)
- Sodium acetate (Sigma, W302406)
- Cellic® CTec2 (Novozymes A/S Bagsværd, Denmark)
- Azide sodium (Sigma Aldrich, 769320)
2.2. Equipment
- Confocal laser scanning microscope (Leica TCS SP8 inverted, Wetzlar, Germany) equipped with a laser diode at 405 nm
- Oil immersion objective 63× with a N.A. = 1.4 (Leica, Germany)
- Sliding microtome (Stemi 1000, Zeiss, Oberkochen, Germany)
- Lauda heater (oil bath) (Lauda-Königshofen, Germany)
- Petri dish 35 mm × 10 mm (Greiner Bio-one, Frickenhausen, Germany)
- Spectrophotometer (Shimadzu UV 2401PC, Kyoto, Japan)
- Microscope adapted incubator chamber (H301-Mini-OKOLab, Pozzuoli, Italy)
- Weight balance (Mettler Teledo PE 2000, Columbus, OH, USA)
- Vacuum pump (Vacuubrand 1C, Wertheim, Germany)
- Poplar blocks (Poplus nigra x deltoides, INRA Estrées-Mons, France)
- Filter paper Whatman (1001090)
- Razor blade (Agar Scientific, Stansted, UK)
- Teflon cups and covers (Parr, Moline, IL, USA)
- Mineralization bombs (Parr, USA)
- Microtome blade (Agar Scientific, UK)
- Gene frame® 65 µL (Thermo Scientific, Waltham, MA, USA)
- Microscopy cover glass (MARIENFELD®, Lauda-Königshofen, Germany, NO. 1.5H)
- Microscopy slide (Starfrost®, Braunschweig, Germany)
- Lens cleaning tissue (Fisher Scientific)
- Vacuum dessicator (Fisher Scientific)
- Brush
2.3. Software
3. Procedure
3.1. Reagent Setup
- Buffer: sodium citrate buffer (0.05 M, pH 5). Weigh 21.01 g of citric acid and dissolve in 1000 mL. Weigh 29.41 g of sodium citrate and dissolve in 1000 mL of ultrapure water. Mix 205 mL of 0.1 M solution of citric acid and 295 mL of 0.1 M solution of sodium citrate. Adjust the final volume with ultrapure water volume to 1000 mL.
- Buffer: sodium acetate buffer (0.05 M, pH 5). Weight 5.77 g of acetic acid and dissolve in 1000 mL. Weigh 8.2 g of sodium citrate and dissolve in 1000 mL of ultrapure water. Mix 148 mL of the 0.1 M solution of acetic acid and 352 mL of 0.1 M solution of sodium citrate. Adjust the final volume with ultrapure water to 1000 mL.
