
Understanding the Liver’s Role in the Clearance of Aβ40

 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Materials
2.3. Multiple Indicator Dilution Method
2.4. Molecular Kinetic Modelling
2.5. Crone–Renkin Early Extraction
2.6. Hepatocyte and LSEC Isolation
2.7. Hepatocyte Uptake of Radiolabelled Substances
2.8. Liver Sinusoidal Endothelial Cell Uptake of Radiolabelled Substances
2.9. Liver Uptake of Aβ40 via Direct Injection
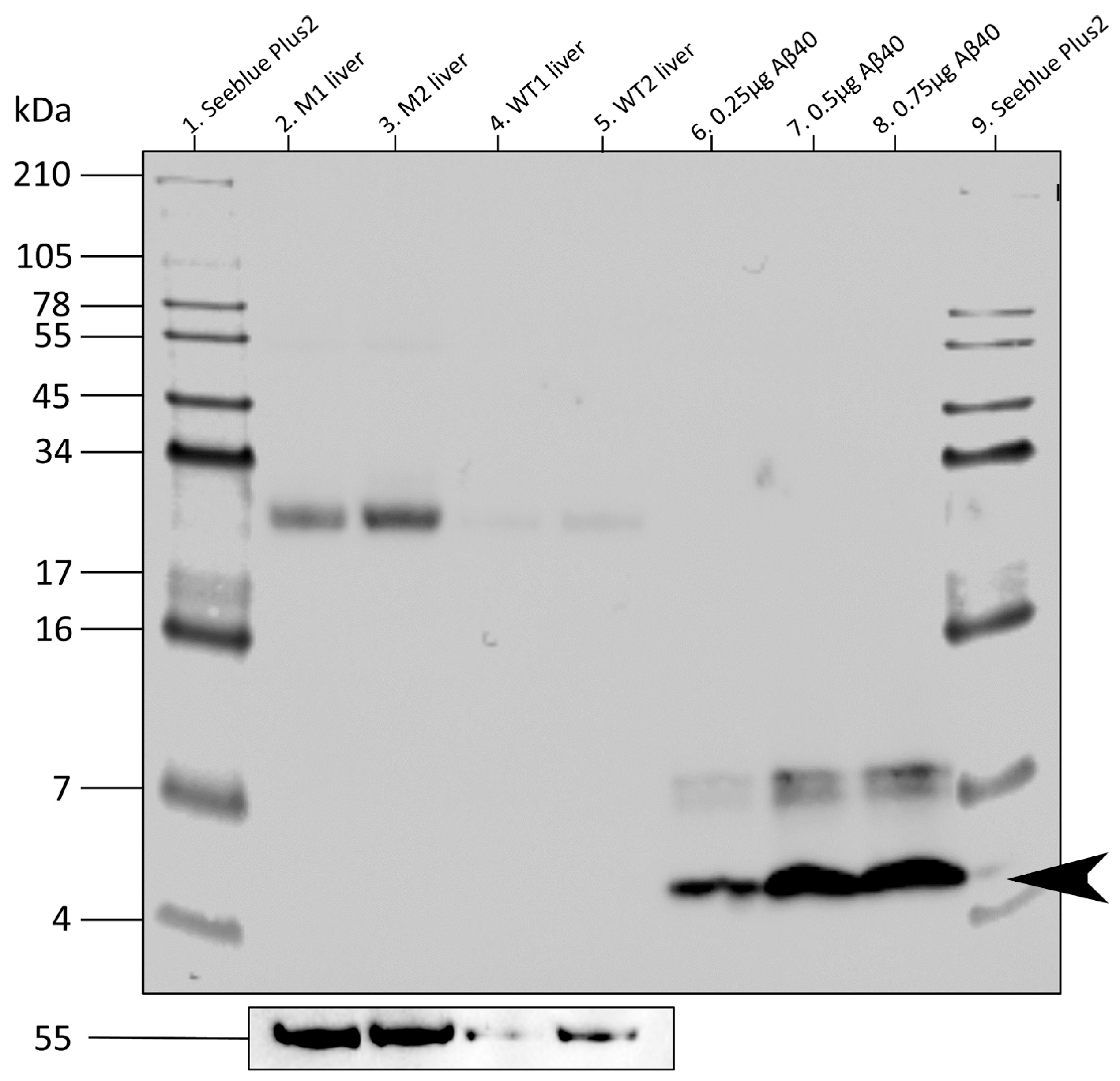
2.10. Western Blots
2.11. Immunofluorescent Staining of Primary Hepatocyte Aβ40 Uptake
2.12. Whole Body Uptake of 3H-Aβ40 in Mice
3. Results
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hunt, N.J.; Kang, S.W.S.; Lockwood, G.P.; Le Couteur, D.G.; Cogger, V.C. Hallmarks of Aging in the Liver. Comput. Struct. Biotechnol. J. 2019, 17, 1151–1161. [Google Scholar] [CrossRef]
- Sengoku, R. Aging and Alzheimer’s disease pathology. Neuropathology 2020, 40, 22–29. [Google Scholar] [CrossRef] [PubMed]
- Musiek, E.S.; Bennett, D.A. Aducanumab and the “post-amyloid” era of Alzheimer research? Neuron 2021, 109, 3045–3047. [Google Scholar] [CrossRef] [PubMed]
- Van Dyck, C.H.; Swanson, C.J.; Aisen, P.; Bateman, R.J.; Chen, C.; Gee, M.; Kanekiyo, M.; Li, D.; Reyderman, L.; Cohen, S.; et al. Lecanemab in Early Alzheimer’s Disease. N. Engl. J. Med. 2023, 388, 9–21. [Google Scholar] [CrossRef] [PubMed]
- Winblad, B.; Amouyel, P.; Andrieu, S.; Ballard, C.; Brayne, C.; Brodaty, H.; Cedazo-Minguez, A.; Dubois, B.; Edvardsson, D.; Feldman, H.; et al. Defeating Alzheimer’s disease and other dementias: A priority for European science and society. Lancet Neurol. 2016, 15, 455–532. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J.; Allsop, D. Amyloid deposition as the central event in the aetiology of Alzheimer’s disease. Trends Pharmacol. Sci. 1991, 12, 383–388. [Google Scholar] [CrossRef] [PubMed]
- Morley, J.E.; Farr, S.A.; Nguyen, A.D.; Xu, F. Editorial: What is the Physiological Function of Amyloid-Beta Protein? J. Nutr. Health Aging 2019, 23, 225–226. [Google Scholar] [CrossRef] [PubMed]
- Tamaki, C.; Ohtsuki, S.; Terasaki, T. Insulin Facilitates the Hepatic Clearance of Plasma Amyloid β-Peptide (1–40) by Intracellular Translocation of Low-Density Lipoprotein Receptor-Related Protein 1 (LRP-1) to the Plasma Membrane in Hepatocytes. Mol. Pharmacol. 2007, 72, 850–855. [Google Scholar] [CrossRef] [PubMed]
- Ghiso, J.; Shayo, M.; Calero, M.; Ng, D.; Tomidokoro, Y.; Gandy, S.; Rostagno, A.; Frangione, B. Systemic catabolism of Alzheimer’s Abeta40 and Abeta42. J. Biol. Chem. 2004, 279, 45897–45908. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, L.A.; Qosa, H.; Kaddoumi, A. Age-Related Decline in Brain and Hepatic Clearance of Amyloid-Beta is Rectified by the Cholinesterase Inhibitors Donepezil and Rivastigmine in Rats. ACS Chem. Neurosci. 2015, 6, 725–736. [Google Scholar] [CrossRef]
