
Deciphering the Molecular Interaction Process of Gallium Maltolate on SARS-CoV-2 Main and Papain-Like Proteases: A Theoretical Study

 , , , , , , and
, , , , , , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Reactivity Indexes and Molecular Electrostatic Potential (MEP) Analysis
2.2. Molecular Docking
2.3. Non-Covalent Interactions
3. Results
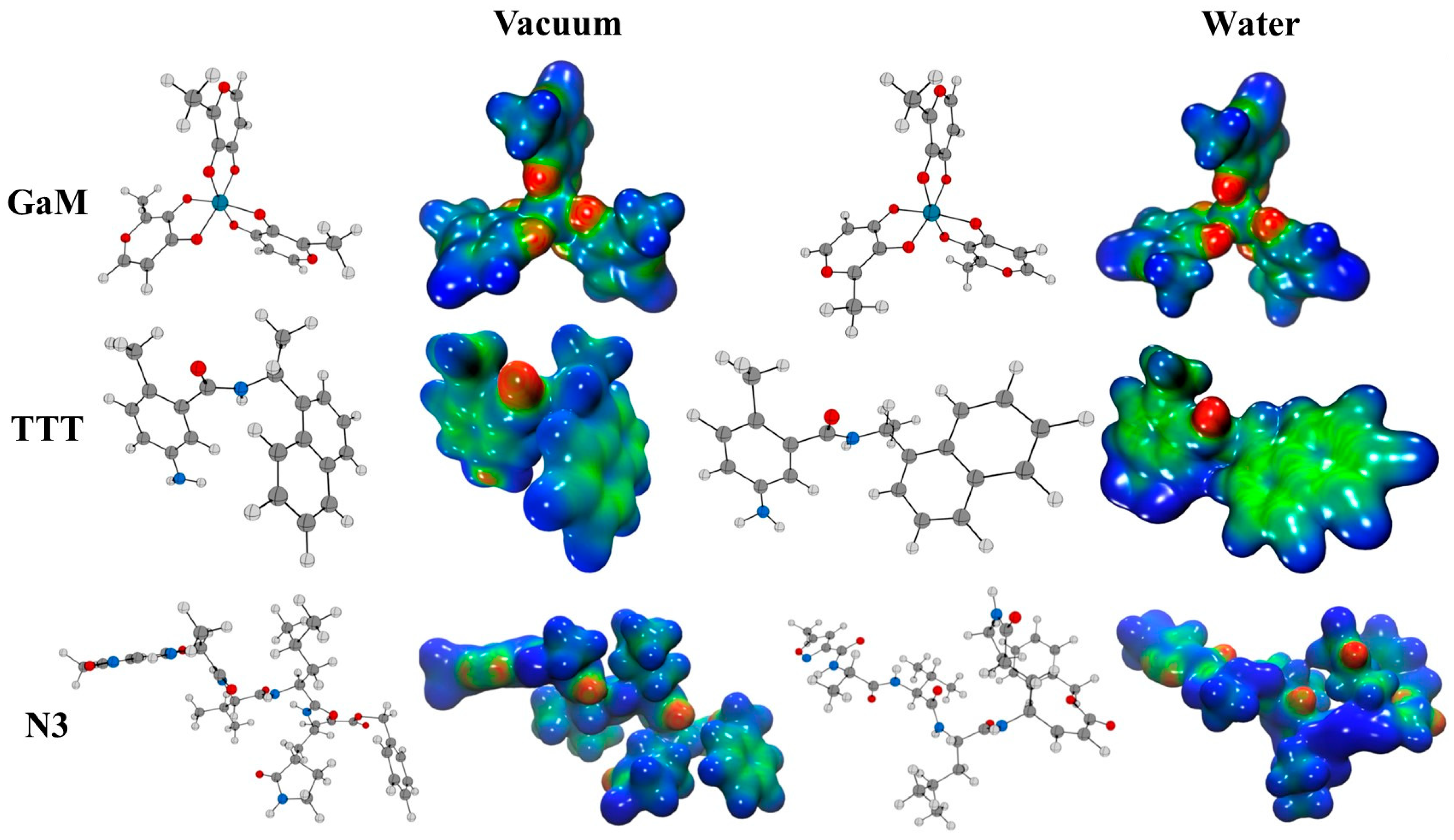
3.1. Reactivity Indexes and Molecular Electrostatic Potential Analysis
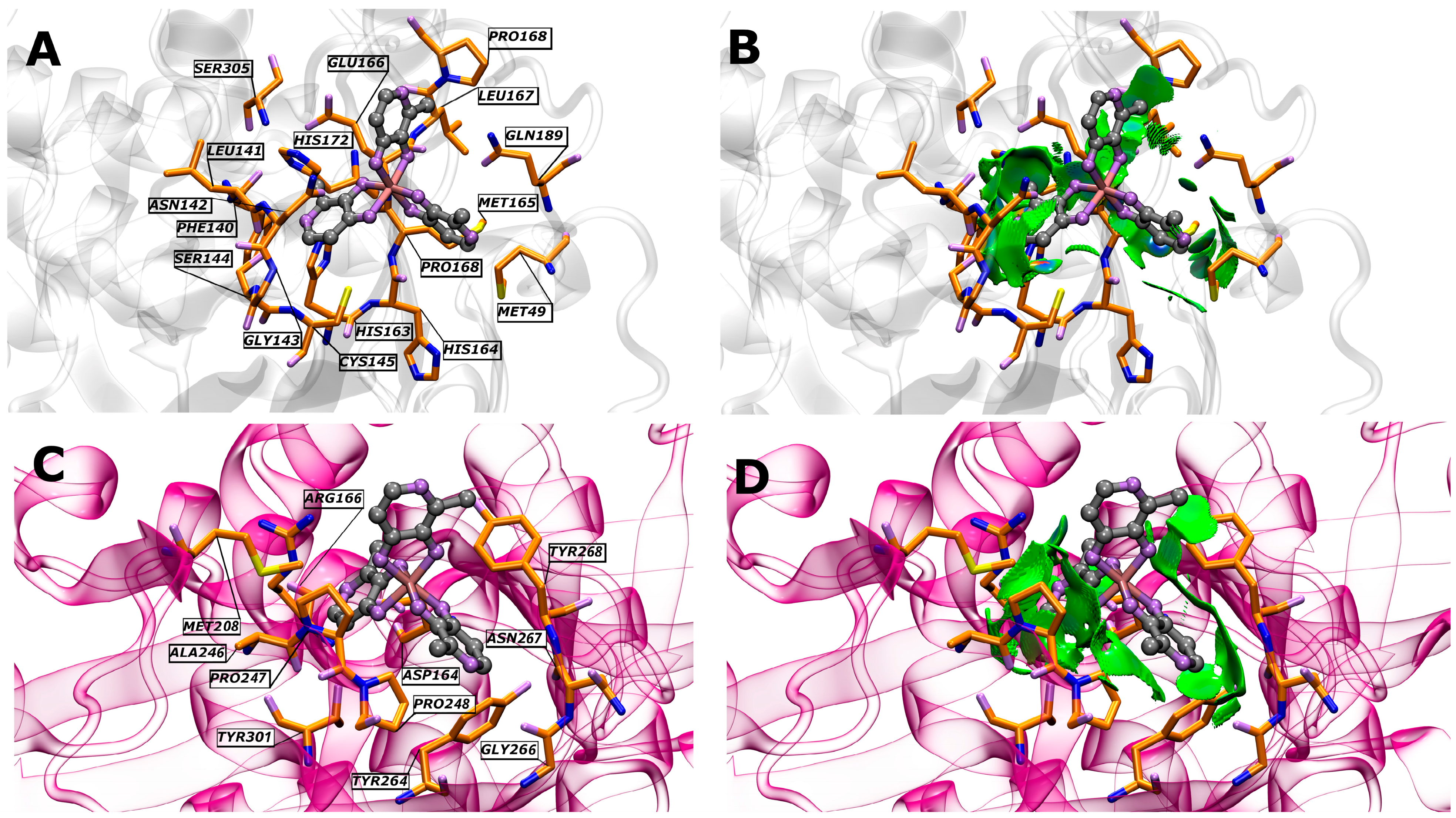
3.2. Molecular Docking Analysis
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| DFT | Density functional theory |
| COVID-19 | Coronavirus disease 2019 |
| GaM | Gallium maltolate |
| SARS-CoV-2 | Severe acute respiratory syndrome coronavirus 2 |
| Mpro | Main protease |
| PLpro | Papain-like protease |
| FMO | Frontier molecular orbital |
| HOMO | Highest occupied molecular orbital |
| LUMO | Lowest unoccupied molecular orbital |
| MEP | Molecular electrostatic potential |
| NCI | Non-covalent interaction |
| RDG | Reduced density gradient |
| TTT | 5-amino-2-methyl-N-[(1R)-1-naphthalen-1-ylethyl] benzamide |
| N3 | N-[(5-methyl-1,2-oxazol-3-yl)carbonyl]-L-alanyl-L-valyl-N-{(2S,3E)-5-(benzyloxy)-5-oxo-1-[(3S)-2-oxopyrrolidin-3-yl]pent-3-en-2-yl}-L-leucinamide |
| η | Global hardness |
| χ | Electronegativity |
| ω | Electrophilicity |
| ω+ | Electron acceptor |
| ω− | Electron donator |
| Δω± | Net electrophilicity |
| ΔEbinding | Binding energy from molecular docking calculations |
References
- Žákovská, A.; Nováková, O.; Balcarová, Z.; Bierbach, U.; Farrell, N.; Brabec, V. DNA Interactions of Antitumor Trans-[PtCl2(NH3)(Quinoline)]. Eur. J. Biochem. 1998, 254, 547–557. [Google Scholar] [CrossRef] [PubMed]
- Kostova, I. Platinum Complexes as Anticancer Agents. Recent Pat. Anti-Cancer Drug Discov. 2006, 1, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez Gómez, M. Uso de Cisplatino y Derivados de Platino En Quimioterapia. 2017. Available online: https://hdl.handle.net/20.500.14352/20714 (accessed on 24 January 2024).
