
Terpenes and Terpenoids: Building Blocks to Produce Biopolymers

 ,
, 

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
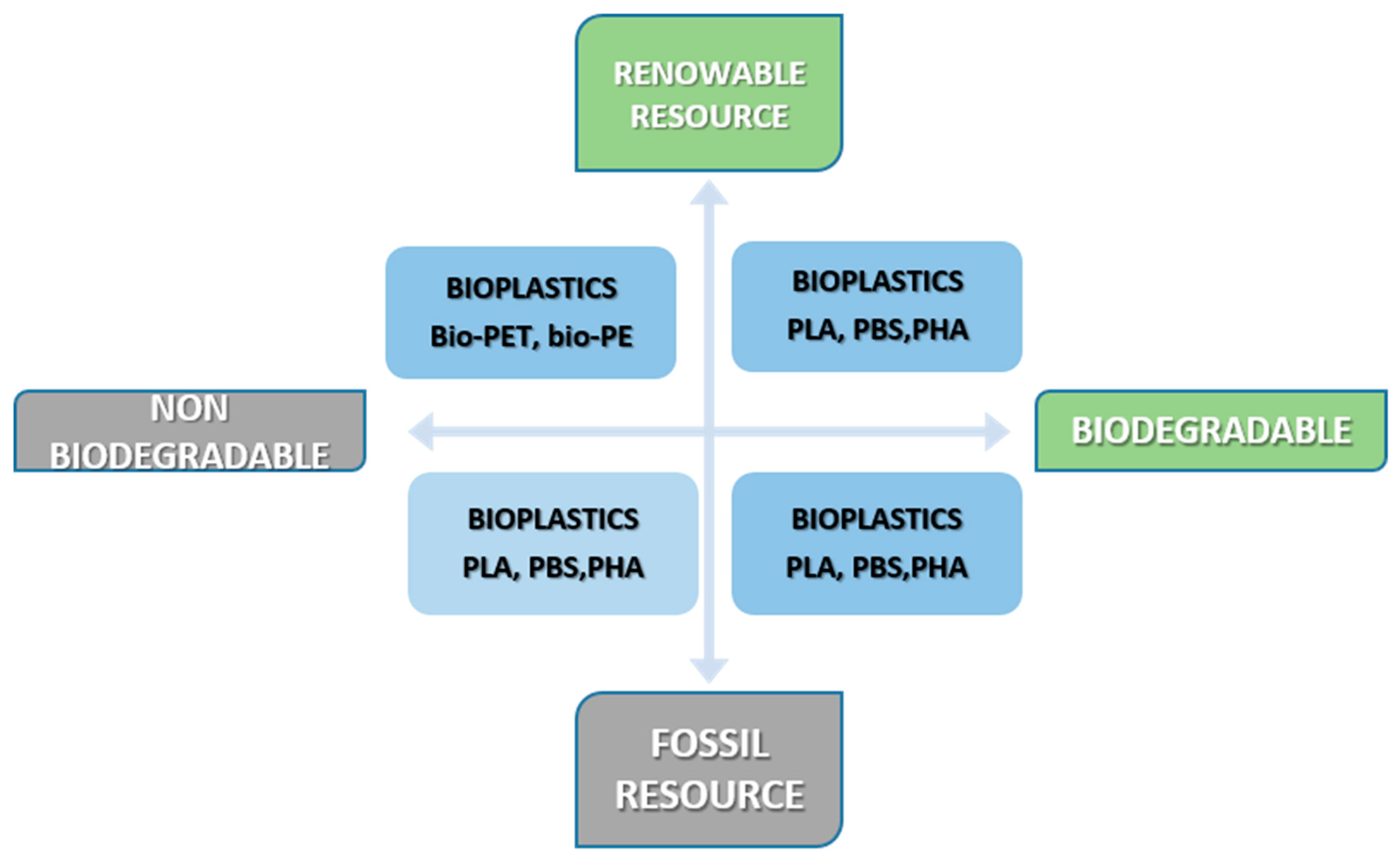
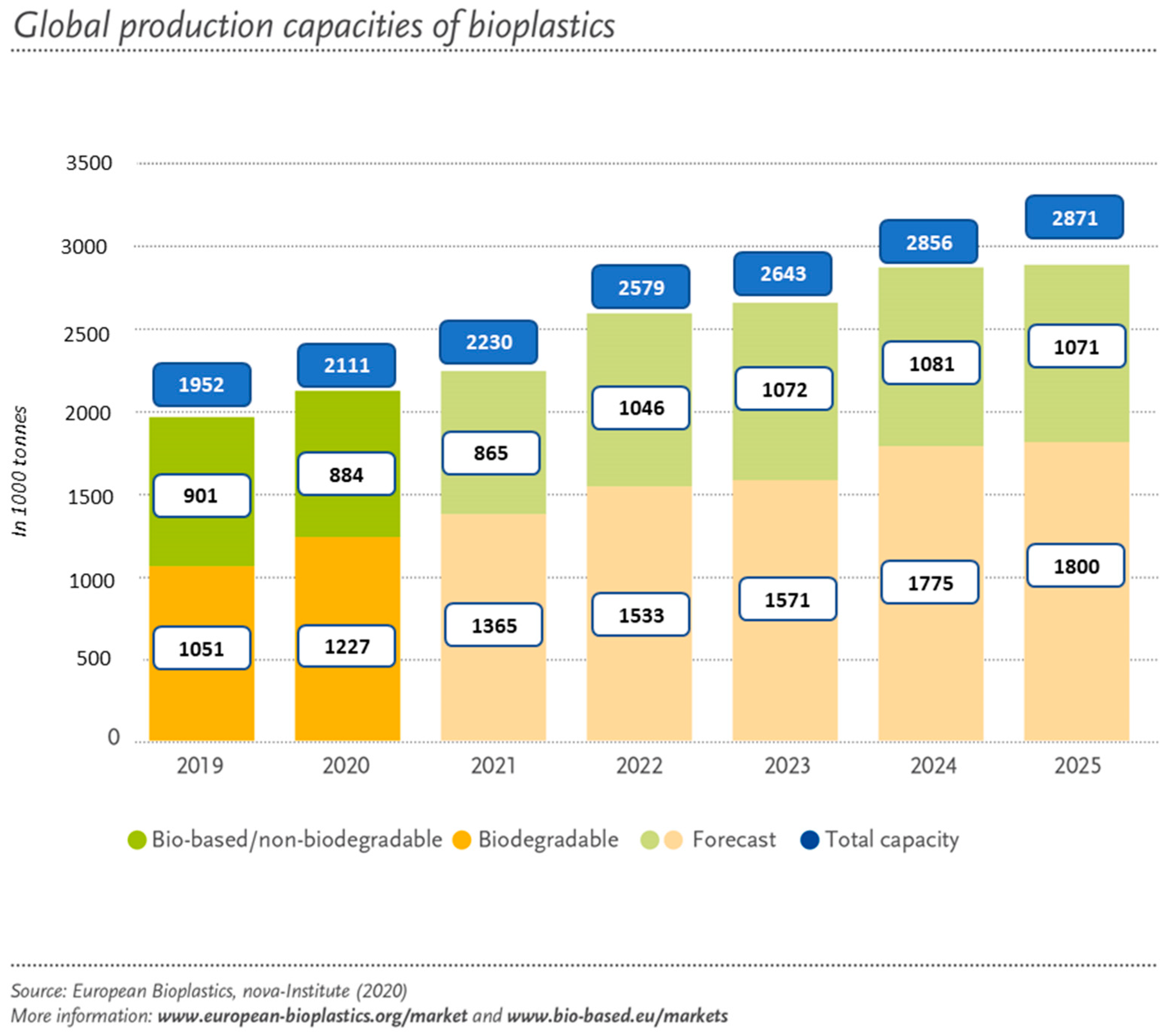
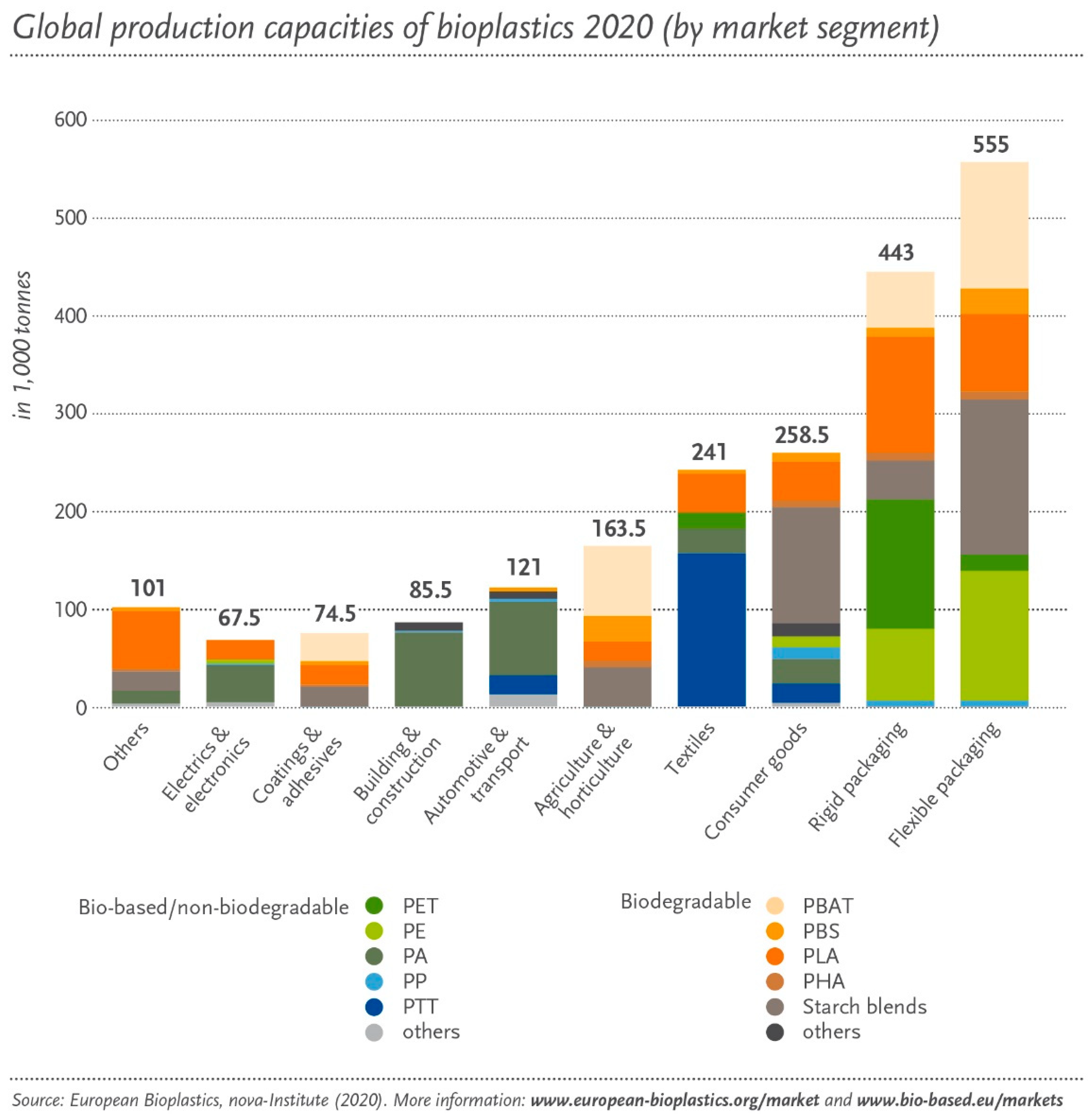
2. Biopolymers and Bioplastics
2.1. General Considerations of Biopolymers and Bioplastics
2.2. Types and Properties of Bioplastics
2.2.1. Cellulose and Starch
2.2.2. Polyesters
2.2.3. Polyvinyl Alcohol
2.2.4. Bio-Based Polymers Analogous to Petroleum-Derived Ones
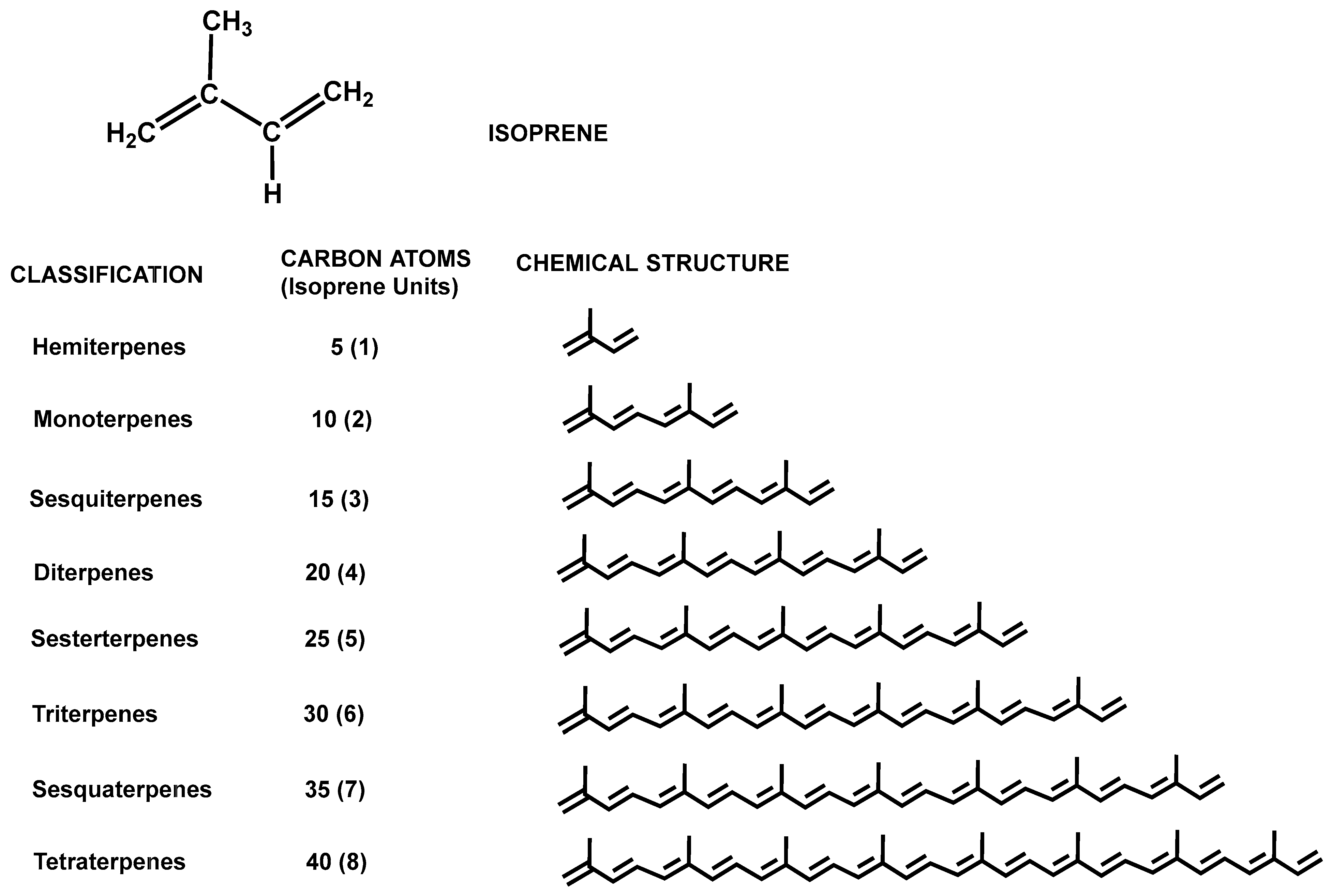
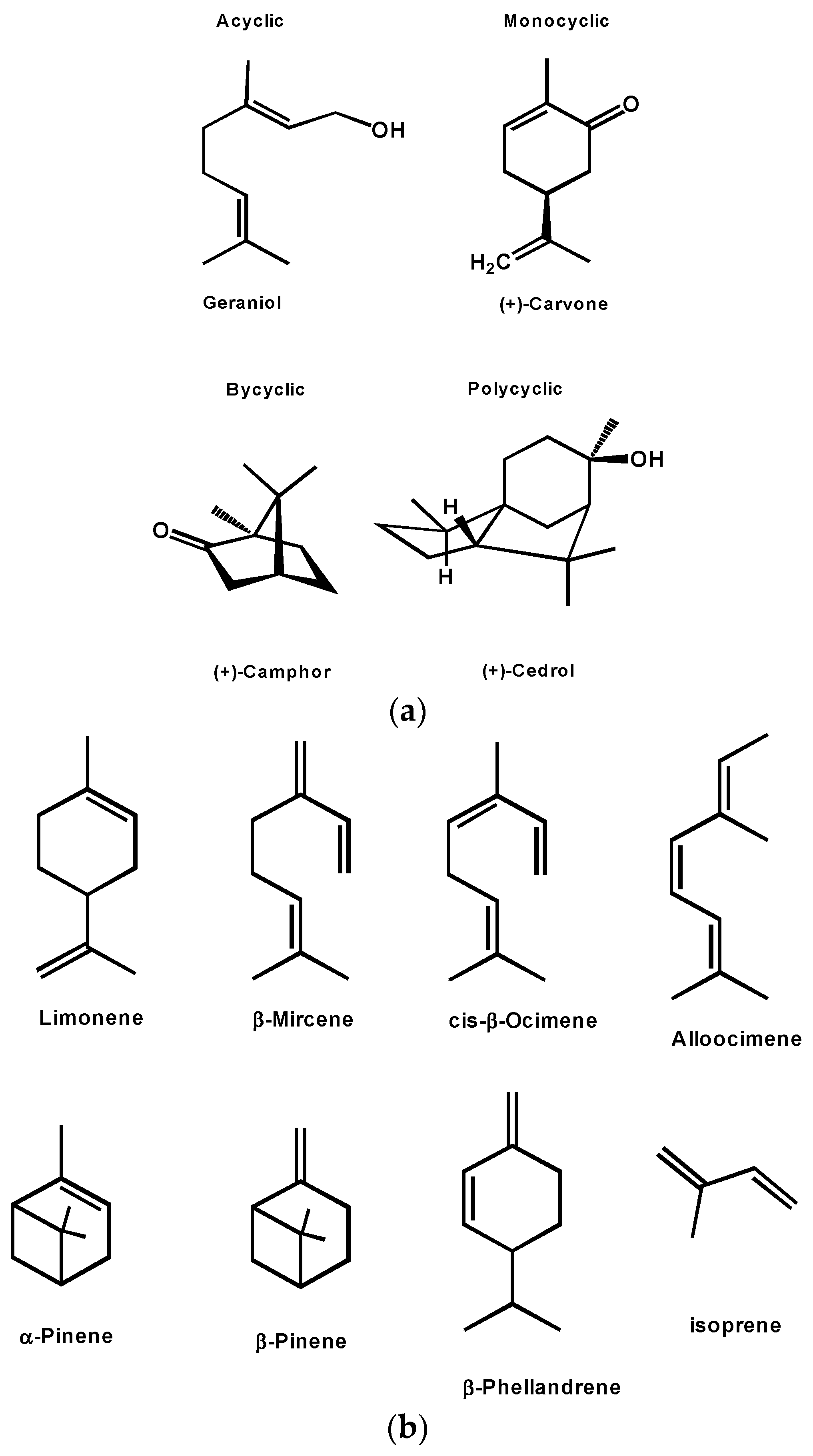
3. Terpenes and Terpenoids
4. Polymerization of Terpenes and Terpenoids
4.1. Polymerization of Acyclic Terpenes
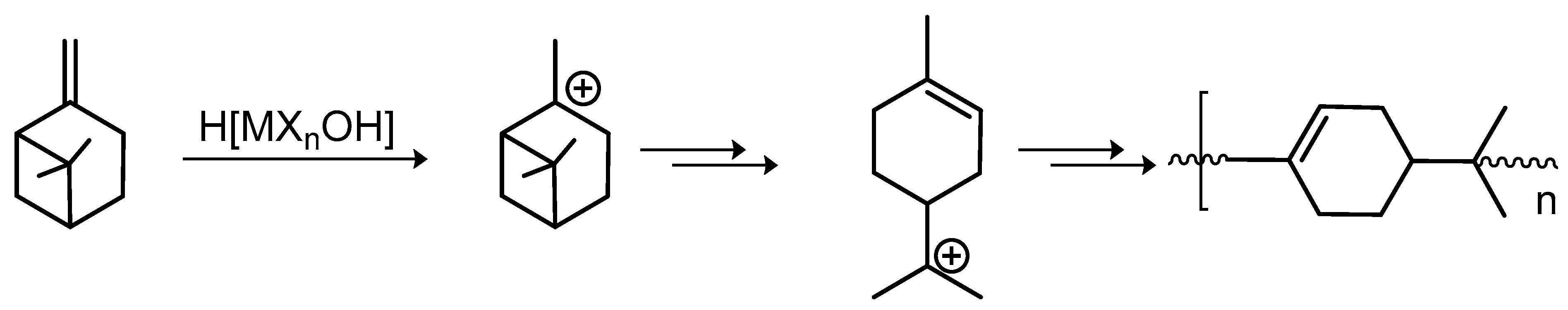
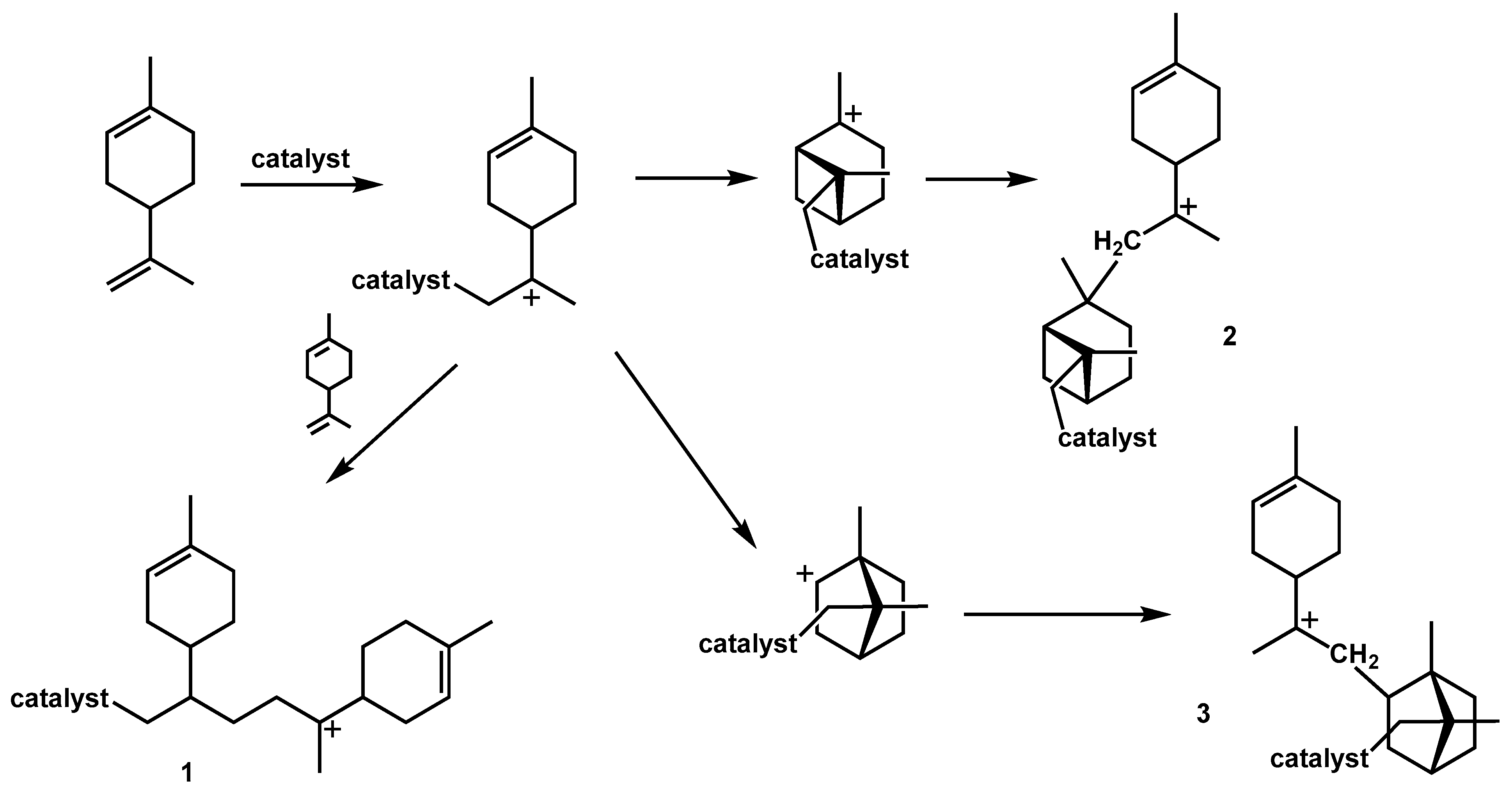
4.2. Polymerization of Cyclic Terpenes
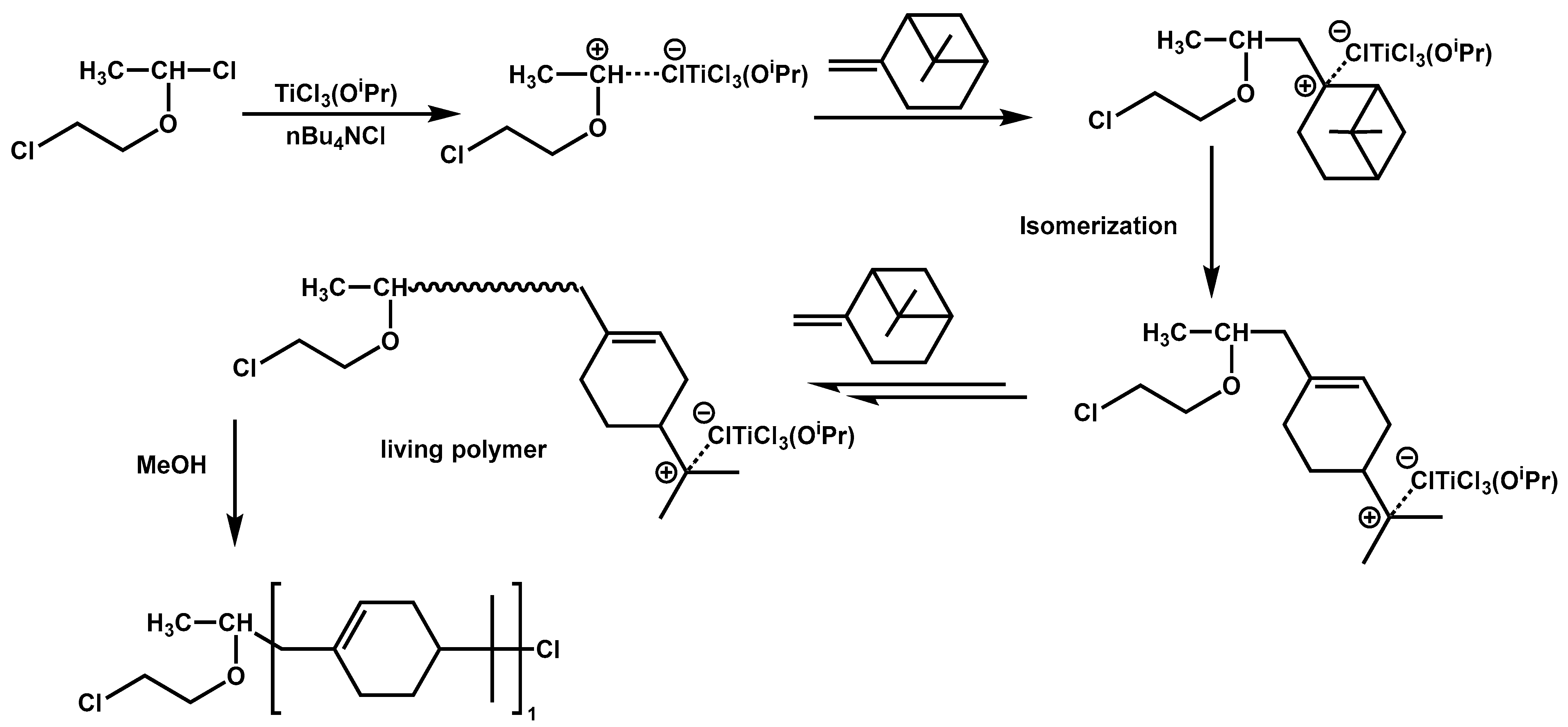
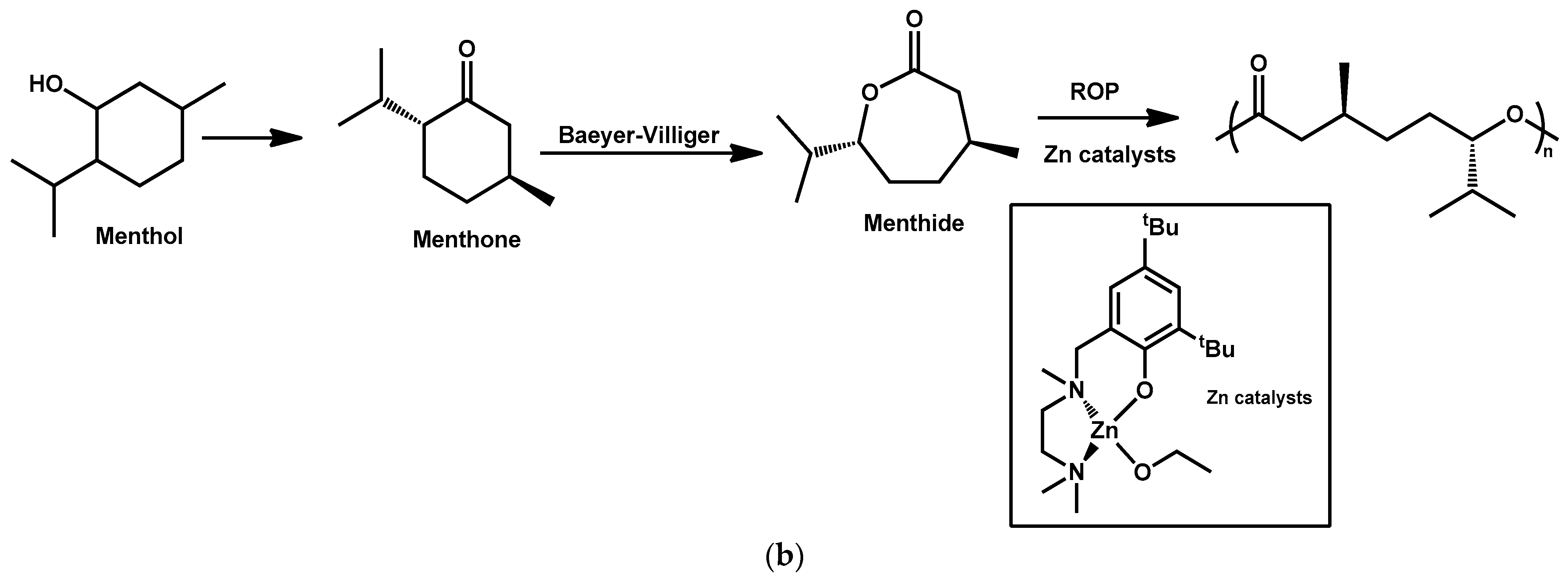
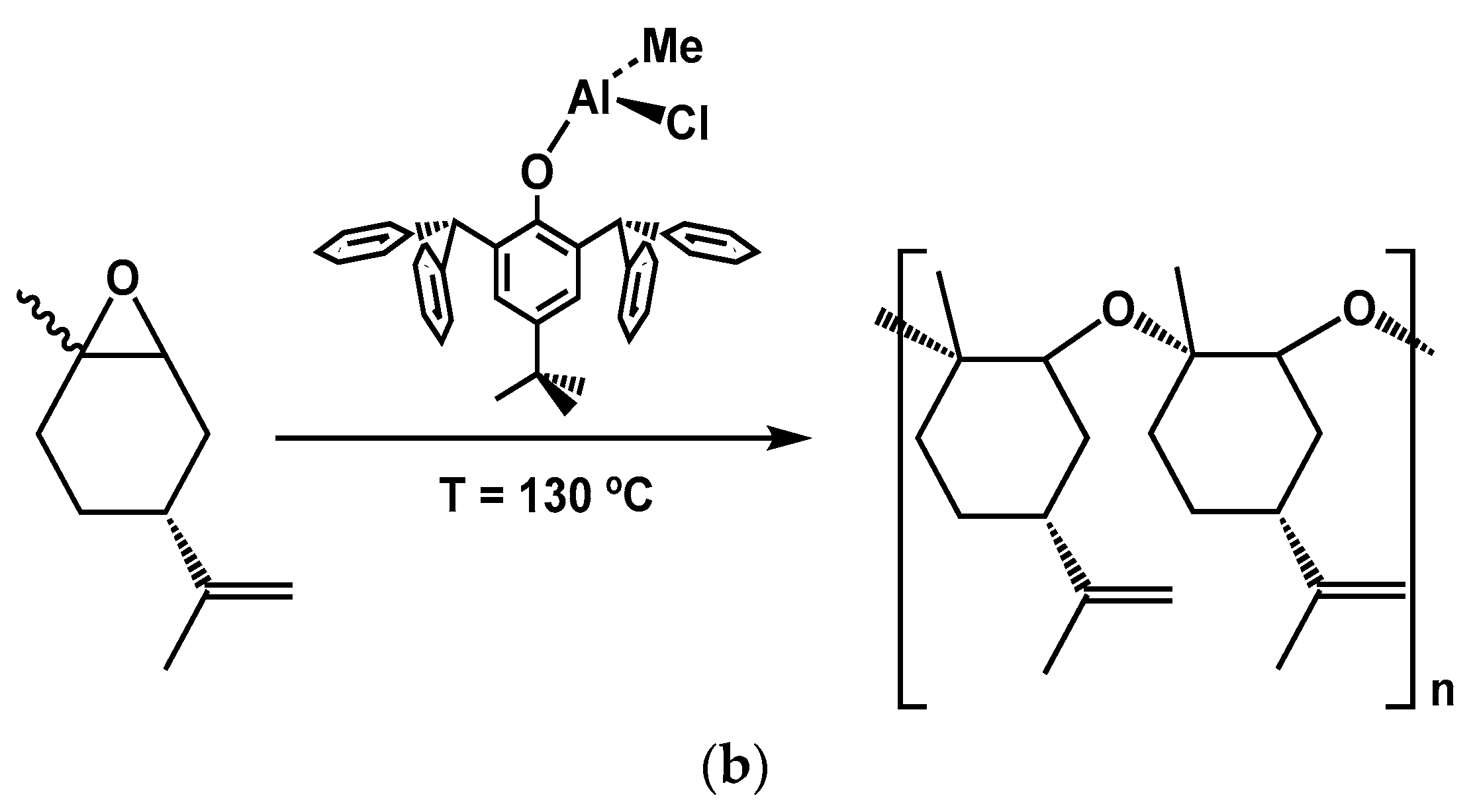
4.3. Polymerization of Terpenoids
4.4. Co-Polymerization of Terpenoids
5. Reactive Extrusion to ROP Polymerization
Reactive Extrusion with Terpenes and Terpenoids
6. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ROP | Ring Opening Polymerization |
