
Biophysical Breakthroughs Projected for the Phage Therapy of Bacterial Disease

 , ,
, , 
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Phages to Use and Not to Use for Phage Therapy
2.1. Known Past Basics
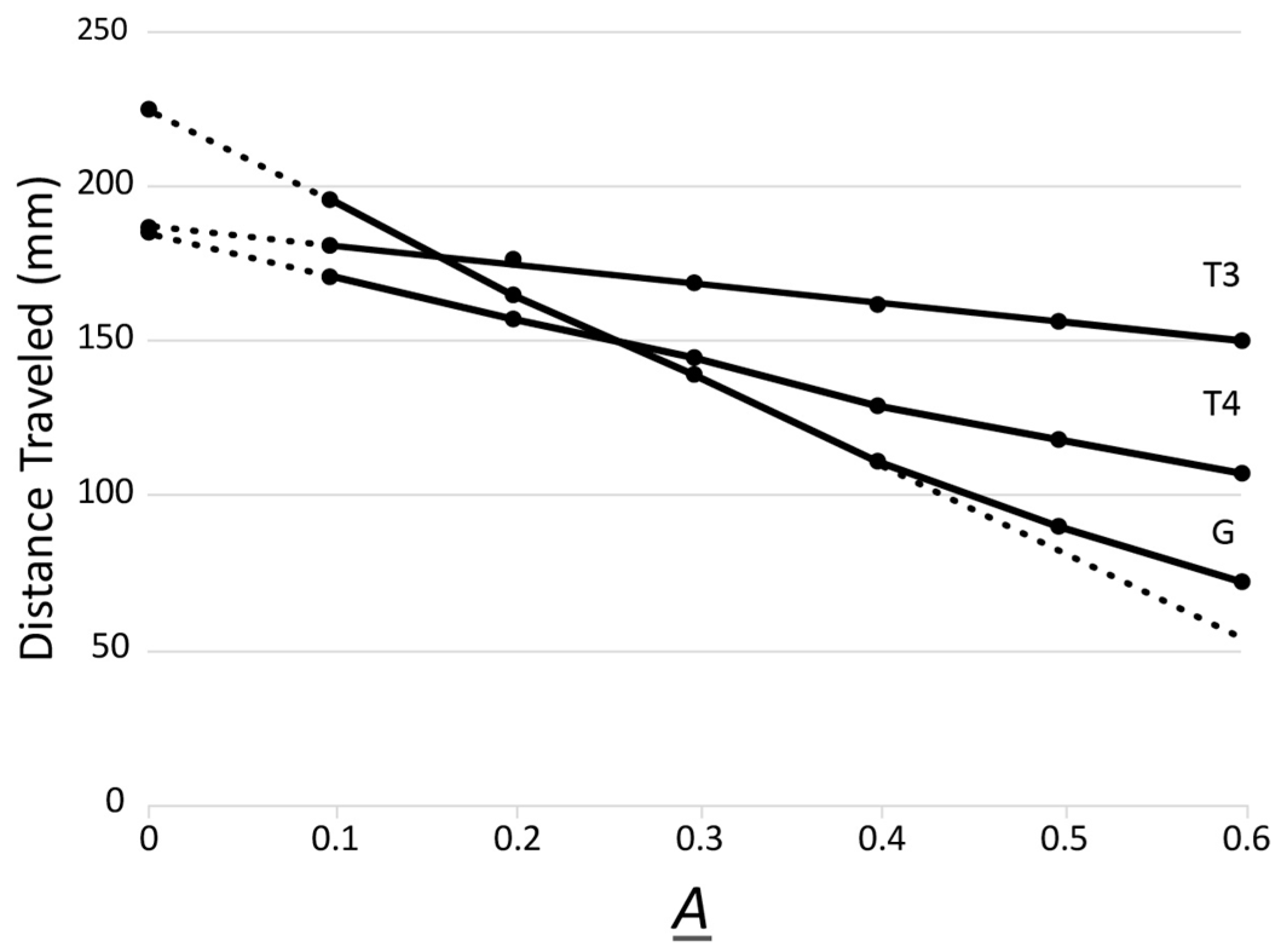
2.2. Next-Generation Biophysical Screening of Phages: Average Electrical Surface Charge Density (σ)
3. Biophysical Technique
3.1. Basics of Native Agarose Gel Electrophoresis (AGE)
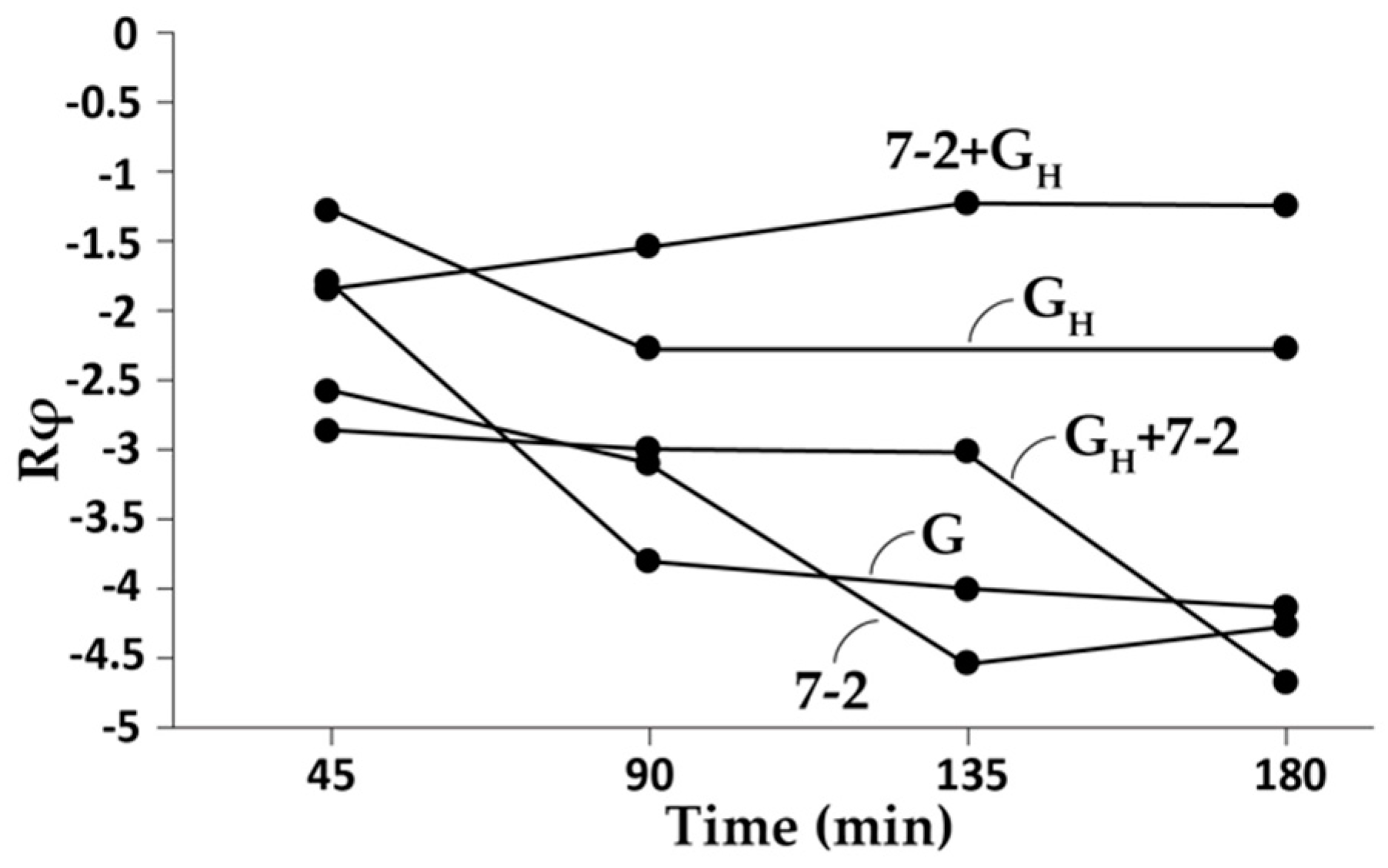
3.2. Further Phage Characterization by AGE
4. The Potential Future of Dry Phage Therapy Cocktails: An Improved Source of Phages
5. Enhancing Phage Persistence: Future Quantification of Persistence
6. Implications for FDA Clearance: Safety and Effectiveness
7. Contextual Significance of Phage Therapy
8. Conclusions (Take-Home Lesson)
9. Patents
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gajic, I.; Jovicevic, M.; Popadic, V.; Trudic, A.; Kabic, J.; Kekic, D.; Ilic, A.; Klasnja, S.; Hadnadjev, M.; Popadic, D.; et al. The emergence of multi-drug-resistant bacteria causing healthcare-associated infections in COVID-19 patients: A retrospective multi-centre study. J. Hosp. Infect. 