- Liu, W.-P.; Ye, Q.-S.; Yu, Y.; Chen, X.-Z.; Hou, S.-Q.; Lou, L.-G.; Yang, Y.-P.; Wang, Y.-M.; Su, Q. Novel Lipophilic Platinum(II) Compounds of Salicylate Derivatives. Platin. Met. Rev. 2008, 52, 163–171. [Google Scholar] [CrossRef]
- Was, H.; Borkowska, A.; Bagues, A.; Tu, L.; Liu, J.Y.H.; Lu, Z.; Rudd, J.A.; Nurgali, K.; Abalo, R. Mechanisms of Chemotherapy-Induced Neurotoxicity. Front. Pharmacol. 2022, 13, 750507. [Google Scholar] [CrossRef] [PubMed]
- Eryazici, I.; Moorefield, C.N.; Newkome, G.R. Square-Planar Pd(II), Pt(II), and Au(III) Terpyridine Complexes: Their Syntheses, Physical Properties, Supramolecular Constructs, and Biomedical Activities. Chem. Rev. 2008, 108, 1834–1895. [Google Scholar] [CrossRef]
- Roberts, J.J.; Thomson, A.J. The Mechanism of Action of Antitumor Platinum Compounds. In Progress in Nucleic Acid Research and Molecular Biology; Cohn, W.E., Ed.; Academic Press: Cambridge, MA, USA, 1979; Volume 22, pp. 71–133. ISBN 0079-6603. [Google Scholar]
- Lippard, S.J. New Chemistry of an Old Molecule: Cis-[Pt(NH3)2Cl2]. Science 1982, 218, 1075–1082. [Google Scholar] [CrossRef]
- Kowol, C.R.; Heffeter, P.; Miklos, W.; Gille, L.; Trondl, R.; Cappellacci, L.; Berger, W.; Keppler, B.K. Mechanisms Underlying Reductant-Induced Reactive Oxygen Species Formation by Anticancer Copper(II) Compounds. J. Biol. Inorg. Chem. 2012, 17, 409–423. [Google Scholar] [CrossRef]
- Li, Z.; Yang, X.; Dong, S.; Li, X. DNa Breakage Induced by Piceatannol and Copper(II): Mechanism and Anticancer Properties. Oncol. Lett. 2012, 3, 1087–1094. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ulukaya, E.; Ari, F.; Dimas, K.; Ikitimur, E.I.; Guney, E.; Yilmaz, V.T. Anti-Cancer Activity of a Novel Palladium(II) Complex on Human Breast Cancer Cells in Vitro and in Vivo. Eur. J. Med. Chem. 2011, 46, 4957–4963. [Google Scholar] [CrossRef]
- Ferraz, K.S.O.; Ferandes, L.; Carrilho, D.; Pinto, M.C.X.; Leite, M.d.F.; Souza–Fagundes, E.M.; Speziali, N.L.; Mendes, I.C.; Beraldo, H. 2-Benzoylpyridine-N(4)-Tolyl Thiosemicarbazones and Their Palladium(II) Complexes: Cytotoxicity against Leukemia Cells. Bioorg. Med. Chem. 2009, 17, 7138–7144. [Google Scholar] [CrossRef]