| REX | Reactive Extrusion |
| CCTP | Coordinative Chain Transfer Polymerization |
| PLA | PolyLactic Acid |
| bio-PET | bio-PolyEthylene Terephthalate |
| PHA | PolyHydroxiAlkanoates |
| PVA | PolyVinyl Alcohol |
| bio-PUR | bio-PolyURethane |
| bio-PA | bio-PolyAmides |
| PBS | Poly(Butylene Succinate) |
| bio-PET | bio-Poly(ethylene Terephtalic acid) |
| PHB | PolyHydroxy Butyrate |
| PHV | PolyHydroxy Valerate |
| PHH | PolyHydroxy Hexanoate |
| PCL | PolyCaprolactone |
| PGA | PolyGlycolic Acid |
| PBSA | PolyButylene Succinate Adipate |
| PTT | PolyTrimethylene Terephthalate |
| PBTA | PolyButadiene Adipate Terephthalate |
| PLLA | Poly L-Lactide |
| PVA | PolyVinyl Alcohol |
| PUR | PolyURethane |
| PA | PolyAmides |
| PET | PolyEthylene Terephthalate |
| EG | Ethylene Glycol |
| MAO | MethylAluminOxane |
References
- Dupont-Inglis, J.; Borg, A. Destination bioeconomy—The path towards a smarter, more sustainable future. New Biotechnol. 2018, 40, 140–143. [Google Scholar] [CrossRef]
- Gallezot, P. Conversion of biomass to selected chemical products. Chem. Soc. Rev. 2012, 41, 1538–1558. [Google Scholar] [CrossRef]