- Buffer: sodium phosphate buffer (0.05 M, pH 5). Weigh 13.9 g of monobasic sodium phosphate and dissolve in 1000 mL of ultrapure water. Weigh 26.82 g of dibasic sodium phosphate and dissolve in 1000 mL of ultrapure water. Mix 93.5 mL of the 0.1 M solution of monobasic sodium phosphate and 6.5 mL of 0.1 M solution of dibasic sodium phosphate. Adjust the final volume with ultrapure water to 1000 mL.
- Using a sensitive pH-meter, check that the pH is close to 5.0, and then store the buffers at 4 °C.
- Sulfuric acid (2%) (v/v), add 24 mL of sulfuric acid (96%) to 977.91 mL of ultrapure water.
- Sodium azide (0.02%), add 40 µL to 199.96 mL of distilled water.
- Ethanol (50%) (v/v), add 500 mL of ethanol absolute to 500 mL of distilled water.
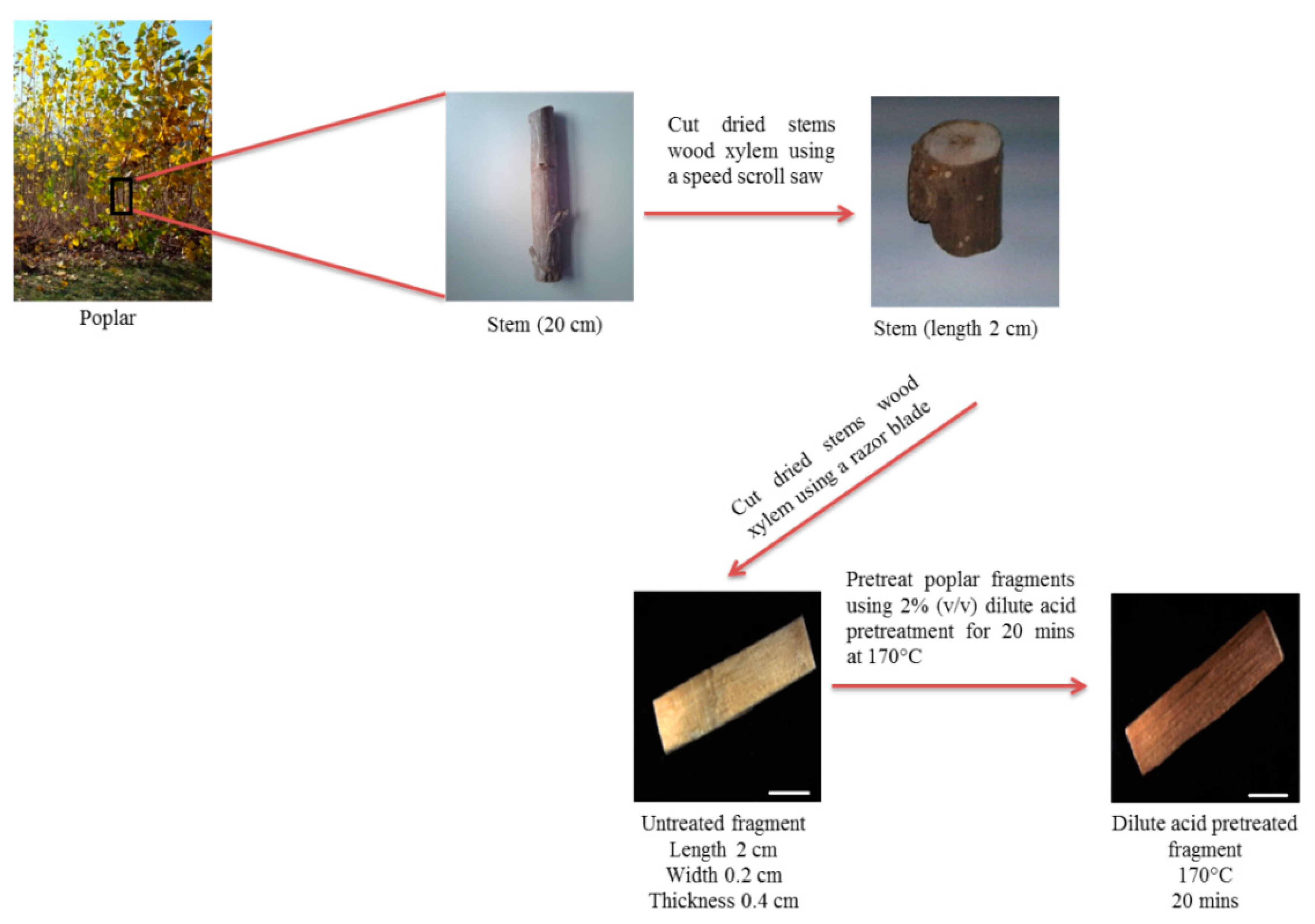
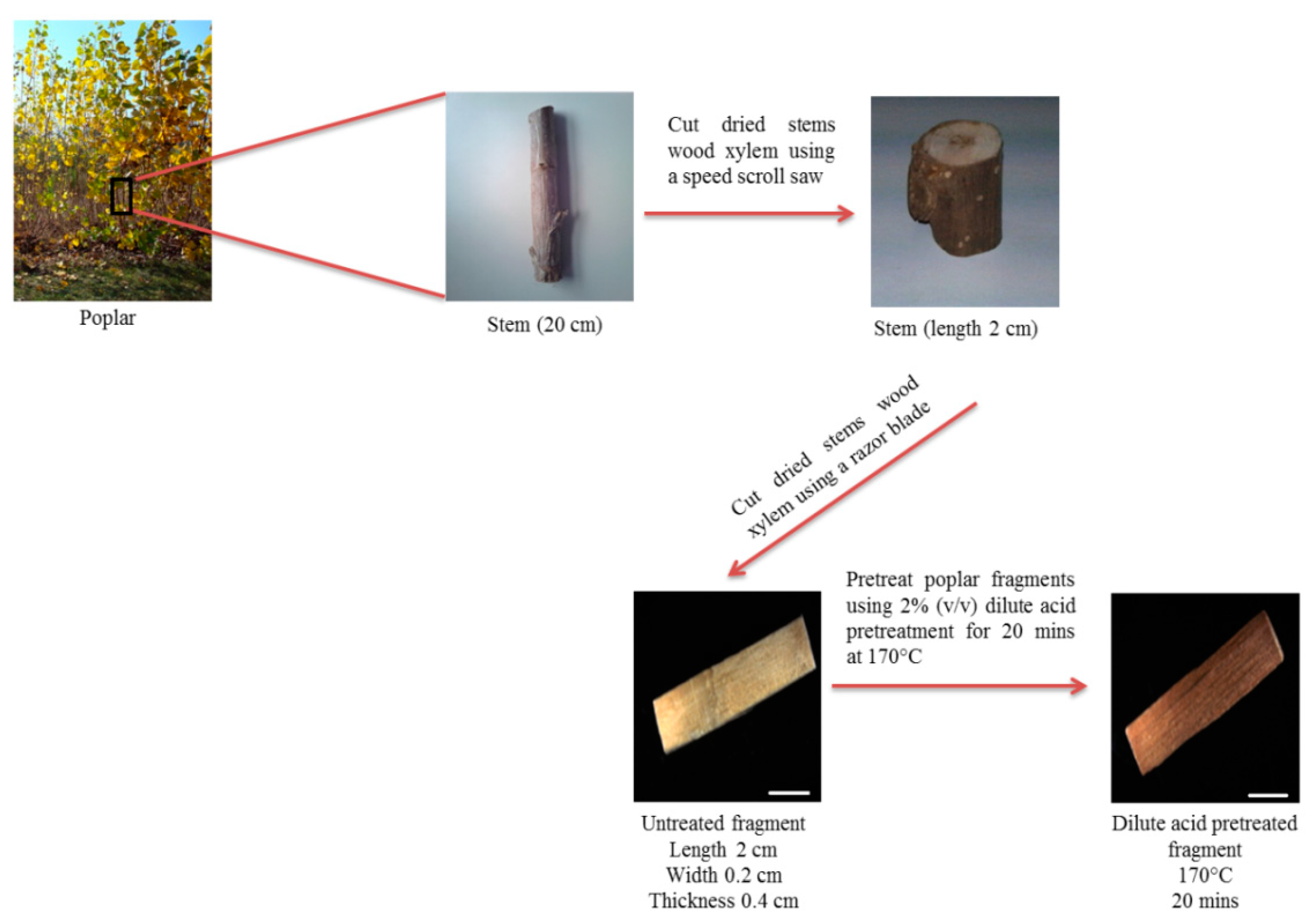
3.2. Sample Preparation
- i.
- Cut dried stems wood xylem of poplar into segments of 2 cm long using a speed scroll saw.
- ii.
- Cut dried stems into fragments of 0.2 cm wide and 0.4 cm thickness using a razor blade.
- iii.
- Place poplar fragments (500 mg) in glass vials containing ultrapure water and put them in a vacuum desiccator and vacuum infiltrate for 3 × 15 min and then incubate them overnight at 4 °C in order to ensure a better diffusion of the reagents during the pretreatment.
- iv.
- Remove the fragments from the vials and wipe them with a paper towel.
- v.
- Place poplar fragments in 2% (v/v) sulfuric acid into mineralization bombs (500 mg per bomb) equipped with Teflon cups and cover them for pretreatment.
- vi.
- Incubate mineralization bombs at 170 °C for 20 min in an oil bath.
- vii.
- Cool down the mineralization bombs in ice for 5 min to stop the acid hydrolysis.
- viii.
- Wash the pretreated fragments three times with a 50% (v/v) ethanol solution and three times with deionized water until the pH of the wash reached 7.0.
- ix.
- Dry the fragments for 48 h at room temperature and store at room temperature until needed.
- x.