2023, 137, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Domingues, C.P.F.; Rebelo, J.S.; Dionisio, F.; Nogueira, T. Multi-drug resistance in bacterial genomes—A comprehensive bioinformatic analysis. Int. J. Mol. Sci. 2023, 24, 11438. [Google Scholar] [CrossRef] [PubMed]
- Cohen, K.A.; Manson, A.L.; Desjardins, C.A.; Abeel, T.; Earl, A.M. Deciphering drug resistance in Mycobacterium tuberculosis using whole-genome sequencing: Progress, promise, and challenges. Genome Med. 2019, 11, 45. [Google Scholar] [CrossRef] [PubMed]
- Ntshanka, N.G.M.; Msagati, T.A.M. Trends and Progress on antibiotic-resistant mycobacterium tuberculosis and genes in relation to human immunodeficiency virus. Can. J. Infect. Dis. Med Microbiol. 2023, 2023, 6659212. [Google Scholar] [CrossRef] [PubMed]
- Coyne, A.J.K.; El Ghali, A.; Holger, D.; Rebold, N.; Rybak, M.J. Therapeutic Strategies for emerging multidrug-resistant pseudomonas aeruginosa. Infect. Dis. Ther. 2022, 11, 661–682. [Google Scholar] [CrossRef] [PubMed]
- Giovagnorio, F.; De Vito, A.; Madeddu, G.; Parisi, S.G.; Geremia, N. Resistance in Pseudomonas aeruginosa: A Narrative Review of Antibiogram Interpretation and Emerging Treatments. Antibiotics 2023, 12, 1621. [Google Scholar] [CrossRef] [PubMed]
- Shrivastava, S.R.; Shrivastava, P.S.; Ramasamy, J. Responding to the challenge of antibiotic resistance: World Health Organization. J. Res. Med Sci. 2018, 23, 21. [Google Scholar] [CrossRef] [PubMed]
- AR Threats Report. Centers for Disease Control and Infection, US Department of Health and Human Services. Available online: www.cdc.gov/DrugResistance/Biggest-Threats.html (accessed on 26 January 2024).
- Centers for Disease Control and Infection, US Department of Health and Human Services. Antibiotic Resistance Threats in the United States. Available online: https://www.cdc.gov/drugresistance/pdf/threats-report/2019-ar-threats-report-508.pdf (accessed on 26 January 2024).
- Antimicrobial resistance collaborators. Global burden of bacterial antimicrobial resistance in 2019: A systematic analysis. Lancet 2022, 399, 629–655.