- Zhu, Y.Q.; Romain, C.; Williams, C.K. Sustainable polymers from renewable resources. Nature 2016, 540, 354–362. [Google Scholar] [CrossRef] [PubMed]
- Xu, F. Structure, Ultrastructure, and Chemical Composition. In Cereal Straw as a Resource for Sustainable Biomaterials and Biofuels; Sun, R.C., Ed.; Elsevier: Amsterdam, The Netherlands, 2010; pp. 9–47. [Google Scholar]
- Duwe, A.; Tippkötter, N.; Ulber, R. Lignocellulose-Biorefinery: Ethanol-Focused. In Biorefineries; Advances in Biochemical Engineering/Biotechnology; Springer: Cham, Switzerland, 2017; pp. 177–216. [Google Scholar]
- Tajmirriahi, M.; Momayez, F.; Karimi, K. The critical impact of rice straw extractives on biogas and bioethanol production. Bioresour. Technol. 2021, 319, 124167. [Google Scholar] [CrossRef] [PubMed]
- de Paula, R.G.; Antoniêto, A.C.; Ribeiro, L.F.; Srivastava, N.; O’Donovan, A.; Mishra, P.K.; Gupta, V.K.; Silva, R.N. Engineered microbial host selection for value-added bioproducts from lignocellulose. Biotechnol. Adv. 2019, 37, 107347. [Google Scholar] [CrossRef] [PubMed]