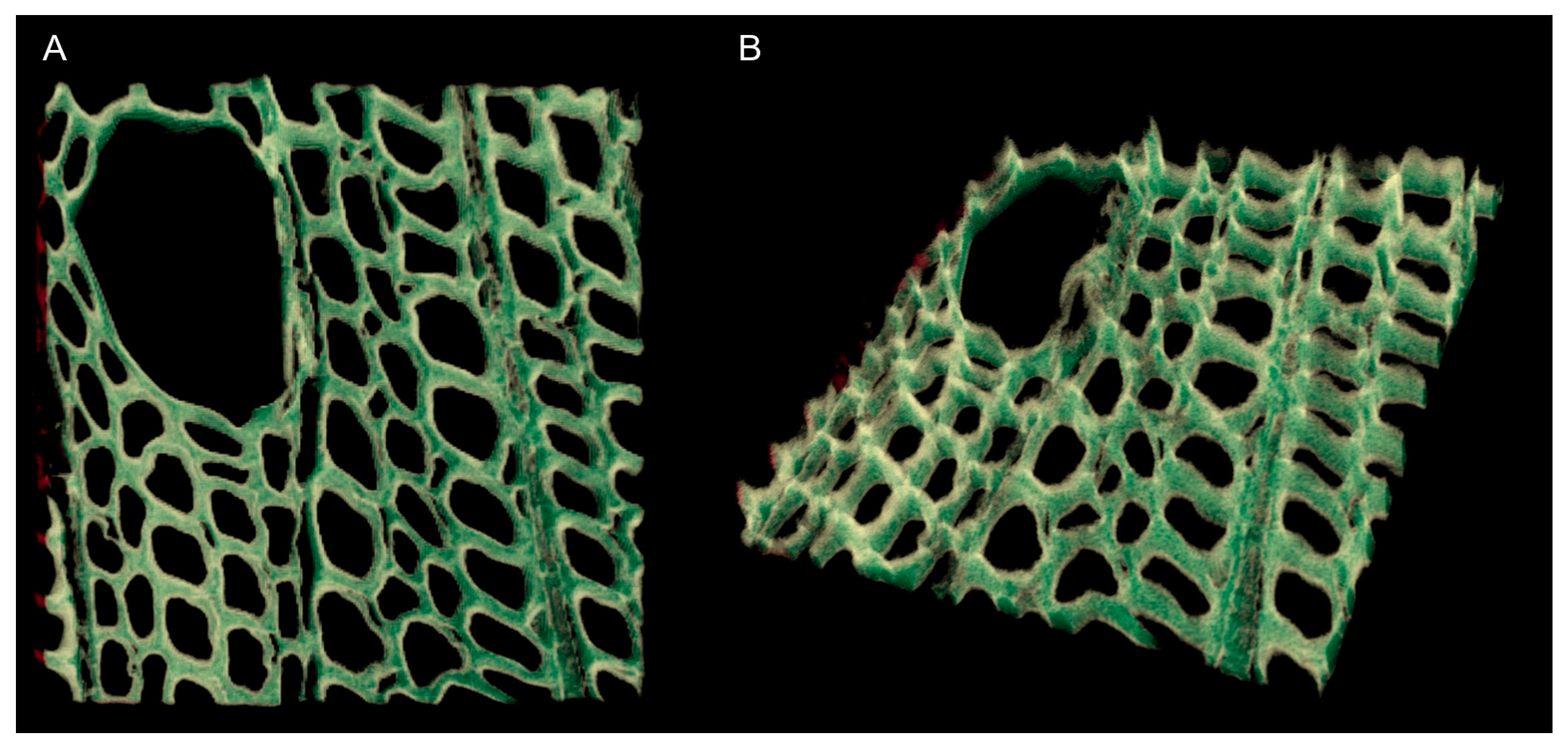
- Cut the dried fragments in the transverse plane from the xylem using a sliding microtome 40 µm thickness for microscopy images acquisition (Figure 1).
3.3. Design of the Customized Incubation Chamber
- i.
- Incubate untreated or pretreated poplar sections in acetate buffer (0.05 M, pH 5) for 30 min in a Petri dish.
- ii.
- Clean the microscope slide and the cover slide with 70% (v/v) ethanol using a lens cleaning tissue and let it dry.
- iii.
- Heat the microscope slide, cover slide and the buffer at 50 °C using a hotplate.
- iv.
- Carefully remove the section from the Petri dish using a brush.
- v.
- Wipe the section with a cotton swab.
- vi.
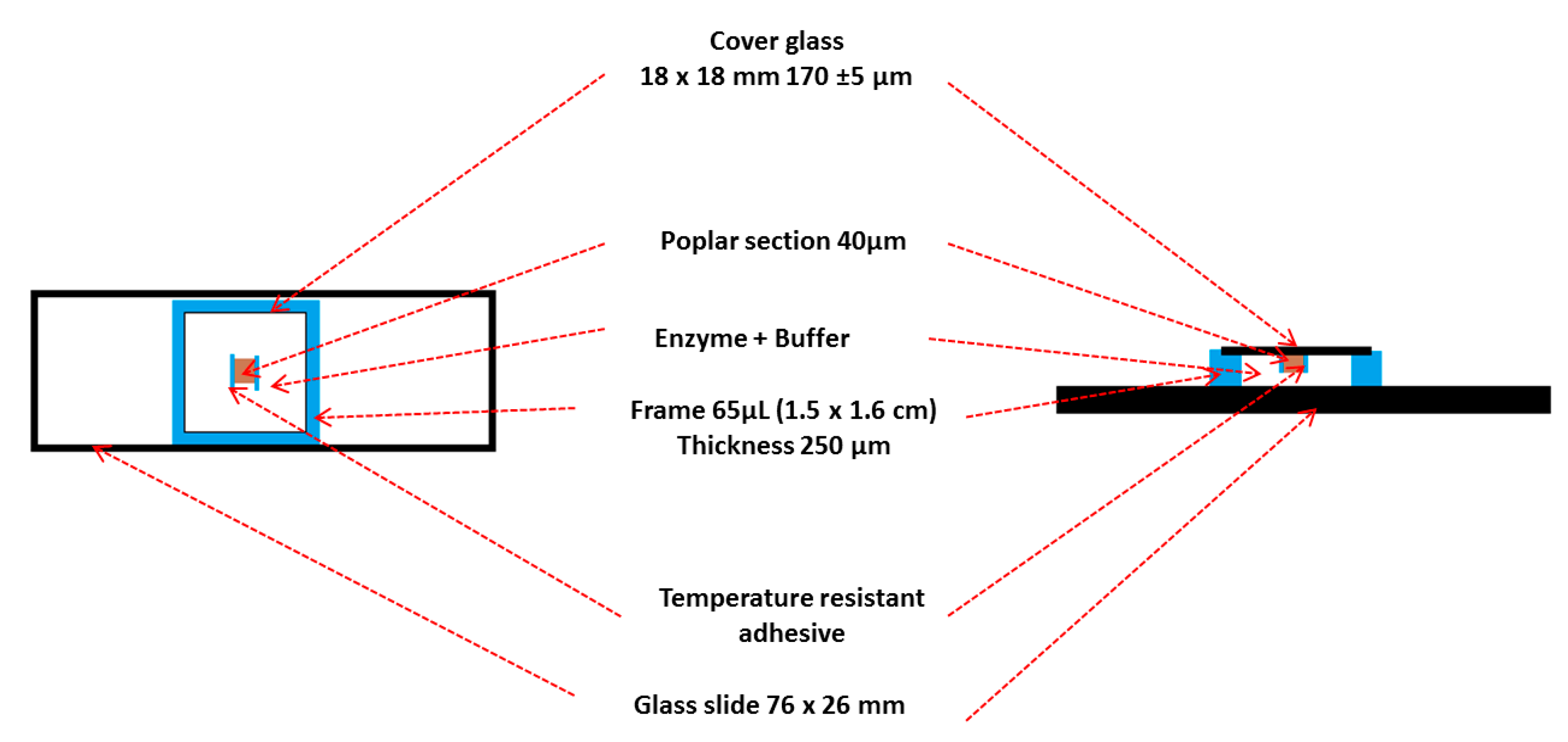
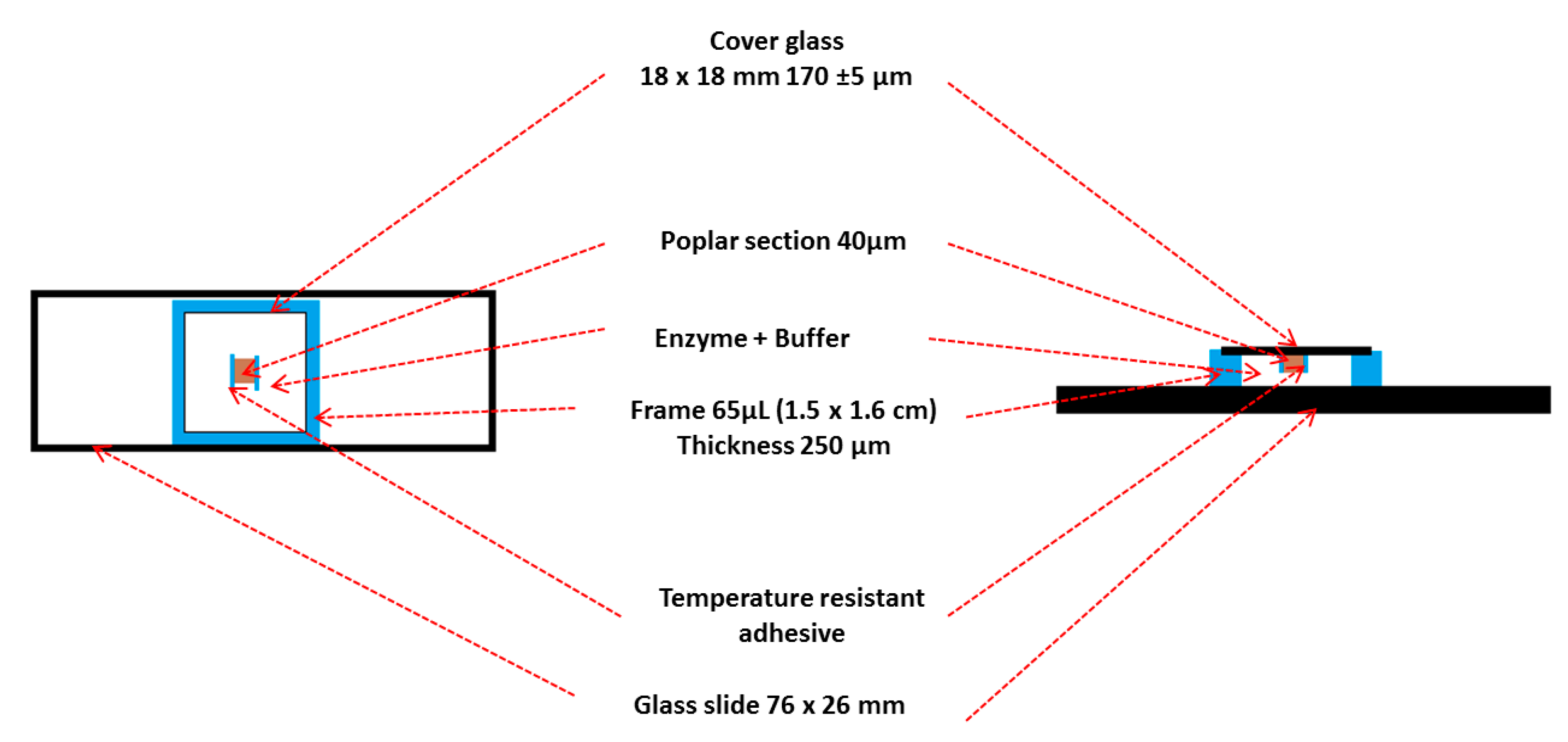
- Fix the section onto a cover slide with a temperature resistant adhesive (Gene frame® was cut into 0.5 cm long strips) to avoid the movement of the samples in the reaction mixtures during image acquisition.
- vii.
- Remove the thick polyester sheet from the frame and apply it to a microscope slide and press the frame to improve the adhesion.
- viii.
- Remove the thin polyester backing sheet from the Gene frame®.
- ix.
- Add 60 µL of the reaction mixture (buffer, sodium azide and enzymes) at the end of the frame (Figure 2).
- x.