- Burnham, J.P.; Olsen, M.A.; Kollef, M.H. Re-estimating annual deaths due to multidrug-resistant organism infections. Infect. Control. Hosp. Epidemiol. 2019, 40, 112–113. [Google Scholar] [CrossRef] [PubMed]
- Hatfull, G.F.; Dedrick, R.M.; Schooley, R.T. Phage therapy for antibiotic-resistant bacterial infections. Annu. Rev. Med. 2022, 73, 197–211. [Google Scholar] [CrossRef]
- Azevedo, M.M.; Pina-Vaz, C.; Rodrigues, A.G. The role of phage therapy in burn wound infections management: Advantages and pitfalls. J. Burn. Care Res. 2022, 43, 336–342. [Google Scholar] [CrossRef]
- Onallah, H.; Hazan, R.; Nir-Paz, R.; Yerushalmy, O.; Rimon, A.; Braunstein, R.; Gelman, D.; Alkalay, S.; Abdalrhman, M.; Stuczynski, D.; et al. Compassionate use of bacteriophages for failed persistent infections during the first 5 years of the israeli phage therapy center. Open Forum Infect. Dis. 2023, 10, ofad221. [Google Scholar] [CrossRef]
- Diallo, K.; Dublanchet, A. A Century of Clinical Use of Phages: A Literature Review. Antibiotics 2023, 12, 751. [Google Scholar] [CrossRef] [PubMed]
- Dąbrowska, K. Phage therapy: What factors shape phage pharmacokinetics and bioavailability? Systematic and critical review. Med. Res. Rev. 2019, 39, 2000–2025. [Google Scholar] [CrossRef] [PubMed]
- Dedrick, R.M.; E Smith, B.; Cristinziano, M.; Freeman, K.G.; Jacobs-Sera, D.; Belessis, Y.; Brown, A.W.; A Cohen, K.; Davidson, R.M.; van Duin, D.; et al. Phage therapy of Mycobacterium infections: Compassionate use of phages in 20 patients with drug-resistant mycobacterial disease. Clin. Infect. Dis. 2023, 76, 103–112. [Google Scholar] [CrossRef] [PubMed]
- Adesanya, O.; Oduselu, T.; Akin-Ajani, O.; Adewumi, O.M.; Ademowo, O.G. An exegesis of bacteriophage therapy: An emerging player in the fight against anti-microbial resistance. AIMS Microbiol. 2020, 6, 204–230. [Google Scholar] [CrossRef]
- McCallin, S.; Sacher, J.C.; Zheng, J.; Chan, B.K. Current state of compassionate phage therapy. Viruses 2019, 11, 343. [Google Scholar] [CrossRef]
- Górski, A.; Borysowski, J.; Międzybrodzki, R. Phage therapy: Towards a successful clinical trial. Antibiotics 2020, 9, 827. [Google Scholar] [CrossRef]
- Ali, Y.; Inusa, I.; Sanghvi, G.; Mandaliya, V.B.; Bishoyi, A.K. The current status of phage therapy and its advancement towards establishing standard antimicrobials for combating multi drug-resistant bacterial pathogens. Microb. Pathog. 2023, 181, 106199. [Google Scholar] [CrossRef] [PubMed]
- Jault, P.; Leclerc, T.; Jennes, S.; Pirnay, J.P.; Que, Y.-A.A.; Resch, G.; Rousseau, A.F.; Ravat, F.; Carsin, H.; Le Floch, R.; et al. Efficacy and tolerability of a cocktail of bacteriophages to treat burn wounds infected by Pseudomonas aeruginosa (PhagoBurn): A randomised, controlled, double-blind phase 1/2 trial. Lancet Infect. Dis. 2019, 19, 35–45. [Google Scholar] [CrossRef]
- Schooley, R.T.; Biswas, B.; Gill, J.J.; Hernandez-Morales, A.; Lancaster, J.; Lessor, L.; Barr, J.J.; Reed, S.L.; Rohwer, F.; Benler, S.; et al. Development and use of personalized bacteriophage-based therapeutic cocktails to treat a patient with a disseminated resistant Acinetobacter baumannii infection. Antimicrob. Agents Chemother. 2017, 61, e00954-17. [Google Scholar] [CrossRef] [PubMed]