- Machado, G.; Santos, F.; Lourega, R.; Mattia, J.; Faria, D.; Eichler, P.; Auler, A. Biopolymers from Lignocellulosic Biomass (Feedstocks, Production Processes, and Applications); Lignocellulosic Biorefining Technologies; John Wiley & Sons: Hoboken, NJ, USA, 2020; pp. 125–158. [Google Scholar]
- Bela Torok, T.D. Green Chemistry, An Inclusive Approach; Elsevier: Cambridge, MA, USA, 2018. [Google Scholar]
- Shen, L.; Worrell, E.; Patel, M. Present and future development in plastics from biomass. Biofuels Bioprod. Biorefin. Innov. Sustain. Econ. 2010, 4, 25–40. [Google Scholar] [CrossRef]
- Narancic, T.; Cerrone, F.; Beagan, N.; O’Connor, K.E. Recent Advances in Bioplastics: Application and Biodegradation. Polymers 2020, 12, 920. [Google Scholar] [CrossRef] [Green Version]
- UNE-CEN/TR 15932:2010 IN: Recommendation for Terminology and Characterisation of Biopolymers and Bioplastics. Available online: https://infostore.saiglobal.com/en-gb/Standards/CEN-TR-15932-2010-335831_SAIG_CEN_CEN_770974/ (accessed on 5 March 2021).
- Fact Sheet, European Bioplastics: What are the Bioplastics? Available online: https://docs.european-bioplastics.org/publications/fs/EuBP_FS_What_are_bioplastics.pdf (accessed on 3 February 2021).