- Carefully place the coverslip over the frame at the end where the reaction mixture has been pipetted.
- xi.
- Slowly press the coverslip over the frame for 10 s to spread the reaction mixture homogeneously within the frame and be sure that the adhesion is optimal to avoid evaporation during the enzymatic hydrolysis (preventing the sample from drying out). The customized incubation chamber is now ready to use (Figure 3).
- xii.
- Place the customized incubation chamber on a hotplate at 50 °C for 2 min to avoid cooling the chamber.
- xiii.
- Fix the customized incubation chamber inside a microscope adapted incubator, previously calibrated, in which the temperature is constant at 50 °C for at least 30 min.
- xiv.
- An objective heater must be used to avoid dissipation of heat; if not, sample temperature will drop due to oil contact with the cold objective.
3.4. Imaging Samples Using Confocal Microscopy
- i.
- Place the incubator containing the customized chamber on the microscope stage equipped with an oil-immersion objective with a magnification of 63× (Figure 4).
- ii.
- Adjust the intensity of the laser at 405 nm to 4% to collect the autofluorescence (between 415 and 700 nm).
- iii.
- Adjust the detectors sensitivity, the fluorescence emission range to (415–700 nm).
- iv.
- Acquire images (256 × 256 pixels) at 400 Hz (acquisition speed).
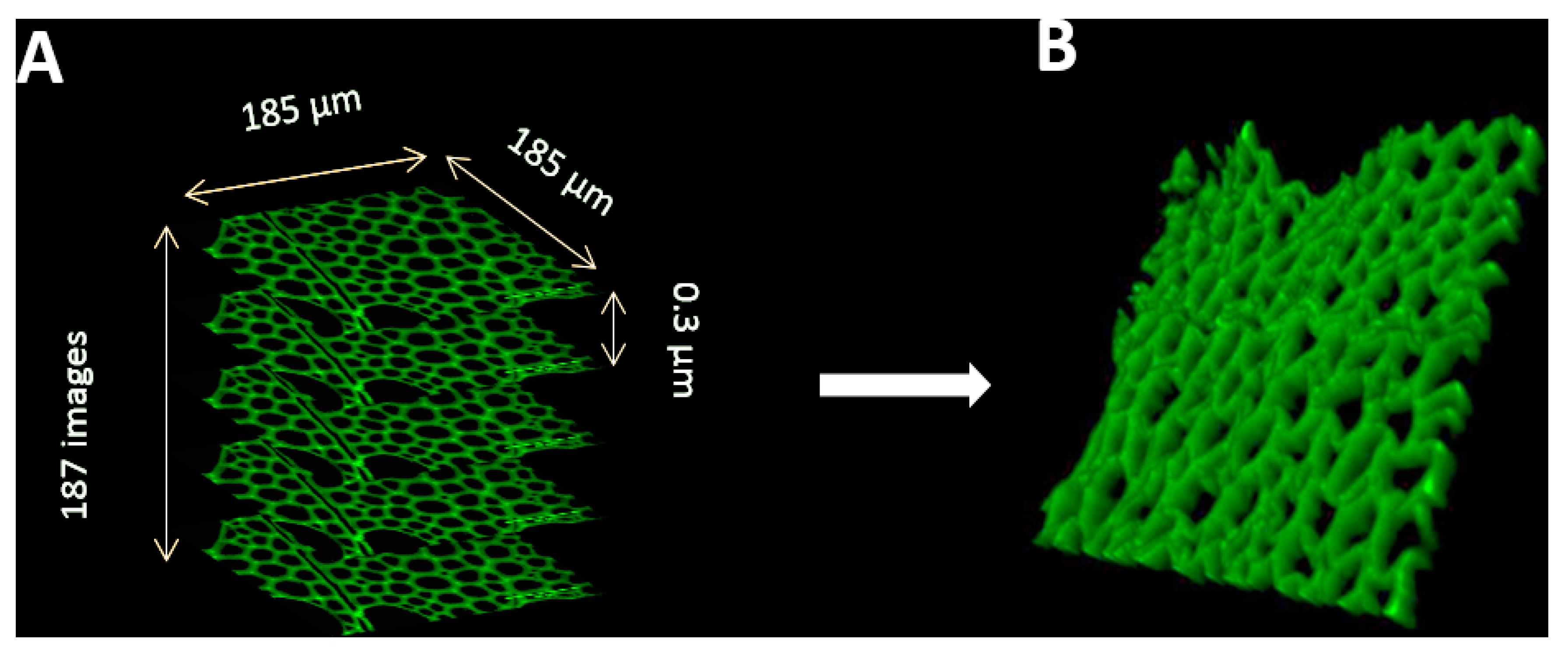
- v.