- Hyman, P. Phages for phage therapy: Isolation, characterization, and host range breadth. Pharmaceuticals 2019, 12, 35. [Google Scholar] [CrossRef] [PubMed]
- Howard-Varona, C.; Hargreaves, K.R.; Abedon, S.T.; Sullivan, M.B. Lysogeny in nature: Mechanisms, impact and ecology of temperate phages. ISME J. 2017, 11, 1511–1520. [Google Scholar] [CrossRef]
- Desranleau, J.M. Progress in the treatment of typhoid fever with Vi bacteriophages. Can. J. Public Health 1949, 40, 473–478. [Google Scholar] [PubMed]
- Oechslin, F. Resistance development to bacteriophages occurring during bacteriophage therapy. Viruses 2018, 10, 351. [Google Scholar] [CrossRef] [PubMed]
- Egido, J.E.; Costa, A.R.; Aparicio-Maldonado, C.; Haas, P.-J.; Brouns, S.J.J. Mechanisms and clinical importance of bacteriophage resistance. FEMS Microbiol. Rev. 2022, 46, fuab048. [Google Scholar] [CrossRef] [PubMed]
- Levin, B.R.; Bull, J.J. Population and evolutionary dynamics of phage therapy. Nat. Rev. Microbiol. 2004, 2, 166–173. [Google Scholar] [CrossRef] [PubMed]
- Marchi, J.; Zborowsky, S.; Debarbieux, L.; Weitz, J.S. The dynamic interplay of bacteriophage, bacteria and the mammalian host during phage therapy. iScience 2023, 26, 106004. [Google Scholar] [CrossRef] [PubMed]
- Van Belleghem, J.D.; Dąbrowska, K.; Vaneechoutte, M.; Barr, J.J.; Bollyky, P.L. Interactions between bacteriophage, bacteria, and the mammalian immune system. Viruses 2018, 11, 10. [Google Scholar] [CrossRef]
- Roach, D.R.; Leung, C.Y.; Henry, M.; Morello, E.; Singh, D.; Santo, P.D.; Weitz, J.S.; Debarbieux, L.; Unit, I.I. Immunophage synergy is essential for eradicating pathogens that provoke acute respiratory infections. Cell Host Microbe 2017, 22, 38–47. [Google Scholar] [CrossRef]
- Serwer, P.; Wright, E.T.; De La Chapa, J.; Gonzales, C.B. Basics for improved use of phages for therapy. Antibiotics 2021, 10, 723. [Google Scholar] [CrossRef] [PubMed]
- Issinger, O.G.; Falk, H. Comparative studies on the structure proteins of T3 and T7 phages. Arch. Virol. 1976, 52, 217–231. [Google Scholar] [CrossRef] [PubMed]
- Serwer, P.; Watson, R.H.; Hayes, S.J.; Allen, J.L. Comparison of the physical properties and assembly pathways of the related bacteriophages T7, T3 and II. J. Mol. Biol. 1983, 170, 447–469. [Google Scholar] [CrossRef] [PubMed]
- Shaw, D.J. Electrophoresis; Academic Press: London, UK, 1969; pp. 4–26. [Google Scholar]
- Stellwagen, E.; Lu, Y.; Stellwagen, N.C. Unified description of electrophoresis and diffusion for DNA and other polyions. Biochemistry 2003, 42, 11745–11750. [Google Scholar] [CrossRef] [PubMed]
- Jan, K.-M.; Chien, S. Role of surface electric charge in red blood cell interactions. J. Gen. Physiol. 1973, 61, 638–654. [Google Scholar] [CrossRef] [PubMed]
- Serwer, P. Improvements in procedures for electrophoresis in dilute agarose gels. Anal. Biochem. 1981, 112, 351–356. [Google Scholar] [CrossRef] [PubMed]
- Zhang, A.; Yang, X.; Yang, F.; Zhang, C.; Zhang, Q.; Duan, G.; Jiang, S. Research progress of the ion activity co-efficient of polyelectrolytes: A review. Molecules 2023, 28, 2042. [Google Scholar] [CrossRef] [PubMed]