- Michael Thielen, B.M. Fachagentur Nachwachsende Rohstoffe e.V. (FNR); Agency for Renewable Resources: Gülzow, Germany, 2014. [Google Scholar]
- Benavides, P.T.; Lee, U.; Zare-Mehrjerdi, O. Life cycle greenhouse gas emissions and energy use of polylactic acid, bio-derived polyethylene, and fossil-derived polyethylene. J. Clean. Prod. 2020, 277, 124010. [Google Scholar] [CrossRef]
- Batori, V.; Akesson, D.; Zamani, A.; Taherzadeh, M.J.; Horvath, I.S. Anaerobic degradation of bioplastics: A review. Waste Manag. 2018, 80, 406–413. [Google Scholar] [CrossRef]
- Arikan, E.B.; Ozsoy, H.D. A Review: Investigation of Bioplastics. J. Civ. Eng. Arch. 2015, 9, 188–192. [Google Scholar] [CrossRef]
- Reichert, C.L.; Bugnicourt, E.; Coltelli, M.-B.; Cinelli, P.; Lazzeri, A.; Canesi, I.; Braca, F.; Martínez, B.M.; Alonso, R.; Agostinis, L.; et al. Bio-Based Packaging: Materials, Modifications, Industrial Applications and Sustainability. Polymers 2020, 12, 1558. [Google Scholar] [CrossRef] [PubMed]
- Bioplastics Market Data. Available online: https://www.european-bioplastics.org (accessed on 5 March 2021).
- Nakajima, H.; Dijkstra, P.; Loos, K. The Recent Developments in Biobased Polymers toward General and Engineering Applications: Polymers that Are Upgraded from Biodegradable Polymers, Analogous to Petroleum-Derived Polymers, and Newly Developed. Polymers 2017, 9, 523. [Google Scholar] [CrossRef] [PubMed]
- Biopolymers. Available online: https://www.eubia.org/cms/wiki-biomass/bio-based-products/biopolymers/ (accessed on 5 March 2021).
- Imre, B.; Pukanszky, B. Compatibilization in bio-based and biodegradable polymer blends. Eur. Polym. J. 2013, 49, 1215–1233. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, H.; Hashim, A.; Abduljalil, H.M. Analysis of Structural, Electrical and Electronic Properties of (Polymer Nanocomposites/Silicon Carbide) for Antibacterial Application. Egypt. J. Chem. 2019, 62, 1167–1176. [Google Scholar] [CrossRef]
- Steinbüchel, S.E. Microbial Succinic Acid, Its Polymer Poly(butylene succinate), and Applications. In Prokaryotic Symbionts in Plants; Microbiology Monographs 2009, Plastics from bacteria; Springer: Berlin, Germany, 2009; pp. 347–388. [Google Scholar] [CrossRef]
- Woodruff, M.A.; Hutmacher, D.W. The return of a forgotten polymer-Polycaprolactone in the 21st century. Prog. Polym. Sci. 2010, 35, 1217–1256. [Google Scholar] [CrossRef] [Green Version]
- Kabir, E.; Kaur, R.; Lee, J.; Kim, K.H.; Kwon, E.E. Prospects of biopolymer technology as an alternative option for non-degradable plastics and sustainable management of plastic wastes. J. Clean. Prod. 2020, 258, 120536. [Google Scholar] [CrossRef]
- Siracusa, V.; Blanco, I. Bio-Polyethylene (Bio-PE), Bio-Polypropylene (Bio-PP) and Bio-Poly(ethylene terephthalate) (Bio-PET): Recent Developments in Bio-Based Polymers Analogous to Petroleum-Derived Ones for Packaging and Engineering Applications. Polymers 2020, 12, 1641. [Google Scholar] [CrossRef]
- Shen, J.H.; Patel, M.K. Product Overview and Market Protection of Emerging Bio-Based Plastics, PRO-BIP 2009; Utrecht University: Utrecht, The Netherlands, 2009. [Google Scholar]
- Gershenzon, J.; Dudareva, N. The function of terpene natural products in the natural world. Nat. Chem. Biol. 2007, 3, 408–414. [Google Scholar] [CrossRef]
- Tholl, D. Biosynthesis and Biological Functions of Terpenoids in Plants. In Biotechnology of Isoprenoids; Schrader, J., Bohlmann, J., Eds.; Advances in Biochemical Engineering/Biotechnology; Springer: Berlin, Germany, 2015; Volume 148, pp. 63–106. [Google Scholar]
- Kännaste, A.; Copolovici, L.; Niinemets, Ü. Plant Isoprenoids: Methods and Protocols, Methods in Molecular Biology; Springer: New York, NY, USA, 2014; pp. 1–5. [Google Scholar]
- Lombard, J.; Moreira, D. Origins and Early Evolution of the Mevalonate Pathway of Isoprenoid Biosynthesis in the Three Domains of Life. Mol. Biol. Evol. 2011, 28, 87–99. [Google Scholar] [CrossRef] [Green Version]
- Lohr, M.; Schwender, J.; Polle, J.E. Isoprenoid biosynthesis in eukaryotic phototrophs: A spotlight on algae. Plant Sci. 2012, 185, 9–22. [Google Scholar] [CrossRef]
- Schrader, J.; Bohlmann, J. (Eds.) Biotechnology of Isoprenoids; Springer: Berlin, Germany, 2015. [Google Scholar]
- Moser, S.; Pichler, H. Identifying and engineering the ideal microbial terpenoid production host. Appl. Microbiol. Biotechnol. 2019, 103, 5501–5516. [Google Scholar] [CrossRef] [Green Version]
- Connolly, J.D.; Hill, R.A. Dictionary of Terpenoids; Chapman & Hall: London, UK, 1991. [Google Scholar]
- Croteau, R.O.; Johnson, M.A. Biosynthesis and Biodegradation of Wood Components; Academic Press: Cambridge, MA, USA, 1985; pp. 379–439. [Google Scholar]
- Guenther, A.; Karl, T.; Harley, P.; Wiedinmyer, C.; Palmer, P.I.; Geron, C. Estimates of global terrestrial isoprene emissions using MEGAN (Model of Emissions of Gases and Aerosols from Nature). Atmos. Chem. Phys. 2006, 6, 3181–3210. [Google Scholar] [CrossRef] [Green Version]
- Ryan, A.C.; Hewitt, C.N.; Possell, M.; Vickers, C.E.; Purnell, A.; Mullineaux, P.M.; Davies, W.J.; Dodd, I.C. Isoprene emission protects photosynthesis but reduces plant productivity during drought in transgenic tobacco (Nicotiana tabacum) plants. New Phytol. 2014, 201, 205–216. [Google Scholar] [CrossRef] [PubMed]
- Cseke, L.J.; Kirakosyan, A.; Kaufman, P.B.; Warber, S.; Duke, J.A.; Brielmann, H.L. Natural Products from Plants; Taylor and Francis: London, UK, 2006. [Google Scholar] [CrossRef]
- Ludwiczuk, A.S.; Georgiev, M.I. Chapter 11: Terpenoids. In Pharmacognosy; Boston Academic: Boston, MA, USA, 2017; pp. 233–266. [Google Scholar]
- Trombetta, D.; Castelli, F.; Sarpietro, M.G.; Venuti, V.; Cristani, M.; Daniele, C.; Saija, A.; Mazzanti, G.; Bisignano, G. Mechanisms of antibacterial action of three monoterpenes. Antimicrob. Agents Chemother. 2005, 49, 2474–2478. [Google Scholar] [CrossRef] [Green Version]
- Sobral, M.V.; Xavier, A.L.; Lima, T.C.; de Sousa, D.P. Antitumor activity of monoterpenes found in essential oils. Sci. World J. 2014, 2014, 953451. [Google Scholar] [CrossRef] [PubMed]
- Singh, G. Chemistry of Terpenoids and Carotenoids; Discovery: New Delhi, India, 2007. [Google Scholar]
- Wei, L.J.; Zhong, Y.T.; Nie, M.Y.; Liu, S.C.; Hua, Q. Biosynthesis of alpha-Pinene by Genetically Engineered Yarrowia lipolytica from Low-Cost Renewable Feedstocks. J. Agric. Food. Chem. 2021, 69, 275–285. [Google Scholar] [CrossRef]
- Villasenor, I.M. Bioactivities of iridoids. Anti-Inflamm. Anti-Allergy Agents Med. Chem. 2007, 6, 307–314. [Google Scholar] [CrossRef]
- Dinda, B.; Debnath, S.; Harigaya, Y. Naturally occurring iridoids. A review, part 1. Chem. Pharm. Bull. 2007, 55, 159–222. [Google Scholar] [CrossRef] [Green Version]
- Misawa, N. Pathway engineering for functional isoprenoids. Curr. Opin. Biotechnol. 2011, 22, 627–633. [Google Scholar] [CrossRef]
- Chan, W.K.; Tan, L.T.; Chan, K.G.; Lee, L.H.; Goh, B.H. Nerolidol: A Sesquiterpene Alcohol with Multi-Faceted Pharmacological and Biological Activities. Molecules 2016, 21, 529. [Google Scholar] [CrossRef] [Green Version]
- de Veras, B.O.; de Oliveira, M.B.; da Silva Oliveira, F.G.; Dos Santos, Y.Q.; de Oliveira, J.R.; de Menezes Lima, V.L.; da Silva Almeida, J.R.; Navarro, D.M.; de Oliveira Farias, J.C.; dos Santos Aguiar, J.; et al. Chemical composition and evaluation of the antinociceptive, antioxidant and antimicrobial effects of essential oil from Hymenaea cangaceira (Pinto, Mansano & Azevedo) native to Brazil: A natural medicine. J. Ethnopharmacol. 2020, 247, 112265. [Google Scholar] [CrossRef]
- Kutyna, D.R.; Borneman, A.R. Heterologous Production of Flavour and Aroma Compounds in Saccharomyces cerevisiae. Genes 2018, 9, 326. [Google Scholar] [CrossRef] [Green Version]