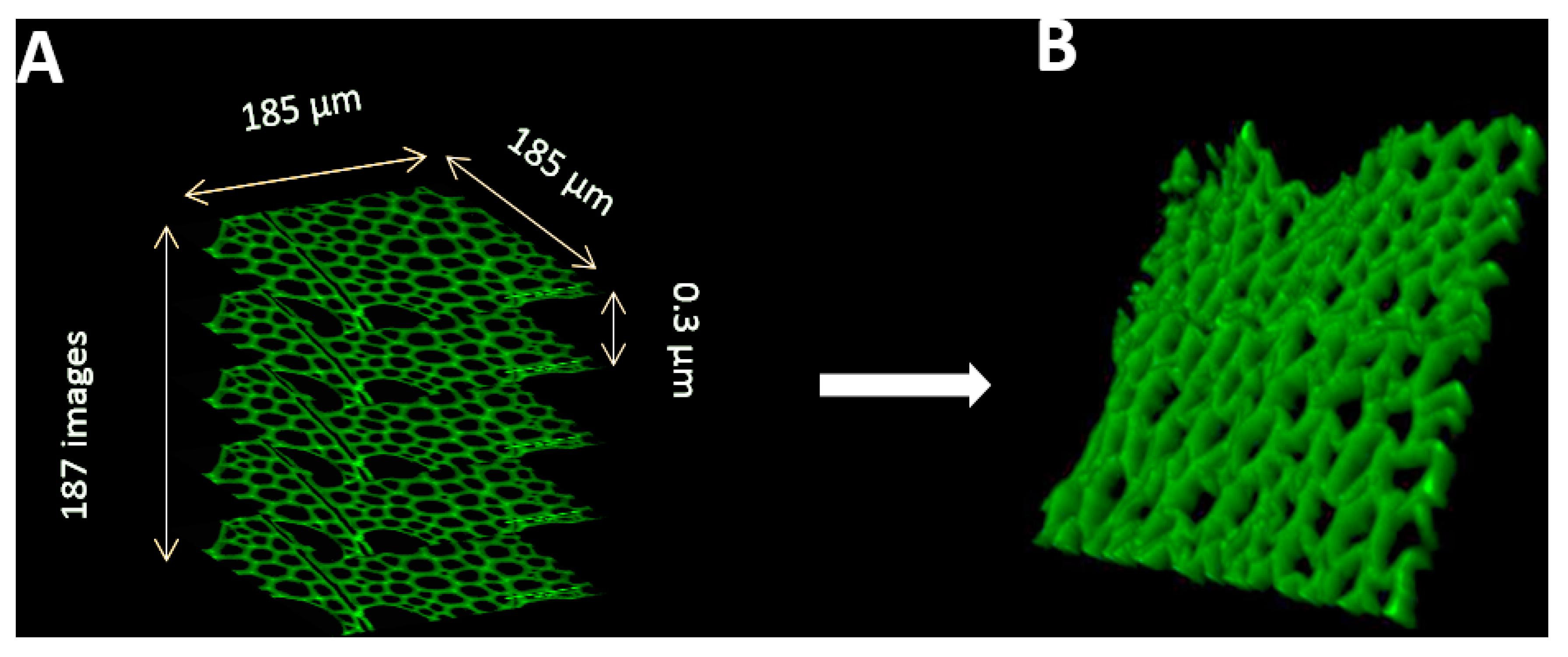
- To acquire confocal z-stacks, first define the top and the bottom of the microscope stage. Set the z step to 0.3µm and pinhole to 1 Airy unit (Figure 5).
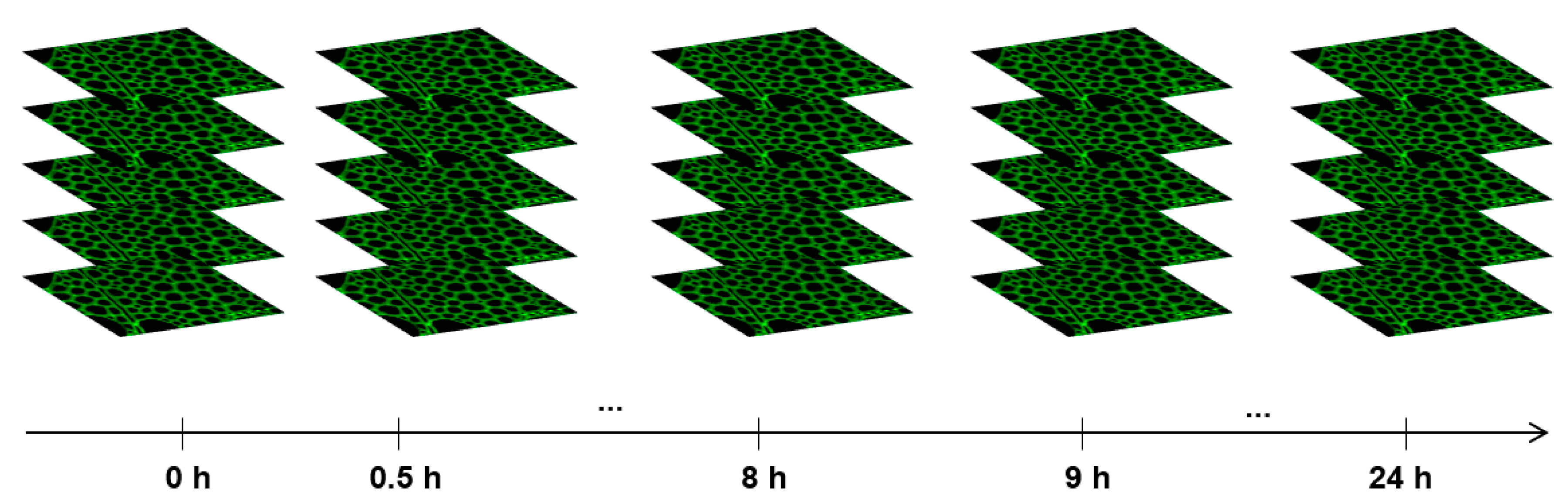
- vi.