- Abu-Surrah, A.S.; Abu Safieh, K.A.; Ahmad, I.M.; Abdalla, M.Y.; Ayoub, M.T.; Qaroush, A.K.; Abu-Mahtheieh, A.M. New Palladium(II) Complexes Bearing Pyrazole-Based Schiff Base Ligands: Synthesis, Characterization and Cytotoxicity. Eur. J. Med. Chem. 2010, 45, 471–475. [Google Scholar] [CrossRef] [PubMed]
- Casini, A.; Messori, L. Molecular Mechanisms and Proposed Targets for Selected Anticancer Gold Compounds. Curr. Top. Med. Chem. 2011, 11, 2647–2660. [Google Scholar] [CrossRef]
- Lin, I.W.-S.; Lok, C.-N.; Yan, K.; Che, C.-M. A Silver Complex of N,N′-Disubstituted Cyclic Thiourea as an Anti-Inflammatory Inhibitor of IκB Kinase. Chem. Commun. 2013, 49, 3297–3299. [Google Scholar] [CrossRef]
- Banti, C.N.; Giannoulis, A.D.; Kourkoumelis, N.; Owczarzak, A.M.; Poyraz, M.; Kubicki, M.; Charalabopoulos, K.; Hadjikakou, S.K. Mixed Ligand–Silver(i) Complexes with Anti-Inflammatory Agents Which Can Bind to Lipoxygenase and Calf-Thymus DNA, Modulating Their Function and Inducing Apoptosis†. Metallomics 2012, 4, 545–560. [Google Scholar] [CrossRef] [PubMed]
- Bergamo, A.; Gaiddon, C.; Schellens, J.H.M.; Beijnen, J.H.; Sava, G. Approaching Tumour Therapy beyond Platinum Drugs: Status of the Art and Perspectives of Ruthenium Drug Candidates. J. Inorg. Biochem. 2012, 106, 90–99. [Google Scholar] [CrossRef] [PubMed]
- Bergamo, A.; Sava, G. Ruthenium Anticancer Compounds: Myths and Realities of the Emerging Metal-Based Drugs. Dalton Trans. 2011, 40, 7817–7823. [Google Scholar] [CrossRef] [PubMed]
- Cunha, L.C.; Lage, D.P.; Ferreira, L.S.; Saboia-Vahia, L.; Coelho, E.A.F.; Belo, V.S.; Teixeira-Neto, R.G.; Soares, L.F.; Chagas, R.C.R.; da Silva, E.S. Leishmanicidal Activity of Ibuprofen and Its Complexes with Ni(II), Mn(II) and Pd(II). Inorg. Chem. Commun. 2020, 113, 107756. [Google Scholar] [CrossRef]
- Medici, S.; Peana, M.; Nurchi, V.M.; Lachowicz, J.I.; Crisponi, G.; Zoroddu, M.A. Noble Metals in Medicine: Latest Advances. Coord. Chem. Rev. 2015, 284, 329–350. [Google Scholar] [CrossRef]