- Jin, W.S.; Shen, L.L.; Bu, X.L.; Zhang, W.W.; Chen, S.H.; Huang, Z.L.; Xiong, J.X.; Gao, C.Y.; Dong, Z.; He, Y.N.; et al. Peritoneal dialysis reduces amyloid-beta plasma levels in humans and attenuates Alzheimer-associated phenotypes in an APP/PS1 mouse model. Acta Neuropathol. 2017, 134, 207–220. [Google Scholar] [CrossRef]
- Mawuenyega, K.G.; Sigurdson, W.; Ovod, V.; Munsell, L.; Kasten, T.; Morris, J.C.; Yarasheski, K.E.; Bateman, R.J. Decreased Clearance of CNS β-Amyloid in Alzheimer’s Disease. Science 2010, 330, 1774. [Google Scholar] [CrossRef]
- Liu, Y.H.; Wang, Y.R.; Xiang, Y.; Zhou, H.D.; Giunta, B.; Manucat-Tan, N.B.; Tan, J.; Zhou, X.F.; Wang, Y.J. Clearance of amyloid-beta in Alzheimer’s disease: Shifting the action site from center to periphery. Mol. Neurobiol. 2015, 51, 1–7. [Google Scholar] [CrossRef]
- Mohamad, M.; Mitchell, S.J.; Wu, L.E.; White, M.Y.; Cordwell, S.J.; Mach, J.; Solon-Biet, S.M.; Boyer, D.; Nines, D.; Das, A.; et al. Ultrastructure of the liver microcirculation influences hepatic and systemic insulin activity and provides a mechanism for age-related insulin resistance. Aging Cell 2016, 15, 706–715. [Google Scholar] [CrossRef]
- Xiang, Y.; Bu, X.L.; Liu, Y.H.; Zhu, C.; Shen, L.L.; Jiao, S.S.; Zhu, X.Y.; Giunta, B.; Tan, J.; Song, W.H.; et al. Physiological amyloid-beta clearance in the periphery and its therapeutic potential for Alzheimer’s disease. Acta Neuropathol. 2015, 130, 487–499. [Google Scholar] [CrossRef]
- Wang, Y.R.; Wang, Q.H.; Zhang, T.; Liu, Y.H.; Yao, X.Q.; Zeng, F.; Li, J.; Zhou, F.Y.; Wang, L.; Yan, J.C.; et al. Associations Between Hepatic Functions and Plasma Amyloid-Beta Levels-Implications for the Capacity of Liver in Peripheral Amyloid-Beta Clearance. Mol. Neurobiol. 2017, 54, 2338–2344. [Google Scholar] [CrossRef]
- Tamaki, C.; Ohtsuki, S.; Iwatsubo, T.; Hashimoto, T.; Yamada, K.; Yabuki, C.; Terasaki, T. Major involvement of low-density lipoprotein receptor-related protein 1 in the clearance of plasma free amyloid beta-peptide by the liver. Pharm. Res. 2006, 23, 1407–1416. [Google Scholar] [CrossRef]
- Mohamed, L.A.; Kaddoumi, A. In vitro investigation of amyloid-beta hepatobiliary disposition in sandwich-cultured primary rat hepatocytes. Drug Metab. Dispos. 2013, 41, 1787–1796. [Google Scholar] [CrossRef]
- Cheng, Y.; He, C.-Y.; Tian, D.-Y.; Chen, S.-H.; Ren, J.-R.; Sun, H.-L.; Xu, M.-Y.; Tan, C.-R.; Fan, D.-Y.; Jian, J.-M.; et al. Physiological β-amyloid clearance by the liver and its therapeutic potential for Alzheimer’s disease. Acta Neuropathol. 2023, 145, 717–731. [Google Scholar] [CrossRef]