- Ramawat, K.G.; Mérillon, J.M. Diterpenes for Therapeutic Use. In Natural Products: Phytochemistry, Botany and Metabolism of Alkaloids, Phenolics and Terpenes; Springer: Berlin/Heidelberg, Germany, 2013; pp. 3171–3191. [Google Scholar]
- Stromgaard, K.; Nakanishi, K. Chemistry and biology of terpene trilactones from Ginkgo biloba. Angew. Chem. Int. Ed. 2004, 43, 1640–1658. [Google Scholar] [CrossRef]
- Topcu, G.; Gören, A.C. Biological Activity of Diterpenoids Isolated from Anatolian Lamiaceae Plants. Rec. Nat. Prod. 2007, 1, 1–16. [Google Scholar]
- Guerra-Bubb, J.; Croteau, R.; Williams, R.M. The early stages of taxol biosynthesis: An interim report on the synthesis and identification of early pathway metabolites. Nat. Prod. Rep. 2012, 29, 683–696. [Google Scholar] [CrossRef]
- Liu, Y.; Wang, L.; Jung, J.H.; Zhanga, S. Sesterterpenoids. Nat. Prod. Rep. 2007, 24, 1401–1429. [Google Scholar] [CrossRef]
- Ebada, S.S.; Lin, W.; Proksch, P. Bioactive Sesterterpenes and Triterpenes from Marine Sponges: Occurrence and Pharmacological Significance. Mar. Drugs 2010, 8, 313–346. [Google Scholar] [CrossRef] [Green Version]
- Silva, F.S.; Oliveira, P.J.; Duarte, M.F. Oleanolic, Ursolic, and Betulinic Acids as Food Supplements or Pharmaceutical Agents for Type 2 Diabetes: Promise or Illusion? J. Agric. Food. Chem. 2016, 64, 2991–3008. [Google Scholar] [CrossRef] [PubMed]
- Blomhoff, R.; Blomhoff, H.K. Overview of retinoid metabolism and function. J. Neurobiol. 2006, 66, 606–630. [Google Scholar] [CrossRef]
- Domonkos, I.; Kis, M.; Gombos, Z.; Ughy, B. Carotenoids, versatile components of oxygenic photosynthesis. Prog. Lipid Res. 2013, 52, 539–561. [Google Scholar] [CrossRef]
- Barredo, J.L.; Garcia-Estrada, C.; Kosalkova, K.; Barreiro, C. Biosynthesis of Astaxanthin as a Main Carotenoid in the Heterobasidiomycetous Yeast Xanthophyllomyces dendrorhous. J. Fungi 2017, 3, 44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mortensen, A. Carotenoids and other pigments as natural colorants. Pure Appl. Chem. 2006, 78, 1477–1491. [Google Scholar] [CrossRef]
- Nonomura, A.M. Industrial biosynthesis of carotenoids. In Carotenoids: Chemistry and Biology; Plenum Press: New York, NY, USA, 1989; pp. 365–375. [Google Scholar]
- Rodriguez-Saiz, M.; Luis de la Fuente, J.; Luis Barredo, J. Xanthophyllomyces dendrorhous for the industrial production of astaxanthin. Appl. Microbiol. Biotechnol. 2010, 88, 645–658. [Google Scholar] [CrossRef]
- Bohlmann, J.; Keeling, C.I. Terpenoid biomaterials. Plant J. 2008, 54, 656–669. [Google Scholar] [CrossRef]
- Zerbe, P.; Bohlmann, J. Recent Advances in Phytochemistry. In Phytochemicals—Biosynthesis, Function and Application; Springer: Berlin, Germany, 2014; Volume 44, pp. 85–108. [Google Scholar]
- Tetali, S.D. Terpenes and isoprenoids: A wealth of compounds for global use. Planta 2019, 249, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Kuehlborn, J.; Gross, J.; Opatz, T. Making natural products from renewable feedstocks: Back to the roots? Nat. Prod. Rep. 2020, 37, 380–424. [Google Scholar] [CrossRef] [Green Version]
- Behr, A.; Johnen, L. Myrcene as a Natural Base Chemical in Sustainable Chemistry: A Critical Review. Chemsuschem 2009, 2, 1072–1095. [Google Scholar] [CrossRef] [PubMed]
- Palenzuela, M.; Sánchez-Roa, D.; Damián, J.; Sessini, V.; Mosquera, M.E. Polymerization of terpenes and terpenoids using metal catalysts. Adv. Organomet. Chem. 2021, 75, 55–93. [Google Scholar] [CrossRef]
- Winnacker, M.; Rieger, B. Recent Progress in Sustainable Polymers Obtained from Cyclic Terpenes: Synthesis, Properties, and Application Potential. Chemsuschem 2015, 8, 2455–2471. [Google Scholar] [CrossRef]
- Swift, K.A. Catalytic transformations of the major terpene feedstocks. Top. Catal. 2004, 27, 143–155. [Google Scholar] [CrossRef]
- Sarkar, P.; Bhowmick, A.K. Synthesis, characterization and properties of a bio-based elastomer: Polymyrcene. RSC Adv. 2014, 4, 61343–61354. [Google Scholar] [CrossRef]
- Aoshima, S.; Kanaoka, S. A Renaissance in Living Cationic Polymerization. Chem. Rev. 2009, 109, 5245–5287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Georges, S.; Toure, A.O.; Visseaux, M.; Zinck, P. Coordinative Chain Transfer Copolymerization and Terpolymerization of Conjugated Dienes. Macromolecules 2014, 47, 4538–4547. [Google Scholar] [CrossRef]
- Avila-Ortega, A.; Aguilar-Vega, M.; Bastarrachea, M.I.; Carrera-Figueiras, C.; Campos-Covarrubias, M. Anionic synthesis of amine omega-terminated beta-myrcene polymers. J. Polym. Res. 2015, 22, 226. [Google Scholar] [CrossRef]
- Arroo, R. Terpenes—Flavors, Fragrances, Pharmaca, Pheromones; Wiley-VCH: Weinheim, Germany, 2006. [Google Scholar]
- Puskas, J.E.; Gautriaud, E.; Deffieux, A.; Kennedy, J.P. Natural rubber biosynthesis—A living carbocationic polymerization? Prog. Polym. Sci. 2006, 31, 533–548. [Google Scholar] [CrossRef]
- Matic, A.; Schlaad, H. Thiol-ene photofunctionalization of 1,4-polymyrcene. Polym. Int. 2018, 67, 500–505. [Google Scholar] [CrossRef]
- Runckel, W.J.; Goldblatt, L.A. Inhibition of myrcene polymerization during storage. Ind. Eng. Chem. 1946, 38, 749–751. [Google Scholar] [CrossRef]
- Rummelsburg, A.L. TERPENE RESIN. U.S. Patent US-2354776-A, 14 June 1941. [Google Scholar]
- Marvel, C.S.; Hwa, C.C. Polymyrcene. J. Polym. Sci. 1960, 45, 25–34. [Google Scholar] [CrossRef]
- Newmark, R.A.; Majumdar, R.N. C-13-NMR spectra of cis-polymyrcene and cis-polyfarnesene. J. Polym. Sci. A Polym. Chem. 1988, 26, 71–77. [Google Scholar] [CrossRef]
- Stepto, R.F. Dispersity in polymer science. Pure Appl. Chem. 2009, 81, 351–353. [Google Scholar] [CrossRef] [Green Version]
- Cawse, J.L.; Stanford, J.L.; Still, R.H. Polymers from renewable sources.4. polyurethane elastomers based on myrcene polyols. J. Appl. Polym. Sci. 1986, 31, 1549–1565. [Google Scholar] [CrossRef]
- Cawse, J.L.; Stanford, J.L.; Still, R.H. Polymers from renewable sources. 5. Myrcene-based polyols as rubber-toughening agents in glassy polyurethanes. Polymer 1987, 28, 368–374. [Google Scholar] [CrossRef]
- Lamparelli, D.H.; Paradiso, V.; Della Monica, F.; Proto, A.; Guerra, S.; Giannini, L.; Capacchione, C. Toward More Sustainable Elastomers: Stereoselective Copolymerization of Linear Terpenes with Butadiene. Macromolecules 2020, 53, 1665–1673. [Google Scholar] [CrossRef]
- Loughmari, S.; Hafid, A.; Bouazza, A.; El Bouadili, A.; Zinck, P.; Visseaux, M. Highly stereoselective coordination polymerization of beta-myrcene from a lanthanide-based catalyst: Access to bio-sourced elastomers. J. Polym. Sci. Part A Polym. Chem. 2012, 50, 2898–2905. [Google Scholar] [CrossRef]
- Valente, A.; Mortreux, A.; Visseaux, M.; Zinck, P. Coordinative Chain Transfer Polymerization. Chem. Rev. 2013, 113, 3836–3857. [Google Scholar] [CrossRef]
- Naddeo, M.; Buonerba, A.; Luciano, E.; Grassi, A.; Proto, A.; Capacchione, C. Stereoselective polymerization of biosourced terpenes beta-myrcene and beta-ocimene and their copolymerization with styrene promoted by titanium catalysts. Polymer 2017, 131, 151–159. [Google Scholar] [CrossRef]
- Kuhlman, R.L.; Wenzel, T.T. Investigations of chain shuttling olefin polymerization using deuterium labeling. Macromolecules 2008, 41, 4090–4094. [Google Scholar] [CrossRef]
- Camara, J.M.; Petros, R.A.; Norton, J.R. Zirconium-Catalyzed Carboalumination of alpha-Olefins and Chain Growth of Aluminum Alkyls: Kinetics and Mechanism. J. Am. Chem. Soc. 2011, 133, 5263–5273. [Google Scholar] [CrossRef] [PubMed]
- Paradiso, V.; Capaccio, V.; Lamparelli, D.H.; Capacchione, C. OSSO-bisphenolate metal complexes: A powerful and versatile tool in polymerization catalysis. Coord. Chem. Rev. 2021, 429, 213644. [Google Scholar] [CrossRef]