- Choroba, K.; Machura, B.; Szlapa-Kula, A.; Malecki, J.G.; Raposo, L.; Roma-Rodrigues, C.; Cordeiro, S.; Baptista, P.V.; Fernandes, A.R. Square Planar Au(III), Pt(II) and Cu(II) Complexes with Quinoline-Substituted 2,2′:6′,2″-Terpyridine Ligands: From in Vitro to in Vivo Biological Properties. Eur. J. Med. Chem. 2021, 218, 113404. [Google Scholar] [CrossRef]
- Karges, J.; Cohen, S.M. Metal Complexes as Antiviral Agents for SARS-CoV-2. ChemBioChem 2021, 22, 2600–2607. [Google Scholar] [CrossRef]
- Jaros, S.W.; Król, J.; Bażanów, B.; Poradowski, D.; Chrószcz, A.; Nesterov, D.S.; Kirillov, A.M.; Smoleński, P. Antiviral, Antibacterial, Antifungal, and Cytotoxic Silver(I) BioMOF Assembled from 1,3,5-Triaza-7-Phoshaadamantane and Pyromellitic Acid. Molecules 2020, 25, 2119. [Google Scholar] [CrossRef] [PubMed]
- Aprajita; Choudhary, M. Design, Synthesis and Characterization of Novel Ni(II) and Cu(II) Complexes as Antivirus Drug Candidates against SARS-CoV-2 and HIV Virus. J. Mol. Struct. 2022, 1263, 133114. [Google Scholar] [CrossRef]
- De Paiva, R.E.F.; Marçal Neto, A.; Santos, I.A.; Jardim, A.C.G.; Corbi, P.P.; Bergamini, F.R.G. What Is Holding Back the Development of Antiviral Metallodrugs? A Literature Overview and Implications for SARS-CoV-2 Therapeutics and Future Viral Outbreaks. Dalton Trans. 2020, 49, 16004–16033. [Google Scholar] [CrossRef] [PubMed]
- Haribabu, J.; Srividya, S.; Mahendiran, D.; Gayathri, D.; Venkatramu, V.; Bhuvanesh, N.; Karvembu, R. Synthesis of Palladium(II) Complexes via Michael Addition: Antiproliferative Effects through ROS-Mediated Mitochondrial Apoptosis and Docking with SARS-CoV-2. Inorg. Chem. 2020, 59, 17109–17122. [Google Scholar] [CrossRef]
- Rasey, J.S.; Nelson, N.J.; Larson, S.M. Tumor Cell Toxicity of Stable Gallium Nitrate: Enhancement by Transferrin and Protection by Iron. Eur. J. Cancer Clin. Oncol. 1982, 18, 661–668. [Google Scholar] [CrossRef]
- Baran, E.J. Metalofármacos: Una Nueva Perspectiva Para La Farmacología y La Medicina. Anales Acad. Nac. Cs. Ex. Fís. Nat. 2014, 66, 5–21. [Google Scholar]
- Yin, H.Y.; Gao, J.J.; Chen, X.; Ma, B.; Yang, Z.S.; Tang, J.; Wang, B.W.; Chen, T.; Wang, C.; Gao, S.; et al. A Gallium(III) Complex That Engages Protein Disulfide Isomerase A3 (PDIA3) as an Anticancer Target. Angew. Chem.-Int. Ed. 2020, 59, 20147–20153. [Google Scholar] [CrossRef]