- Braet, F.; Wisse, E. Structural and functional aspects of liver sinusoidal endothelial cell fenestrae: A review. Comp. Hepatol. 2002, 1, 1. [Google Scholar] [CrossRef]
- Straub, A.C.; Stolz, D.B.; Ross, M.A.; Hernández-Zavala, A.; Soucy, N.V.; Klei, L.R.; Barchowsky, A. Arsenic stimulates sinusoidal endothelial cell capillarization and vessel remodeling in mouse liver. Hepatology 2007, 45, 205–212. [Google Scholar] [CrossRef] [PubMed]
- Cogger, V.C.; Muller, M.; Fraser, R.; McLean, A.J.; Khan, J.; Le Couteur, D.G. The effects of oxidative stress on the liver sieve. J. Hepatol. 2004, 41, 370–376. [Google Scholar] [CrossRef] [PubMed]
- Steffan, A.M.; Pereira, C.A.; Bingen, A.; Valle, M.; Martin, J.P.; Koehren, F.; Royer, C.; Gendrault, J.L.; Kirn, A. Mouse hepatitis virus type 3 infection provokes a decrease in the number of sinusoidal endothelial cell fenestrae both in vivo and in vitro. Hepatology 1995, 22, 395–401. [Google Scholar] [CrossRef] [PubMed]
- Le Couteur, D.G.; Cogger, V.C.; Markus, A.M.; Harvey, P.J.; Yin, Z.L.; Ansselin, A.D.; McLean, A.J. Pseudocapillarization and associated energy limitation in the aged rat liver. Hepatology 2001, 33, 537–543. [Google Scholar] [CrossRef] [PubMed]
- Hilmer, S.N.; Cogger, V.C.; Fraser, R.; McLean, A.J.; Sullivan, D.; Le Couteur, D.G. Age-related changes in the hepatic sinusoidal endothelium impede lipoprotein transfer in the rat. Hepatology 2005, 42, 1349–1354. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, S.J.; Huizer-Pajkos, A.; Cogger, V.C.; McLachlan, A.J.; Le Couteur, D.G.; Jones, B.; de Cabo, R.; Hilmer, S.N. Age-related pseudocapillarization of the liver sinusoidal endothelium impairs the hepatic clearance of acetaminophen in rats. J. Gerontol. A Biol. Sci. Med. Sci. 2011, 66, 400–408. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, S.J.; Huizer-Pajkos, A.; Cogger, V.C.; McLachlan, A.J.; Le Couteur, D.G.; Jones, B.; De Cabo, R.; Hilmer, S.N. The Influence of Old Age and Poloxamer-407 on the Hepatic Disposition of Diazepam in the Isolated Perfused Rat Liver. Pharmacology 2012, 90, 233–241. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, S.J.; Kane, A.E.; Hilmer, S.N. Age-related changes in the hepatic pharmacology and toxicology of paracetamol. Curr. Gerontol. Geriatr. Res. 2011, 2011, 624156. [Google Scholar] [CrossRef] [PubMed]
- Goresky, C.A. A linear method for determining liver sinusoidal and extravascular volumes. Am. J. Physiol. 1963, 204, 626–640. [Google Scholar] [CrossRef] [PubMed]
- Goresky, C.A. Kinetic interpretation of hepatic multiple-indicator dilution studies. Am. J. Physiol. 1983, 245, G1–G12. [Google Scholar] [CrossRef]