- Gonzalez-Zapata, J.L.; Enriquez-Medrano, F.J.; Lopez Gonzalez, H.R.; Revilla-Vazquez, J.; Carrizales, R.M.; Georgouvelas, D.; Valencia, L.; Diaz de Leon Gomez, R.E. Introducing random bio-terpene segments to high cis-polybutadiene: Making elastomeric materials more sustainable. RSC Adv. 2020, 10, 44096–44102. [Google Scholar] [CrossRef]
- You, F.; Zhai, J.; So, Y.M.; Shi, X. Rigid Acridane-Based Pincer Supported Rare-Earth Complexes for cis-1,4-Polymerization of 1,3-Conjugated Dienes. Inorg. Chem. 2021, 60, 1797–1805. [Google Scholar] [CrossRef] [PubMed]
- Winnacker, M. Pinenes: Abundant and Renewable Building Blocks for a Variety of Sustainable Polymers. Angew. Chem. Int. Ed. 2018, 57, 14362–14371. [Google Scholar] [CrossRef] [PubMed]
- Yao, K.; Tang, C. Controlled Polymerization of Next-Generation Renewable Monomers and Beyond. Macromolecules 2013, 46, 1689–1712. [Google Scholar] [CrossRef]
- Yu, P.; Li, A.L.; Liang, H.; Lu, J. Polymerization of beta-pinene with Schiff-Base nickel complexes catalyst: Synthesis of relatively high molecular weight poly(beta-pinene) at high temperature with high productivity. J. Polym. Sci. A Polym. Chem. 2007, 45, 3739–3746. [Google Scholar] [CrossRef]
- Roberts, W.J.; Day, A.R. A study of the polymerization of alpha-pinene and beta-pinene with friedel crafts type catalysts. J. Am. Chem. Soc. 1950, 72, 1226–1230. [Google Scholar] [CrossRef]
- Kennedy, J.P.; Liao, T.P.; Guhaniyogi, S.; Chang, V.S. New telechelic polymers and sequential copolymers by polyfunctional initiator-transfer agents (inifers). 20. Poly(beta-pinenes) carrying one, two, and three functional end groups. II. Syntheses and characterization. J. Polym. Sci. Part A Polym. Chem. 1982, 20, 3229–3240. [Google Scholar] [CrossRef]
- Lu, J.; Kamigaito, M.; Sawamoto, M.; Higashimura, T.; Deng, Y.X. Cationic polymerization of beta-pinene with the AlC3/SbCl3 binary catalyst: Comparison with alpha-pinene polymerization. J. Appl. Polym. Sci. 1996, 61, 1011–1016. [Google Scholar] [CrossRef]
- Mathers, R.T.; Lewis, S.P. Monoterpenes as Polymerization Solvents and Monomers in Polymer Chemistry. In Green Polymerization Methods: Renewable Starting Materials, Catalysis and Waste Reduction; Mathers, R.T., Meier, M.A., Eds.; Wiley-VCH: Weinheim, Germany, 2011; pp. 91–128. [Google Scholar]
- Satoh, K. Controlled/living polymerization of renewable vinyl monomers into bio-based polymers. Polym. J. 2015, 47, 527–536. [Google Scholar] [CrossRef]
- Lu, J.; Kamigaito, M.; Sawamoto, M.; Higashimura, T.; Deng, Y.X. Living cationic isomerization polymerization of beta-pinene. Initiation with HCl-2-chloroethyl vinyl ether adduct TiCl3(OiPr) in conjunction with nBu(4)NCl. Macromolecules 1997, 30, 22–26. [Google Scholar] [CrossRef]
- Satoh, K.; Sugiyama, H.; Kamigaito, M. Biomass-derived heat-resistant alicyclic hydrocarbon polymers: Poly(terpenes) and their hydrogenated derivatives. Green Chem. 2006, 8, 878–882. [Google Scholar] [CrossRef]
- Satoh, K.; Nakahara, A.; Mukunoki, K.; Sugiyama, H.; Saito, H.; Kamigaito, M. Sustainable cycloolefin polymer from pine tree oil for optoelectronics material: Living cationic polymerization of beta-pinene and catalytic hydrogenation of high-molecular-weight hydrogenated poly(beta-pinene). Polym. Chem. 2014, 5, 3222–3230. [Google Scholar] [CrossRef]
- Sawamoto, M. Modern cationic vinyl polymerization. Prog. Polym. Sci. 1991, 16, 111–172. [Google Scholar] [CrossRef]
- Aoshima, S.; Yoshida, T.; Kanazawa, A.; Kanaoka, S. New stage in living cationic polymerization: An array of effective Lewis acid catalysts and fast living polymerization in seconds. J. Polym. Sci. Part A Polym. Chem. 2007, 45, 1801–1813. [Google Scholar] [CrossRef]
- McCaulay, D.A.; Lien, A.P. Relative basicity of the methylbenzenes. J. Am. Chem. Soc. 1951, 73, 2013–2017. [Google Scholar] [CrossRef]
- Solari, E.; Floriani, C.; Chiesivilla, A.; Guastini, C. Titanium tetrachloride binding and making arenes from acetylenes—The synthesis and X-ray crystal-structure of a titanium(iv) hexamethylbenzene complex. J. Chem. Soc. Chem. Comm. 1989, 1747–1749. [Google Scholar] [CrossRef]
- Ulvenlund, S.; Wheatley, A.; Bengtsson, L.A. Spectroscopic investigation of concentrated-solutions of gallium(iii) chloride in mesitylene and benzene. J. Chem. Soc. Dalton Trans. 1995, 255–263. [Google Scholar] [CrossRef]
- Laurence, C.; Graton, J.; Berthelot, M.; Besseau, F.; Le Questel, J.Y.; Lucon, M.; Ouvrard, C.; Planchat, A.; Renault, E. An Enthalpic Scale of Hydrogen-Bond Basicity. 4. Carbon pi Bases, Oxygen Bases, and Miscellaneous Second-Row, Third-Row, and Fourth-Row Bases and a Survey of the 4-Fluorophenol Affinity Scale. J. Org. Chem. 2010, 75, 4105–4123. [Google Scholar] [CrossRef]
- Karasawa, Y.; Kimura, M.; Kanazawa, A.; Kanaoka, S.; Aoshima, S. New initiating systems for cationic polymerization of plant-derived monomers: GaCl3/alkylbenzene-induced controlled cationic polymerization of beta-pinene. Polym. J. 2015, 47, 152–157. [Google Scholar] [CrossRef]
- Kukhta, N.A.; Vasilenko, I.V.; Kostjuk, S.V. Room temperature cationic polymerization of beta-pinene using modified AlCl3 catalyst: Toward sustainable plastics from renewable biomass resources. Green Chem. 2011, 13, 2362–2364. [Google Scholar] [CrossRef]
- Doiuchi, T.; Yamaguchi, H.; Minoura, Y. Cyclo-copolymerization of d-limonene with maleic-anhydride. Eur. Polym. J. 1981, 17, 961–968. [Google Scholar] [CrossRef]
- Andriotis, E.G.; Achilias, D.S. Role of Polylimonene as a Bio-Based Additive in Thermal Oxidation of High Impact Polystyrene. Macromol. Symp. 2013, 331, 173–180. [Google Scholar] [CrossRef]
- Modena, M.; Bates, R.B.; Marvel, C.S. Some low molecular weight polymers of d-limonene and related terpenes obtained by Ziegler-type catalysts. J. Polym Sci. A Polym. Chem. 1965, 3, 949–960. [Google Scholar] [CrossRef]
- Pietila, H.; Sivola, A.; Sheffer, H. Cationic polymerization of beta-pinene, styrene and alpha-methylstyrene. J. Polym. Sci. Part A Polym. Chem. 1970, 8, 727–737. [Google Scholar] [CrossRef]
- Sahu, P.; Bhowmick, A.K.; Kali, G. Terpene Based Elastomers: Synthesis, Properties, and Applications. Processes 2020, 8, 553. [Google Scholar] [CrossRef]
- Sharma, S.; Srivastava, A.K. Alternating copolymers of limonene with methyl methacrylate: Kinetics and mechanism. J. Macromol. Sci. A 2003, A40, 593–603. [Google Scholar] [CrossRef]
- Zhang, Y.; Dube, M.A. Copolymerization of 2-Ethylhexyl Acrylate and D-Limonene. Polym. Plast. Technol. Eng. 2015, 54, 499–505. [Google Scholar] [CrossRef]
- Ishido, Y.; Kanazawa, A.; Kanaoka, S.; Aoshima, S. New Degradable Alternating Copolymers from Naturally Occurring Aldehydes: Well-Controlled Cationic Copolymerization and Complete Degradation. Macromolecules 2012, 45, 4060–4068. [Google Scholar] [CrossRef]
- Zhang, D.H.; Hillmyer, M.A.; Tolman, W.B. Catalytic polymerization of a cyclic ester derived from a “cool” natural precursor. Biomacromolecules 2005, 6, 2091–2095. [Google Scholar] [CrossRef]
- Lowe, J.R.; Tolman, W.B.; Hillmyer, M.A. Oxidized Dihydrocarvone as a Renewable Multifunctional Monomer for the Synthesis of Shape Memory Polyesters. Biomacromolecules 2009, 10, 2003–2008. [Google Scholar] [CrossRef]
- Lowe, J.R.; Martello, M.T.; Tolman, W.B.; Hillmyer, M.A. Functional biorenewable polyesters from carvone-derived lactones. Polym. Chem. 2011, 2, 702–708. [Google Scholar] [CrossRef]
- Nishida, T.; Satoh, K.; Nagano, S.; Seki, T.; Tamura, M.; Li, Y.; Tomishige, K.; Kamigaito, M. Biobased Cycloolefin Polymers: Carvone-Derived Cyclic Conjugated Diene with Reactive exo-Methylene Group for Regioselective and Stereospecific Living Cationic Polymerization. ACS Macro Lett. 2020, 9, 1178–1183. [Google Scholar] [CrossRef]
- Winnacker, M.; Sag, J. Sustainable terpene-based polyamides via anionic polymerization of a pinene-derived lactam. J. Chem. Soc. Chem. Comm. 2018, 54, 841–844. [Google Scholar] [CrossRef] [PubMed]