- Bernstein, L.R.; Zhang, L. Gallium Maltolate Has in Vitro Antiviral Activity against SARS-CoV-2 and Is a Potential Treatment for COVID-19. Antivir. Chem. Chemother. 2020, 28, 1–4. [Google Scholar] [CrossRef]
- Mohapatra, R.K.; Perekhoda, L.; Azam, M.; Suleiman, M.; Sarangi, A.K.; Semenets, A.; Pintilie, L.; Al-Resayes, S.I. Computational Investigations of Three Main Drugs and Their Comparison with Synthesized Compounds as Potent Inhibitors of SARS-CoV-2 Main Protease (Mpro): DFT, QSAR, Molecular Docking, and in Silico Toxicity Analysis. J. King Saud. Univ. Sci. 2021, 33, 101315. [Google Scholar] [CrossRef] [PubMed]
- Adamo, C.; Barone, V. Toward Reliable Density Functional Methods without Adjustable Parameters: The PBE0 Model. J. Chem. Phys. 1999, 110, 6158. [Google Scholar] [CrossRef]
- Weigend, F.; Ahlrichs, R. Balanced Basis Sets of Split Valence, Triple Zeta Valence and Quadruple Zeta Valence Quality for H to Rn: Design and Assessment of Accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian16 Revision B.01; EEUU: Wallingford, CT, USA, 2016. [Google Scholar]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef]
- Pino-Rios, R.; Yañez, O.; Inostroza, D.; Ruiz, L.; Cardenas, C.; Fuentealba, P.; Tiznado, W. Proposal of a Simple and Effective Local Reactivity Descriptor through a Topological Analysis of an Orbital-Weighted Fukui Function. J. Comput. Chem. 2017, 38, 481–488. [Google Scholar] [CrossRef]
- Koopmans, T. Über Die Zuordnung von Wellenfunktionen Und Eigenwerten Zu Den Einzelnen Elektronen Eines Atoms. Physica 1934, 1, 104–113. [Google Scholar] [CrossRef]
- Parr, R.G.; Pearson, R.G. Absolute Hardness: Companion Parameter to Absolute Electronegativity. J. Am. Chem. Soc. 1983, 105, 7512–7516. [Google Scholar] [CrossRef]
- Lewars, E.G. Computational Chemistry-Introduction to the Theory and Applications of Molecular and Quantum Mechanics; Springer: New York, NY, USA, 2011; ISBN 978-90-481-3860-9. [Google Scholar]