- Hilmer, S.N.; Cogger, V.C.; Muller, M.; Le Couteur, D.G. The hepatic pharmacokinetics of doxorubicin and liposomal doxorubicin. Drug Metab. Dispos. 2004, 32, 794–799. [Google Scholar] [CrossRef] [PubMed]
- Crone, C. Capillary permeability to small solutes. In Handbook of Physiology. The Cardiovascular System. Microcirculation, Section 2; American Physiological Society: Rockville, MD, USA, 1984; pp. 411–466. [Google Scholar]
- Crone, C. The Permeability of Capillaries in Various Organs as Determined by Use of the ‘Indicator Diffusion’ Method. Acta Physiol. Scand. 1963, 58, 292–305. [Google Scholar] [CrossRef]
- Le Couteur, D.G.; Yin, Z.L.; McLean, A.J.; Rivory, L.P. Wash-In Methodology and Modeling to Determine Hepatocellular D-Glucose Transport in the Perfused Rat Liver. Jpn. J. Physiol. 2004, 54, 421–429. [Google Scholar] [CrossRef] [PubMed]
- Hunt, N.J.; Lockwood, G.P.; Warren, A.; Mao, H.; McCourt, P.A.G.; Le Couteur, D.G.; Cogger, V.C. Manipulating fenestrations in young and old liver sinusoidal endothelial cells. Am. J. Physiol. Gastrointest. Liver Physiol. 2019, 316, G144–G154. [Google Scholar] [CrossRef] [PubMed]
- Cogger, V.C.; O'Reilly, J.N.; Warren, A.; Le Couteur, D.G. A standardized method for the analysis of liver sinusoidal endothelial cells and their fenestrations by scanning electron microscopy. J. Vis. Exp. 2015, 98, e52698. [Google Scholar]
- Kang, S.W.S.; Cogger, V.C.; Le Couteur, D.G.; Fu, D. Multiple cellular pathways regulate lipid droplet homeostasis for the establishment of polarity in collagen sandwich-cultured hepatocytes. Am. J. Physiol. Cell Physiol. 2019, 317, C942–C952. [Google Scholar] [CrossRef] [PubMed]
- Poduslo, J.F.; Curran, G.L.; Haggard, J.J.; Biere, A.L.; Selkoe, D.J. Permeability and residual plasma volume of human, Dutch variant, and rat amyloid beta-protein 1-40 at the blood-brain barrier. Neurobiol. Dis. 1997, 4, 27–34. [Google Scholar] [CrossRef]
- Hone, E.; Martins, I.J.; Fonte, J.; Martins, R.N. Apolipoprotein E influences amyloid-beta clearance from the murine periphery. J. Alzheimer’s Dis. 2003, 5, 1–8. [Google Scholar] [CrossRef]
- Ghersi-Egea, J.F.; Gorevic, P.D.; Ghiso, J.; Frangione, B.; Patlak, C.S.; Fenstermacher, J.D. Fate of cerebrospinal fluid-borne amyloid beta-peptide: Rapid clearance into blood and appreciable accumulation by cerebral arteries. J. Neurochem. 1996, 67, 880–883. [Google Scholar] [CrossRef]
- Kandimalla, K.K.; Curran, G.L.; Holasek, S.S.; Gilles, E.J.; Wengenack, T.M.; Poduslo, J.F. Pharmacokinetic analysis of the blood-brain barrier transport of 125I-amyloid beta protein 40 in wild-type and Alzheimer’s disease transgenic mice (APP,PS1) and its implications for amyloid plaque formation. J. Pharmacol. Exp. Ther. 2005, 313, 1370–1378. [Google Scholar] [CrossRef]