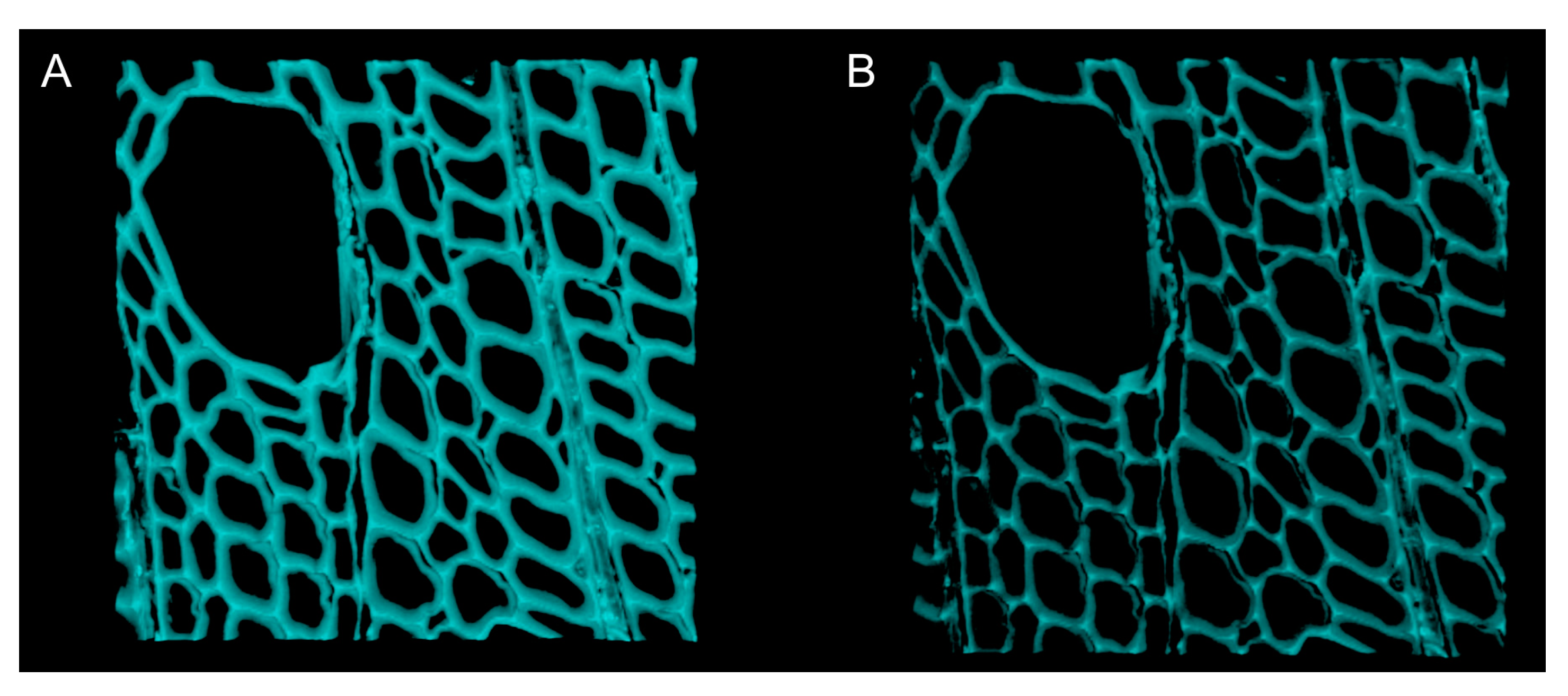
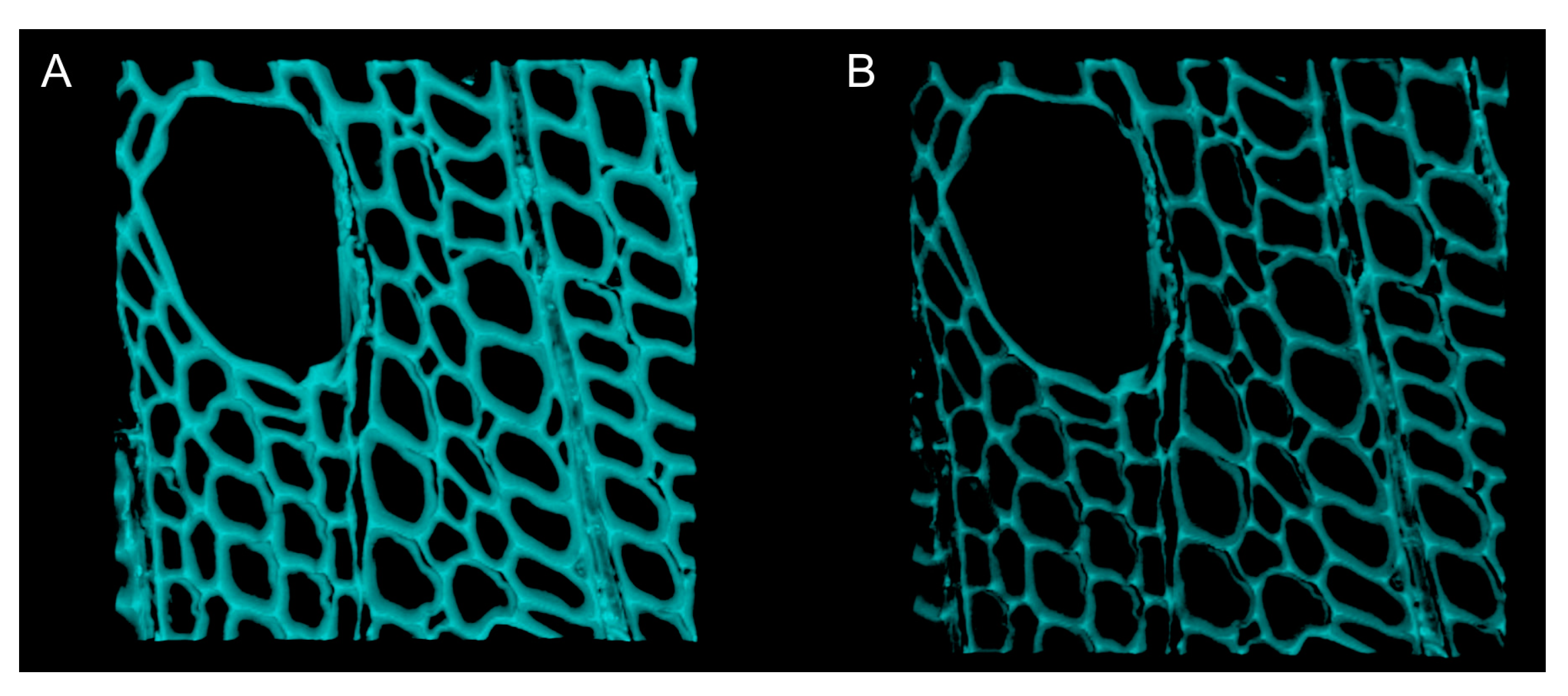
- Acquire and save a confocal z-stack every 30 min for the first 4 h, then every hour for the next 20 h (Figure 6).