- Young, D.C. Computational Chemistry: A Practical Guide for Applying Techniques to Real World Problems; John Wiley & Sons: New York, NY, USA, 2002; ISBN 9780471333685. [Google Scholar]
- Jensen, F. Introduction to Computational Chemistry, 2nd ed.; John Wiley & Sons: New York, NY, USA, 2006. [Google Scholar]
- Cramer, C.J. Essentials of Computational Chemistry: Theories and Models, 2nd ed.; John Wiley & Sons: New York, NY, USA, 2004; ISBN 978-0-470-09182-1. [Google Scholar]
- Pearson, R.G. Hard and Soft Acids and Bases. J. Am. Chem. Soc. 1963, 85, 3533–3539. [Google Scholar] [CrossRef]
- Pearson, R.G. Chemical Hardness; Wiley: New York, NY, USA, 2005; ISBN 9783527294824. [Google Scholar]
- Gázquez, J.L.; Cedillo, A.; Vela, A. Electrodonating and Electroaccepting Powers. J. Phys. Chem. A 2007, 111, 1966–1970. [Google Scholar] [CrossRef]
- Chattaraj, P.K.; Chakraborty, A.; Giri, S. Net Electrophilicity. J. Phys. Chem. A 2009, 113, 10068–10074. [Google Scholar] [CrossRef]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated Docking with Selective Receptor Flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef]
- Hou, X.; Du, J.; Zhang, J.; Du, L.; Fang, H.; Li, M. How to Improve Docking Accuracy of AutoDock4.2: A Case Study Using Different Electrostatic Potentials. J. Chem. Inf. Model. 2013, 53, 188–200. [Google Scholar] [CrossRef]
- Groom, C.R.; Bruno, I.J.; Lightfoot, M.P.; Ward, S.C. The Cambridge Structural Database. Acta Crystallogr. Sect. B 2016, 72, 171–179. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization, and Multithreading. J. Comput. Chem. 2009, 31, 455–461. [Google Scholar] [CrossRef]
- Jin, Z.; Du, X.; Xu, Y.; Deng, Y.; Liu, M.; Zhao, Y.; Zhang, B.; Li, X.; Zhang, L.; Peng, C.; et al. Structure of Mpro from SARS-CoV-2 and Discovery of Its Inhibitors. Nature 2020, 582, 289–293. [Google Scholar] [CrossRef]
- Gao, X.; Qin, B.; Chen, P.; Zhu, K.; Hou, P.; Wojdyla, J.A.; Wang, M.; Cui, S. Crystal Structure of SARS-CoV-2 Papain-like Protease. Acta Pharm. Sin. B 2021, 11, 237–245. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger Release 2024-1; Canvas, Schrödinger, LLC: New York, NY, USA, 2024.