- Marques, M.A.; Kulstad, J.J.; Savard, C.E.; Green, P.S.; Lee, S.P.; Craft, S.; Watson, G.S.; Cook, D.G. Peripheral amyloid-beta levels regulate amyloid-beta clearance from the central nervous system. J. Alzheimers Dis. 2009, 16, 325–329. [Google Scholar] [CrossRef]
- Horn, T.; Henriksen, J.H.; Christoffersen, P. The sinusoidal lining cells in “normal” human liver: A scanning electron microscopic investigation. Liver 1986, 6, 98–110. [Google Scholar] [CrossRef] [PubMed]
- Pardridge, W.M.; Mietus, L.J. Transport of protein-bound steroid hormones into liver in vivo. Am. J. Physiol. 1979, 237, E367–E372. [Google Scholar] [CrossRef]
- Tian, D.Y.; Cheng, Y.; Zhuang, Z.Q.; He, C.Y.; Pan, Q.G.; Tang, M.Z.; Hu, X.L.; Shen, Y.Y.; Wang, Y.R.; Chen, S.H.; et al. Physiological clearance of amyloid-beta by the kidney and its therapeutic potential for Alzheimer’s disease. Mol. Psychiatry 2021, 26, 6074–6082. [Google Scholar] [CrossRef]
- Pappolla, M.; Sambamurti, K.; Vidal, R.; Pacheco-Quinto, J.; Poeggeler, B.; Matsubara, E. Evidence for lymphatic Abeta clearance in Alzheimer’s transgenic mice. Neurobiol. Dis. 2014, 71, 215–219. [Google Scholar] [CrossRef]
- Brubaker, W.D.; Crane, A.; Johansson, J.U.; Yen, K.; Garfinkel, K.; Mastroeni, D.; Asok, P.; Bradt, B.; Sabbagh, M.; Wallace, T.L.; et al. Peripheral complement interactions with amyloid beta peptide: Erythrocyte clearance mechanisms. Alzheimers Dement. 2017, 13, 1397–1409. [Google Scholar] [CrossRef] [PubMed]
- Braet, F.; Bomans, P.H.; Wisse, E.; Frederik, P.M. The observation of intact hepatic endothelial cells by cryo-electron microscopy. J. Microsc. 2003, 212 Pt 2, 175–185. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.H.; Tian, D.Y.; Shen, Y.Y.; Cheng, Y.; Fan, D.Y.; Sun, H.L.; He, C.Y.; Sun, P.Y.; Bu, X.L.; Zeng, F.; et al. Amyloid-beta uptake by blood monocytes is reduced with ageing and Alzheimer’s disease. Transl. Psychiatry 2020, 10, 423. [Google Scholar] [CrossRef] [PubMed]
- Buniatian, G.H.; Weiskirchen, R.; Weiss, T.S.; Schwinghammer, U.; Fritz, M.; Seferyan, T.; Proksch, B.; Glaser, M.; Lourhmati, A.; Buadze, M.; et al. Antifibrotic Effects of Amyloid-Beta and Its Loss in Cirrhotic Liver. Cells 2020, 9, 452. [Google Scholar] [CrossRef]
- Biere, A.L.; Ostaszewski, B.; Stimson, E.R.; Hyman, B.T.; Maggio, J.E.; Selkoe, D.J. Amyloid beta-peptide is transported on lipoproteins and albumin in human plasma. J. Biol. Chem. 1996, 271, 32916–32922. [Google Scholar] [CrossRef]
- Deane, R.; Sagare, A.; Zlokovic, B.V. The role of the cell surface LRP and soluble LRP in blood-brain barrier Abeta clearance in Alzheimer’s disease. Curr. Pharm. Des. 2008, 14, 1601–1605. [Google Scholar] [CrossRef]