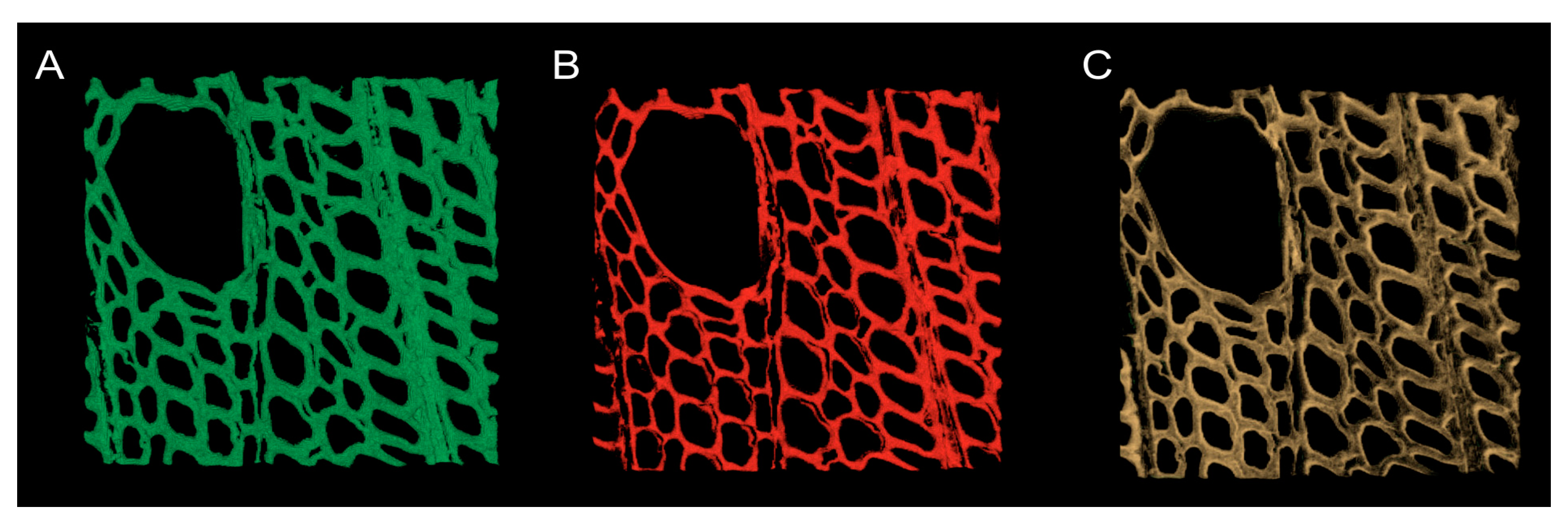
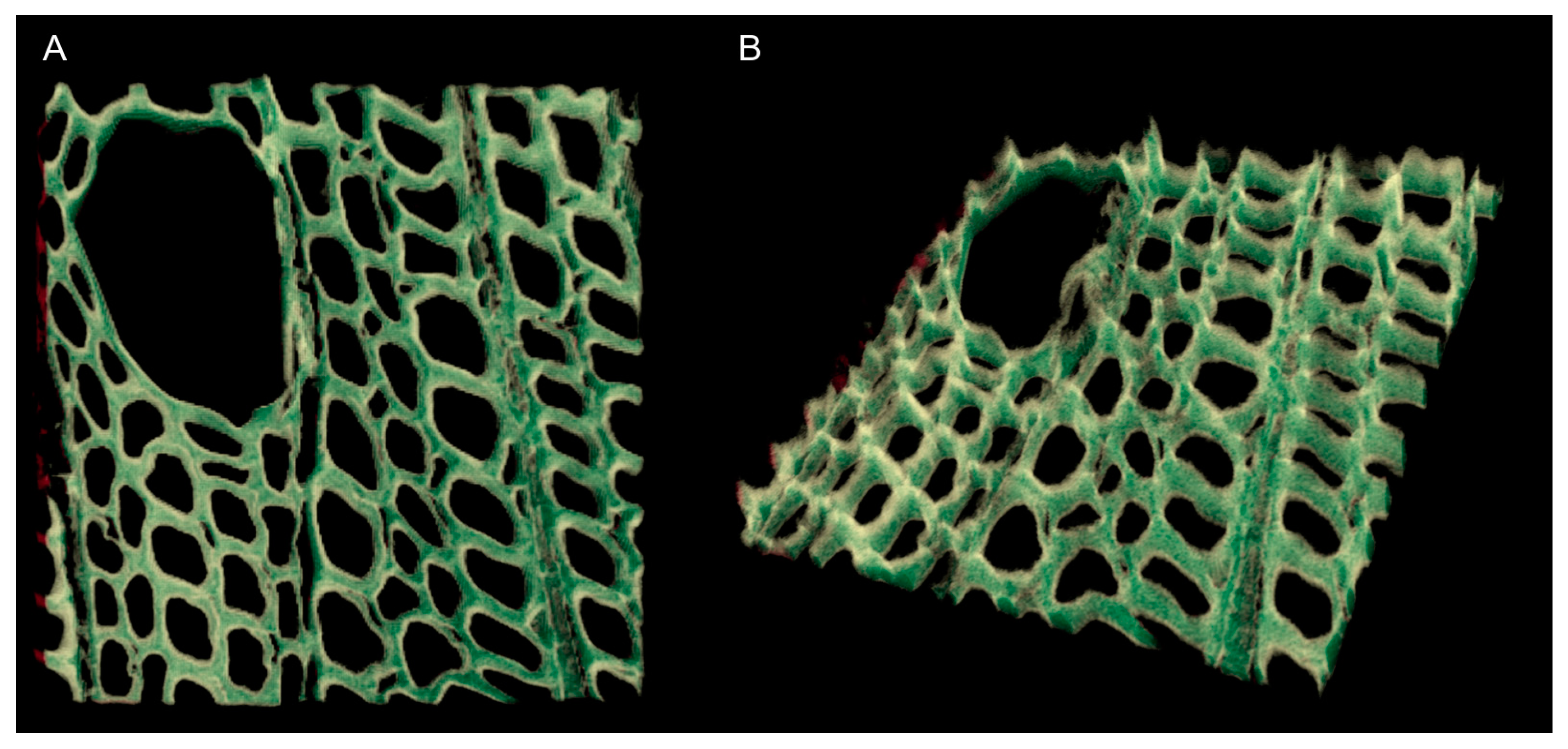
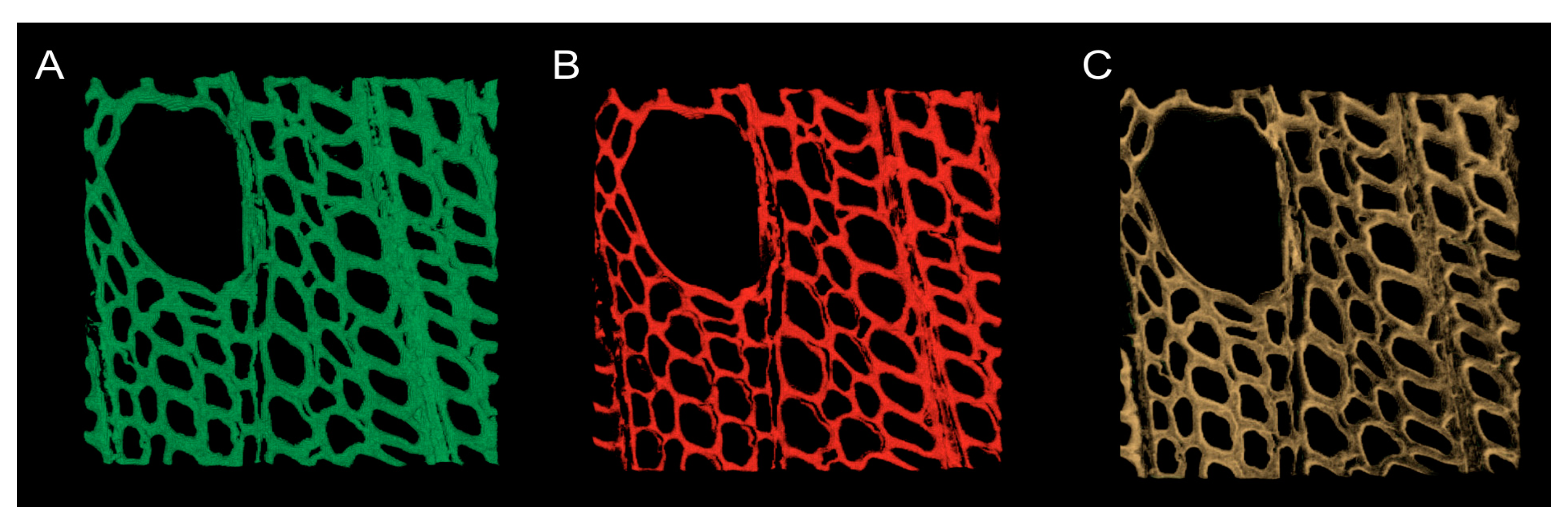
3.5. Analyzing Confocal Images: Three Dimensional Image Registration
4. Results
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hassan, S.S.; Williams, G.A.; Jaiswal, A.K. Moving towards the second generation of lignocellulosic biorefineries in the EU: Drivers, challenges, and opportunities. Renew. Sust. Energ. Rev. 2019, 101, 590–599. [Google Scholar] [CrossRef] [Green Version]
- Dugarova, E.; Gülasan, N. Global Trends: Challenges and Opportunities in the Implementation of the Sustainable Development Goals; United Nations Research Institute for Social Development: New York, NY, USA, 2017. [Google Scholar]
- Claassen, P.A.M.; Van Lier, J.B.; Contreras, A.M.L.; Van Niel, E.W.J.; Sijtsma, L.; Stams, A.J.M.; De Vries, S.S.; Weusthuis, R.A. Utilisation of biomass for the supply of energy carriers. Appl. Microbiol. Biotechnol. 1999, 52, 741–755. [Google Scholar] [CrossRef]
- Zoghlami, M.; Paës, G. Lignocellulosic Biomass: Understanding Recalcitrance and Predicting Hydrolysis. Front. Chem. 2019, 7, 874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Studer, M.H.; DeMartini, J.D.; Davis, M.F.; Sykes, R.W.; Davison, B.; Keller, M.; Tuskan, G.A.; Wyman, C.E. Lignin content in natural Populus variants affects sugar release. Proc. Natl. Acad. Sci. USA 2011, 108, 6300–6305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, H.; Che, X.; Ding, Y.; Kong, Y.; Li, B.; Tian, W. Effect of crystallinity on pretreatment and enzymatic hydrolysis of lignocellulosic biomass based on multivariate analysis. Bioresour. Technol. 2019, 279, 271–280. [Google Scholar] [CrossRef] [PubMed]
- Hallac, B.B.; Ragauskas, A.J. Analyzing cellulose degree of polymerization and its relevancy to cellulosic ethanol. Biofuels Bioprod. Bioref. 2011, 5, 215–225. [Google Scholar] [CrossRef]
- Herbaut, M.; Zoghlami, A.; Paës, G. Dynamical assessment of fluorescent probes mobility in poplar cell walls reveals nanopores govern saccharification. Biotechnol. Biofuels 2018, 11, 271–284. [Google Scholar] [CrossRef]