- Kaminski, G.A.; Friesner, R.A.; Tirado-Rives, J.; Jorgensen, W.L. Evaluation and Reparametrization of the OPLS-AA Force Field for Proteins via Comparison with Accurate Quantum Chemical Calculations on Peptides. J. Phys. Chem. B 2001, 105, 6474–6487. [Google Scholar] [CrossRef]
- Osorio, M.I.; Yáñez, O.; Gallardo, M.; Zuñiga-Bustos, M.; Mulia-Rodríguez, J.; López-Rendón, R.; García-Beltrán, O.; González-Nilo, F.; Pérez-Donoso, J.M. Search for Novel Potent Inhibitors of the SARS-CoV-2 Papain-like Enzyme: A Computational Biochemistry Approach. Pharmaceuticals 2022, 15, 986. [Google Scholar] [CrossRef] [PubMed]
- Yañez, O.; Osorio, M.I.; Uriarte, E.; Areche, C.; Tiznado, W.; Pérez-Donoso, J.M.; García-Beltrán, O.; González-Nilo, F. In Silico Study of Coumarins and Quinolines Derivatives as Potent Inhibitors of SARS-CoV-2 Main Protease. Front. Chem. 2021, 8, 1273. [Google Scholar] [CrossRef]
- Yañez, O.; Osorio, M.I.; Areche, C.; Vasquez-Espinal, A.; Bravo, J.; Sandoval-Aldana, A.; Pérez-Donoso, J.M.; González-Nilo, F.; Matos, M.J.; Osorio, E.; et al. Theobroma Cacao L. Compounds: Theoretical Study and Molecular Modeling as Inhibitors of Main SARS-CoV-2 Protease. Biomed. Pharmacother. 2021, 140, 111764. [Google Scholar] [CrossRef]
- Johnson, E.R.; Keinan, S.; Mori-Sánchez, P.; Contreras-García, J.; Cohen, A.J.; Yang, W. Revealing Noncovalent Interactions. J. Am. Chem. Soc. 2010, 132, 6498–6506. [Google Scholar] [CrossRef]
- Contreras-García, J.; Johnson, E.R.; Keinan, S.; Chaudret, R.; Piquemal, J.P.; Beratan, D.N.; Yang, W. NCIPLOT: A Program for Plotting Noncovalent Interaction Regions. J. Chem. Theory Comput. 2011, 7, 625–632. [Google Scholar] [CrossRef] [PubMed]
- Pichler, W.J. The Important Role of Non-Covalent Drug-Protein Interactions in Drug Hypersensitivity Reactions. Allergy 2022, 77, 404–415. [Google Scholar] [CrossRef] [PubMed]
- Tandon, H.; Yadav, P.; Chakraborty, T.; Suhag, V. Can Chemical Reactivity Descriptors Explain Catalytic Reactivity? J. Organomet. Chem. 2022, 960, 122229. [Google Scholar] [CrossRef]
- Sutanto, F.; Konstantinidou, M.; Dömling, A. Covalent Inhibitors: A Rational Approach to Drug Discovery. RSC Med. Chem. 2020, 11, 876–884. [Google Scholar] [CrossRef] [PubMed]
- Hagar, M.; Ahmed, H.A.; Aljohani, G.; Alhaddad, O.A. Investigation of Some Antiviral N-Heterocycles as COVID-19 Drug: Molecular Docking and DFT Calculations. Int. J. Mol. Sci. 2020, 21, 3922. [Google Scholar] [CrossRef] [PubMed]
- Suárez, D.; Díaz, N. SARS-CoV-2 Main Protease: A Molecular Dynamics Study. J. Chem. Inf. Model. 2020, 60, 5815–5831. [Google Scholar] [CrossRef] [PubMed]
- Ullrich, S.; Nitsche, C. The SARS-CoV-2 Main Protease as Drug Target. Bioorg. Med. Chem. Lett. 2020, 30, 127377. [Google Scholar] [CrossRef] [PubMed]
- Ershov, P.V.; Yablokov, E.O.; Mezentsev, Y.V.; Chuev, G.N.; Fedotova, M.V.; Kruchinin, S.E.; Ivanov, A.S. SARS-COV-2 Coronavirus Papain-like Protease PLpro as an Antiviral Target for Inhibitors of Active Site and Protein–Protein Interactions. Biophysics 2022, 67, 902–912. [Google Scholar] [CrossRef]