- Parrino, F.; Fidalgo, A.; Palmisano, L.; Ilharco, L.M.; Pagliaro, M.; Ciriminna, R. Polymers of Limonene Oxide and Carbon Dioxide: Polycarbonates of the Solar Economy. ACS Omega 2018, 3, 4884–4890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castro-Gomez, F.; Salassa, G.; Kleij, A.W.; Bo, C. A DFT Study on the Mechanism of the Cycloaddition Reaction of CO2 to Epoxides Catalyzed by Zn(Salphen) Complexes. Chem. Eur. J. 2013, 19, 6289–6298. [Google Scholar] [CrossRef]
- Park, H.J.; Ryu, C.Y.; Crivello, J.V. Photoinitiated cationic polymerization of limonene 1,2-oxide and a-pinene oxide. J. Polym. Sci. Part A Polym. Chem. 2013, 51, 109–117. [Google Scholar] [CrossRef]
- Munoz, M.T.; Palenzuela, M.; Cuenca, T.; Mosquera, M.E. Aluminum Aryloxide Compounds as Very Active Catalysts for Glycidyl Methacrylate Selective Ring-Opening Polymerization. Chemcatchem 2018, 10, 936–939. [Google Scholar] [CrossRef]
- Sessini, V.; Palenzuela, M.; Damian, J.; Mosquera, M.E. Bio-based polyether from limonene oxide catalytic ROP as green polymeric plasticizer for PLA. Polymer 2020, 210, 123003. [Google Scholar] [CrossRef]
- Pena Carrodeguas, L.; Martin, C.; Kleij, A.W. Semiaromatic Polyesters Derived from Renewable Terpene Oxides with High Glass Transitions. Macromolecules 2017, 50, 5337–5345. [Google Scholar] [CrossRef]
- Neumann, S.; Leitner, L.C.; Schmalz, H.; Agarwal, S.; Greiner, A. Unlocking the Processability and Recyclability of Biobased Poly(limonene carbonate). ACS Sustain. Chem. Eng. 2020, 8, 6442–6448. [Google Scholar] [CrossRef]
- Zhang, Y.Y.; Wu, G.P.; Darensbourg, D.J. CO2-Based Block Copolymers: Present and Future Designs. Trends Chem. 2020, 2, 750–763. [Google Scholar] [CrossRef]
- Darensbourg, D.J. Chain transfer agents utilized in epoxide and CO2 copolymerization processes. Green Chem. 2019, 21, 2214–2223. [Google Scholar] [CrossRef]
- Bhat, G.A.; Luo, M.; Darensbourg, D.J. Catalysis of carbon dioxide and oxetanes to produce aliphatic polycarbonates. Green Chem. 2020, 22, 7707–7724. [Google Scholar] [CrossRef]
- Darensbourg, D.J.; Mackiewicz, R.M. Role of the cocatalyst in the copolymerization of CO2 and cyclohexene oxide utilizing chromium salen complexes. J. Am. Chem. Soc. 2005, 127, 14026–14038. [Google Scholar] [CrossRef]
- Jeske, R.C.; DiCiccio, A.M.; Coates, G.W. Alternating copolymerization of Epoxides and cyclic anhydrides: An improved route to aliphatic polyesters. J. Am. Chem. Soc. 2007, 129, 11330–11331. [Google Scholar] [CrossRef]
- Robert, C.; de Montigny, F.; Thomas, C.M. Tandem synthesis of alternating polyesters from renewable resources. Nat. Commun. 2011, 2, 586. [Google Scholar] [CrossRef] [PubMed]
- Nejad, E.H.; Paoniasari, A.; van Melis, C.G.; Koning, C.E.; Duchateau, R. Catalytic Ring-Opening Copolymerization of Limonene Oxide and Phthalic Anhydride: Toward Partially Renewable Polyesters. Macromolecules 2013, 46, 631–637. [Google Scholar] [CrossRef]
- Isnard, F.; Lamberti, M.; Pellecchia, C.; Mazzeo, M. Ring-Opening Copolymerization of Epoxides with Cyclic Anhydrides Promoted by Bimetallic and Monometallic Phenoxy-Imine Aluminum complexes. Chemcatchem 2017, 9, 2972–2979. [Google Scholar] [CrossRef]
- de Sarasa Buchaca, M.M.; de la Cruz-Martinez, F.; Martinez, J.; Alonso-Moreno, C.; Fernandez-Baeza, J.; Tejeda, J.; Niza, E.; Castro-Osma, J.A.; Otero, A.; Lara-Sanchez, A. Alternating Copolymerization of Epoxides and Anhydrides Catalyzed by Aluminum Complexes. ACS Omega 2018, 3, 17581–17589. [Google Scholar] [CrossRef]
- Santulli, F.; D’Auria, I.; Boggioni, L.; Losio, S.; Proverbio, M.; Costabile, C.; Mazzeo, M. Bimetallic Aluminum Complexes Bearing Binaphthyl-Based Iminophenolate Ligands as Catalysts for the Synthesis of Polyesters. Organometallics 2020, 39, 1213–1220. [Google Scholar] [CrossRef]
- Sanford, M.J.; Pena Carrodeguas, L.; Van Zee, N.J.; Kleij, A.W.; Coates, G.W. Alternating Copolymerization of Propylene Oxide and Cyclohexene Oxide with Tricyclic Anhydrides: Access to Partially Renewable Aliphatic Polyesters with High Glass Transition Temperatures. Macromolecules 2016, 49, 6394–6400. [Google Scholar] [CrossRef]
- Van Zee, N.J.; Sanford, M.J.; Coates, G.W. Electronic Effects of Aluminum Complexes in the Copolymerization of Propylene Oxide with Tricyclic Anhydrides: Access to Well-Defined, Functionalizable Aliphatic Polyesters. J. Am. Chem. Soc. 2016, 138, 2755–2761. [Google Scholar] [CrossRef]
- Inoue, S.; Koinuma, H.; Tsuruta, T. Copolymerization of carbon dioxide and epoxide. J. Pol. Sci. Part B Polym. Lett. 1969, 7, 287–292. [Google Scholar] [CrossRef]
- Anderson, T.S.; Kozak, C.M. Ring-opening polymerization of epoxides and ring-opening copolymerization of CO2 with epoxides by a zinc amino-bis(phenolate) catalyst. Eur. Polym. J. 2019, 120, 109237. [Google Scholar] [CrossRef]
- Bobbink, F.D.; van Muyden, A.P.; Dyson, P.J. En route to CO2-containing renewable materials: Catalytic synthesis of polycarbonates and non-isocyanate polyhydroxyurethanes derived from cyclic carbonates. J Chem. Soc. Chem. Comm. 2019, 55, 1360–1373. [Google Scholar] [CrossRef] [Green Version]
- Mani, R.; Bhattacharya, M.; Tang, J. Functionalization of polyesters with maleic anhydride by reactive extrusion. J. Polym. Sci. Part A Polym. Chem. 1999, 37, 1693–1702. [Google Scholar] [CrossRef]
- Raquez, J.M.; Degee, P.; Nabar, Y.; Narayan, R.; Dubois, P. Biodegradable materials by reactive extrusion: From catalyzed polymerization to functionalization and blend compatibilization. C. D. Chim. 2006, 9, 1370–1379. [Google Scholar] [CrossRef]
- Fink, J.K. Reactive Polymers: Fundamentals and Applications. A Concise Guide to Industrial Polymers; Plastics Design Library; Elsevier: Amsterdam, The Netherlands, 2018; pp. 449–496. [Google Scholar]
- Raquez, J.M.; Ramy-Ratiarison, R.; Murariu, M.; Dubois, P. Reactive Extrusion of PLA-based Materials: From Synthesis to Reactive melt-blending. Technol. Process. Prop. Addit. Appl. R. Soc. Chem. Polym. Chem. Ser. 2014, 101–122. [Google Scholar] [CrossRef]
- Jacobsen, S.; Fritz, H.G.; Degee, P.; Dubois, P.; Jerome, R. Single-step reactive extrusion of PLLA in a corotating twin-screw extruder promoted by 2-ethylhexanoic acid tin(II) salt and triphenylphosphine. Polymer 2000, 41, 3395–3403. [Google Scholar] [CrossRef] [Green Version]
- Bruester, B.; Adjoua, Y.O.; Dieden, R.; Grysan, P.; Federico, C.E.; Berthe, V.; Addiego, F. Plasticization of Polylactide with Myrcene and Limonene as Bio-Based Plasticizers: Conventional vs. Reactive Extrusion. Polymers 2019, 11, 1363. [Google Scholar] [CrossRef] [Green Version]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mosquera, M.E.G.; Jiménez, G.; Tabernero, V.; Vinueza-Vaca, J.; García-Estrada, C.; Kosalková, K.; Sola-Landa, A.; Monje, B.; Acosta, C.; Alonso, R.; et al. Terpenes and Terpenoids: Building Blocks to Produce Biopolymers. Sustain. Chem. 2021, 2, 467-492. https://doi.org/10.3390/suschem2030026
Mosquera MEG, Jiménez G, Tabernero V, Vinueza-Vaca J, García-Estrada C, Kosalková K, Sola-Landa A, Monje B, Acosta C, Alonso R, et al. Terpenes and Terpenoids: Building Blocks to Produce Biopolymers. Sustainable Chemistry. 2021; 2(3):467-492. https://doi.org/10.3390/suschem2030026
Chicago/Turabian StyleMosquera, Marta. E. G., Gerardo Jiménez, Vanessa Tabernero, Joan Vinueza-Vaca, Carlos García-Estrada, Katarina Kosalková, Alberto Sola-Landa, Belén Monje, Carolina Acosta, Rafael Alonso, and et al. 2021. "Terpenes and Terpenoids: Building Blocks to Produce Biopolymers" Sustainable Chemistry 2, no. 3: 467-492. https://doi.org/10.3390/suschem2030026
APA StyleMosquera, M. E. G., Jiménez, G., Tabernero, V., Vinueza-Vaca, J., García-Estrada, C., Kosalková, K., Sola-Landa, A., Monje, B., Acosta, C., Alonso, R., & Valera, M. Á. (2021). Terpenes and Terpenoids: Building Blocks to Produce Biopolymers. Sustainable Chemistry, 2(3), 467-492. https://doi.org/10.3390/suschem2030026