- Willis, L.; Refahi, Y.; Wightman, R.; Landrein, B.; Teles, J.; Huang, K.C.; Meyerowitz, E.M.; Jönsson, H. Cell size and growth regulation in the Arabidopsis thaliana apical stem cell niche. Proc. Natl. Acad. Sci. USA 2016, 113, 8238–8246. [Google Scholar] [CrossRef] [Green Version]
- Guignard, L.; Fiuza, U.M.; Leggio, B.; Faure, E.; Laussu, J.; Hufnagel, L.; Malandain, G.; Godin, C.; Lemaire, P. Contact-dependent cell communications drive morphological invariance during ascidian embryogenesis. Science 2020, 369, eaar5663. [Google Scholar] [CrossRef] [PubMed]
- Michelin, G.; Refahi, Y.; Wightman, R.; Jönsson, H.; Traas, J.; Godin, C.; Malandain, G. Spatio-temporal registration of 3D microscopy image sequences of Arabidopsis floral meristems. In Proceedings of the 2016 IEEE 13th International Symposium on Biomedical Imaging (ISBI), Prague, Czech Republic, 13–16 April 2016; pp. 1127–1130. [Google Scholar]



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zoghlami, A.; Refahi, Y.; Terryn, C.; Paës, G. Three-Dimensional Imaging of Plant Cell Wall Deconstruction Using Fluorescence Confocal Microscopy. Sustain. Chem. 2020, 1, 75-85. https://doi.org/10.3390/suschem1020007
Zoghlami A, Refahi Y, Terryn C, Paës G. Three-Dimensional Imaging of Plant Cell Wall Deconstruction Using Fluorescence Confocal Microscopy. Sustainable Chemistry. 2020; 1(2):75-85. https://doi.org/10.3390/suschem1020007
Chicago/Turabian StyleZoghlami, Aya, Yassin Refahi, Christine Terryn, and Gabriel Paës. 2020. "Three-Dimensional Imaging of Plant Cell Wall Deconstruction Using Fluorescence Confocal Microscopy" Sustainable Chemistry 1, no. 2: 75-85. https://doi.org/10.3390/suschem1020007
APA StyleZoghlami, A., Refahi, Y., Terryn, C., & Paës, G. (2020). Three-Dimensional Imaging of Plant Cell Wall Deconstruction Using Fluorescence Confocal Microscopy. Sustainable Chemistry, 1(2), 75-85. https://doi.org/10.3390/suschem1020007