- Patel, R.; Prajapati, J.; Rao, P.; Rawal, R.M.; Saraf, M.; Goswami, D. Repurposing the Antibacterial Drugs for Inhibition of SARS-CoV2-PLpro Using Molecular Docking, MD Simulation and Binding Energy Calculation. Mol. Divers. 2022, 26, 2189–2209. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Koopmans’ Theorem | References | |
|---|---|---|
| Global hardness (η) | [37,38,39,40,41,42] | |
| Electronegativity (χ) | [38,43,44] | |
| Electrophilicity (ω) | [45] | |
| Electron acceptor (ω+) | [45] | |
| Electron donator (ω−) | [45] | |
| Net electrophilicity (Δω±) | [46] |
| Compound | χ | η | ω | ω− | ω+ | Δω± | |||
|---|---|---|---|---|---|---|---|---|---|
| GaM (vacuum) | −5.7 | −1.2 | 4.5 | 3.5 | 2.3 | 2.7 | 4.7 | 1.2 | 5.9 |
| GaM (water) | −6.2 | −1.5 | 4.7 | 3.9 | 2.3 | 3.2 | 5.5 | 1.6 | 7.0 |
| TTT (vacuum) | −5.9 | −1.3 | 4.6 | 3.6 | 2.3 | 2.8 | 4.9 | 1.3 | 6.2 |
| TTT (water) | −6.0 | −1.3 | 4.7 | 3.7 | 2.3 | 2.9 | 5.0 | 1.3 | 6.4 |
| N3 (vacuum) | −7.0 | −1.6 | 5.4 | 4.3 | 2.7 | 3.5 | 6.0 | 1.7 | 7.6 |
| N3 (water) | −7.3 | −1.5 | 5.8 | 4.4 | 2.9 | 3.3 | 5.8 | 1.5 | 7.3 |
| Compounds | Mpro | PLpro |
|---|---|---|
| GaM | −6.58 | −5.74 |
| TTT | −8.04 | −10.34 |
| N3 | −7.29 | −5.56 |
| Interacting Amino Acids in the Binding Pockets of Mpro and PLpro | |
|---|---|
| Ligand/Protein | Amino Acids |
| GaM/SARS-CoV-2 Mpro | Ser305, His172, Glu166 *, Pro168, Leu167, Gln189 *, Met165, Pro168, Met49, His163, His164, Cys145 *, Gly143, Ser144, Phe140, Asn142, and Leu141 |
| GaM/SARS-CoV-2 PLpro | Arg166 *, Tyr268, Asn267, Asp164, Pro248, Gly266, Tyr264 *, Tyr301, Pro247, Ala246, and Met208 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Taype-Huanca, K.; Osorio, M.I.; Inostroza, D.; Leyva-Parra, L.; Ruíz, L.; Valderrama-Negrón, A.; Alvarado-Huayhuaz, J.; Yañez, O.; Tiznado, W. Deciphering the Molecular Interaction Process of Gallium Maltolate on SARS-CoV-2 Main and Papain-Like Proteases: A Theoretical Study. Biophysica 2024, 4, 182-194. https://doi.org/10.3390/biophysica4020013
Taype-Huanca K, Osorio MI, Inostroza D, Leyva-Parra L, Ruíz L, Valderrama-Negrón A, Alvarado-Huayhuaz J, Yañez O, Tiznado W. Deciphering the Molecular Interaction Process of Gallium Maltolate on SARS-CoV-2 Main and Papain-Like Proteases: A Theoretical Study. Biophysica. 2024; 4(2):182-194. https://doi.org/10.3390/biophysica4020013
Chicago/Turabian StyleTaype-Huanca, Kevin, Manuel I. Osorio, Diego Inostroza, Luis Leyva-Parra, Lina Ruíz, Ana Valderrama-Negrón, Jesús Alvarado-Huayhuaz, Osvaldo Yañez, and William Tiznado. 2024. "Deciphering the Molecular Interaction Process of Gallium Maltolate on SARS-CoV-2 Main and Papain-Like Proteases: A Theoretical Study" Biophysica 4, no. 2: 182-194. https://doi.org/10.3390/biophysica4020013
APA StyleTaype-Huanca, K., Osorio, M. I., Inostroza, D., Leyva-Parra, L., Ruíz, L., Valderrama-Negrón, A., Alvarado-Huayhuaz, J., Yañez, O., & Tiznado, W. (2024). Deciphering the Molecular Interaction Process of Gallium Maltolate on SARS-CoV-2 Main and Papain-Like Proteases: A Theoretical Study. Biophysica, 4(2), 182-194. https://doi.org/10.3390/biophysica4020013