- Baker, A.M.; Fu, Q.; Hayward, W.; Lindsay, S.M.; Fletcher, T.M. The Myb/SANT domain of the telomere-binding protein TRF2 alters chromatin structure. Nucleic Acids Res. 2009, 37, 5019–5031. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, T.M.; Serwer, P.; Hansen, J.C. Quantitative analysis of macromolecular conformational changes using agarose gel electrophoresis: Application to chromatin folding. Biochemistry 1994, 33, 10859–10863. [Google Scholar] [CrossRef]
- Roberts, S.M.; Aldis, M.; Wright, E.T.; Gonzales, C.B.; Lai, Z.; Weintraub, S.T.; Hardies, S.C.; Serwer, P. Sipho-phage 0105phi7-2 of Bacillus thuringiensis: Novel propagation, DNA, and genome-implied assembly. Int. J. Mol. Sci. 2023, 24, 8941. [Google Scholar] [CrossRef]
- Petithory, T.; Pieuchot, L.; Josien, L.; Ponche, A.; Anselme, K.; Vonna, L. Size-dependent internalization effi-ciency of macrophages from adsorbed nanoparticle-based monolayers. Nanomaterials 2021, 11, 1963. [Google Scholar] [CrossRef]
- Bastin, G.; Gantzer, C.; Sautrey, G. New method to quantify hydrophobicity of non-enveloped virions in aqueous media by capillary zone electrophoresis. Virology 2022, 568, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Griess, G.A.; Moreno, E.T.; Herrmann, R.; Serwer, P. The sieving of rod-shaped viruses during agarose gel elec-trophoresis. I. Comparison with the sieving of spheres. Biopolymers 1990, 29, 1277–1287. [Google Scholar] [CrossRef]
- Serwer, P.; Hayes, S.J.; Watson, R.H.; Khan, S.A. Gel electrophoretic analysis of bacteriophage assembly inter-mediates in bacteriophage plaques. Appl. Theor. Electrophor. 1995, 4, 211–217. [Google Scholar] [PubMed]
- Serwer, P.; Khan, S.A.; Griess, G.A. Non-denaturing gel electrophoresis of biological nanoparticles: Viruses. J. Chromatogr. A 1995, 698, 251–261. [Google Scholar] [CrossRef]
- d’Herelle, F.H.D. The Bacteriophage and Its Behavior; The Williams & Wilkins Company: Baltimore, MD, USA, 1926. [Google Scholar]
- Dublanchet, A.; Fruciano, E. Brève histoire de la phagothérapie A short history of phage therapy. Med. Mal. Infect. 2008, 38, 415–420. [Google Scholar] [CrossRef] [PubMed]
- Serwer, P.; Hayes, S.J.; Zaman, S.; Lieman, K.; Rolando, M.; Hardies, S.C. Improved isolation of undersampled bacteriophages: Finding of distant terminase genes. Virology 2004, 329, 412–424. [Google Scholar] [CrossRef]
- Serwer, P.; Hayes, S.J.; Thomas, J.A.; Hardies, S.C. Propagating the missing bacteriophages: A large bacterio-phage in a new class. Virol. J. 2007, 4, 21. [Google Scholar] [CrossRef]
- Montso, P.K.; Mlambo, V.; Ateba, C.N. Characterization of lytic bacteriophages infecting multidrug-resistant shiga toxigenic atypical Escherichia coli O177 strains isolated from cattle feces. Front. Public Health 2019, 7, 355. [Google Scholar] [CrossRef]
- Letarov, A.; Kulikov, E. The bacteriophages in human- and animal body-associated microbial communities. J. Appl. Microbiol. 2009, 107, 1–13. [Google Scholar] [CrossRef]
- Clokie, M.R.; Millard, A.D.; Letarov, A.V.; Heaphy, S. Phages in nature. Bacteriophage 2011, 1, 31–45. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Mohler, J.; Mahajan, S.D.; Schwartz, S.A.; Bruggemann, L.; Aalinkeel, R. Microbial Biofilm: A Review on Formation, Infection, Antibiotic Resistance, Control Measures, and Innovative Treatment. Microorganisms 2023, 11, 1614. [Google Scholar] [CrossRef] [PubMed]