- Kitaguchi, N.; Kawaguchi, K.; Sakai, K. Removal of blood amyloid as a therapeutic strategy for Alzheimer’s disease: The influence of smoking and nicotine. In Nicotinic Acetylcholine Receptor Signaling in Neuroprotection; Springer: Singapore, 2018; pp. 173–191. [Google Scholar]
- Boada, M.; Lopez, O.L.; Olazaran, J.; Nunez, L.; Pfeffer, M.; Paricio, M.; Lorites, J.; Pinol-Ripoll, G.; Gamez, J.E.; Anaya, F.; et al. A randomized, controlled clinical trial of plasma exchange with albumin replacement for Alzheimer’s disease: Primary results of the AMBAR Study. Alzheimers Dement. 2020, 16, 1412–1425. [Google Scholar] [CrossRef]
- Lam, V.; Takechi, R.; Hackett, M.J.; Francis, R.; Bynevelt, M.; Celliers, L.M.; Nesbit, M.; Mamsa, S.; Arfuso, F.; Das, S.; et al. Synthesis of human amyloid restricted to liver results in an Alzheimer disease-like neurodegenerative phenotype. PLoS Biol. 2021, 19, e3001358. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Young Rat (n = 6) | Old Rat (n = 10) | |
|---|---|---|
| Fractional recovery | ||
| Aβ40 | 0.99 ± 0.03 | 0.86 ± 0.08 ** |
| Evans Blue | 1.02 ± 0.04 | 0.85 ± 0.09 ** |
| Sucrose | 0.98 ± 0.03 | 0.85 ± 0.13 * |
| Ratio of recovery | ||
| Aβ40/Evans Blue | 0.97 ± 0.04 | 1.01 ± 0.07 ns |
| Aβ40/sucrose | 1.01 ± 0.03 | 1.02 ± 0.06 ns |
| Evans Blue/sucrose | 1.05 ± 0.02 | 1.01 ± 0.04 ns |
| Young Rat (n = 6) | Old Rat (n = 10) | |
|---|---|---|
| Apparent volume of distribution | ||
| Aβ40 (mL/g) | 0.16 ± 0.02 | 0.19 ± 0.05 ns |
| Evans Blue (mL/g) | 0.15 ± 0.02 | 0.16 ± 0.04 ns |
| Sucrose (mL/g) | 0.17 ± 0.03 | 0.20 ± 0.05 ns |
| Ratio of apparent volume of distribution | ||
| Aβ40/Evans Blue | 1.05 ± 0.04 | 1.19 ± 0.17 ns |
| Aβ40/Sucrose | 0.91 ± 0.05 | 0.92 ± 0.05 ns |
| Evans Blue/Sucrose | 0.87 ± 0.05 | 0.79 ± 0.13 ns |
| Permeability Surface area product | ||
| Aβ40/Evans Blue (mL/s/g liver) | 0.004 ± 0.001 | 0.008 ± 0.009 ns |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lockwood, G.P.; Hunt, N.J.; Kockx, M.; Kang, S.W.S.; Le Couteur, D.G.; Cogger, V.C. Understanding the Liver’s Role in the Clearance of Aβ40. Livers 2024, 4, 253-267. https://doi.org/10.3390/livers4020018
Lockwood GP, Hunt NJ, Kockx M, Kang SWS, Le Couteur DG, Cogger VC. Understanding the Liver’s Role in the Clearance of Aβ40. Livers. 2024; 4(2):253-267. https://doi.org/10.3390/livers4020018
Chicago/Turabian StyleLockwood, Glen P., Nicholas J. Hunt, Maaike Kockx, Sun Woo Sophie Kang, David G. Le Couteur, and Victoria C. Cogger. 2024. "Understanding the Liver’s Role in the Clearance of Aβ40" Livers 4, no. 2: 253-267. https://doi.org/10.3390/livers4020018
APA StyleLockwood, G. P., Hunt, N. J., Kockx, M., Kang, S. W. S., Le Couteur, D. G., & Cogger, V. C. (2024). Understanding the Liver’s Role in the Clearance of Aβ40. Livers, 4(2), 253-267. https://doi.org/10.3390/livers4020018