- Serwer, P.; E Masker, W.; Allen, J.L. Stability and in vitro DNA packaging of bacteriophages: Effects of dextrans, sugars, and polyols. J. Virol. 1983, 45, 665–671. [Google Scholar] [CrossRef] [PubMed]
- Evilevitch, A.; Fang, L.T.; Yoffe, A.M.; Castelnovo, M.; Rau, D.C.; Parsegian, V.A.; Gelbart, W.M.; Knobler, C.M. Effects of salt concentrations and bending energy on the extent of ejection of phage genomes. Biophys. J. 2008, 94, 1110–1120. [Google Scholar] [CrossRef] [PubMed]
- Grayson, P.; Evilevitch, A.; Inamdar, M.M.; Purohit, P.K.; Gelbart, W.M.; Knobler, C.M.; Phillips, R. The effect of genome length on ejection forces in bacteriophage lambda. Virology 2006, 348, 430–436. [Google Scholar] [CrossRef] [PubMed]
- Flint, R.; Laucirica, D.R.; Chan, H.-K.; Chang, B.J.; Stick, S.M.; Kicic, A. Stability considerations for bacteriophages in liquid formulations designed for nebulization. Cells 2023, 12, 2057. [Google Scholar] [CrossRef] [PubMed]
- Köhler, T.; Luscher, A.; Falconnet, L.; Resch, G.; McBride, R.; Mai, Q.A.; Simonin, J.L.; Chanson, M.; Maco, B.; Galiotto, R.; et al. Personalized aerosolised bacteriophage treatment of a chronic lung infection due to multidrug-resistant Pseudomonas aeruginosa. Nat. Commun. 2023, 14, 3629. [Google Scholar] [CrossRef] [PubMed]
- Clark, W.A.; Geary, D. Proceedings: Preservation of bacteriophages by freezing and freeze-drying. Cryobiol 1973, 10, 351–360. [Google Scholar] [CrossRef]
- Jończyk-Matysiak, E.; Łodej, N.; Kula, D.; Owczarek, B.; Orwat, F.; Międzybrodzki, R.; Neuberg, J.; Bagińska, N.; Weber-Dąbrowska, B.; Górski, A. Factors determining phage stability/activity: Challenges in practical phage application. Expert Rev. Anti-Infect. Ther. 2019, 17, 583–606. [Google Scholar] [CrossRef]
- Malik, D.J.; Sokolov, I.J.; Vinner, G.K.; Mancuso, F.; Cinquerrui, S.; Vladisavljevic, G.T.; Clokie, M.R.J.; Garton, N.J.; Stapley, A.G.F.; Kirpichnikova, A. Formulation, stabilisation and encapsulation of bacteriophage for phage therapy. Adv. Colloid Interface Sci. 2017, 249, 100–133. [Google Scholar] [CrossRef]
- Wdowiak, M.; Paczesny, J.; Raza, S. Enhancing the stability of bacteriophages using physical, chemical, and nano-based approaches: A review. Pharmaceutics 2022, 14, 1936. [Google Scholar] [CrossRef]
- Merril, C.R.; Biswas, B.; Carlton, R.; Jensen, N.C.; Creed, G.J.; Zullo, S.; Adhya, S. Long-circulating bacteriophage as antibacterial agents. Proc. Natl. Acad. Sci. USA 1996, 93, 3188–3192. [Google Scholar] [CrossRef] [PubMed]
- Sig, A.K.; Guney, M.; Guclu, A.U.; Ozmen, E. Medicinal leech therapy—An overall perspective. Integr. Med. Res. 2017, 6, 337–343. [Google Scholar] [CrossRef]
- Hackenberger, P.N.B.; Janis, J.E.M. A Comprehensive review of medicinal leeches in plastic and reconstructive surgery. Plast. Reconstr. Surg.–Glob. Open 2019, 7, e2555. [Google Scholar] [CrossRef] [PubMed]
- El-Beyrouty, C.; Buckler, R.; Mitchell, M.; Phillips, S.; Groome, S. Pneumococcal vaccination—A literature review and practice guideline update. Pharmacotherapy 2022, 42, 724–740. [Google Scholar] [CrossRef]
- Tietz, D.; Aldroubi, A.; Schneerson, R.; Unser, M.; Chrambach, A. The distribution of particles characterized by size and free mobility within polydisperse populations of protein-polysaccharide conjugates, determined from two-dimensional agarose electropherograms. Electrophoresis 1991, 12, 46–54. [Google Scholar] [CrossRef]
- Łusiak-Szelachowska, M.; Weber-Dąbrowska, B.; Żaczek, M.; Borysowski, J.; Górski, A. The presence of bacteriophages in the human body: Good, bad or neutral? Microorganisms 2020, 8, 2012. [Google Scholar] [CrossRef] [PubMed]
- Podlacha, M.; Węgrzyn, G.; Węgrzyn, A. Bacteriophages—Dangerous viruses acting incognito or underestimated saviors in the fight against bacteria? Int. J. Mol. Sci. 2024, 25, 2107. [Google Scholar] [CrossRef]
- Foster, D.M.; Kellum, J.A. Endotoxic septic shock: Diagnosis and treatment. Int. J. Mol. Sci. 2023, 24, 16185. [Google Scholar] [CrossRef]
- Gefen, O.; Chekol, B.; Strahilevitz, J.; Balaban, N.Q. TDtest: Easy detection of bacterial tolerance and persistence in clinical isolates by a modified disk-diffusion assay. Sci. Rep. 2017, 7, 41284. [Google Scholar] [CrossRef]
- Tumpa, A.; Štritof, Z.; Pintarić, S. Prevalence and antimicrobial susceptibility of Enterococcus spp. from urine of dogs and cats in northwestern Croatia. Res. Veter- Sci. 2022, 151, 42–46. [Google Scholar] [CrossRef] [PubMed]
- Chambers, J.P.; Wright, E.T.; Hunter, B.; Serwer, P. Inactivating host bacteria for characterization and use of phages. Biophysica 2023, 3, 558–568. [Google Scholar] [CrossRef]
- Martinho, F.C.; de Rabello, D.G.D.; Ferreira, L.L.; Nascimento, G.G. Participation of endotoxin in root canal infections: A systematic review and meta-analysis. Eur. J. Dent. 2017, 11, 398–406. [Google Scholar] [CrossRef] [PubMed]
- Available online: https://www.pfizer.com/sites/default/files/investors/financial_reports/annual_reports/2022/files/Pfizer_Annual_Review.pdf (accessed on 29 December 2023).
- Available online: https://www.imperial.ac.uk/enterprise/business/industry-partnerships-and-commercialisation/industry-partnerships/featured-partnerships/pfizer/ (accessed on 29 December 2023).
- Hatfull, G.F. Phage therapy for nontuberculous mycobacteria: Challenges and opportunities. Pulm. Ther. 2023, 9, 91–107. [Google Scholar] [CrossRef]


Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chambers, J.P.; Aldis, M.; Thomas, J.A.; Gonzales, C.B.; White, R.A., III; Serwer, P. Biophysical Breakthroughs Projected for the Phage Therapy of Bacterial Disease. Biophysica 2024, 4, 195-206. https://doi.org/10.3390/biophysica4020014
Chambers JP, Aldis M, Thomas JA, Gonzales CB, White RA III, Serwer P. Biophysical Breakthroughs Projected for the Phage Therapy of Bacterial Disease. Biophysica. 2024; 4(2):195-206. https://doi.org/10.3390/biophysica4020014
Chicago/Turabian StyleChambers, James P., Miranda Aldis, Julie A. Thomas, Cara B. Gonzales, Richard Allen White, III, and Philip Serwer. 2024. "Biophysical Breakthroughs Projected for the Phage Therapy of Bacterial Disease" Biophysica 4, no. 2: 195-206. https://doi.org/10.3390/biophysica4020014
APA StyleChambers, J. P., Aldis, M., Thomas, J. A., Gonzales, C. B., White, R. A., III, & Serwer, P. (2024). Biophysical Breakthroughs Projected for the Phage Therapy of Bacterial Disease. Biophysica, 4(2), 195-206. https://doi.org/10.3390/biophysica4020014