Abstract
Cell-death-inducing DNA fragmentation factor-alpha (DFFA)-like effector b (CIDEB) was first identified as an apoptosis-inducing protein. Further research revealed a pivotal role in lipid metabolism, regulating very-low-density lipoprotein (VLDL), lipid droplets (LD), sterol response element-binding protein (SREBP), and chylomicrons. Recent studies have uncovered that rare germline variants in CIDEB protect against liver diseases, including MAFLD, cirrhosis, and viral hepatitis. Furthermore, CIDEB influences steps of the hepatitis C virus (HCV) replication cycle. This review summarizes the current knowledge about CIDEB’s roles in apoptosis, lipid metabolism, and viral hepatitis, and highlights its critical role in liver diseases.
1. Introduction
Chronic liver disease (CLD) and cirrhosis account for two million deaths worldwide each year [1,2]. The primary causes of CLD include metabolic-associated fatty liver disease (MAFLD), hepatitis B virus (HBV), hepatitis C virus (HCV), and alcohol-associated liver disease [3]. MAFLD, characterized by the accumulation of hepatic fat linked to insulin resistance, is identified when steatosis is present in more than 5% of hepatocytes [4].
MAFLD encompasses two conditions with distinct prognoses: metabolic-associated fatty liver, which involves steatosis; and metabolic-associated steatohepatitis (MASH), which ranges in severity from fibrosis and cirrhosis to hepatocellular carcinoma (HCC) [5]. The global prevalence of MAFLD is approximately 30% [6,7], and its incidence is rising alongside the increasing rates of obesity [8]. Consequently, MASH is becoming the leading cause of liver transplantation in the United States and continues to rise [9,10,11].
For most individuals with MAFLD, treatment has traditionally involved diet and regular exercise due to the lack of approved medications for MASH [12]. Just this year, the U.S. Food and Drug Administration (FDA) approved Rezdiffra™, the first treatment for patients with liver scarring, promoting future drug developments against MAFLD [13]. Other therapeutic strategies include the reduction of secondary diseases with a focus on cardiovascular diseases using, for example, glucagon-like peptide 1 receptor agonists [14,15], glitazones [16], and SGLT2 inhibitors [17]. The first two mentioned above also support weight loss, helping the traditional treatment.
Genome-wide association studies have identified loci linked to disease severity, highlighting several genetic risk factors that decrease susceptibility to MASH [18,19,20,21]. Carriers of cell-death-inducing DNA fragmentation factor-alpha (DFFA)-like effector b (CIDEB) germline variants have the greatest risk reduction of MASH and are generally protected from CLD. In this review, we summarize the known functions of CIDEB in lipid metabolism and apoptosis and demonstrate its essential role in liver diseases, including MAFLD and viral hepatitis (Figure 1).
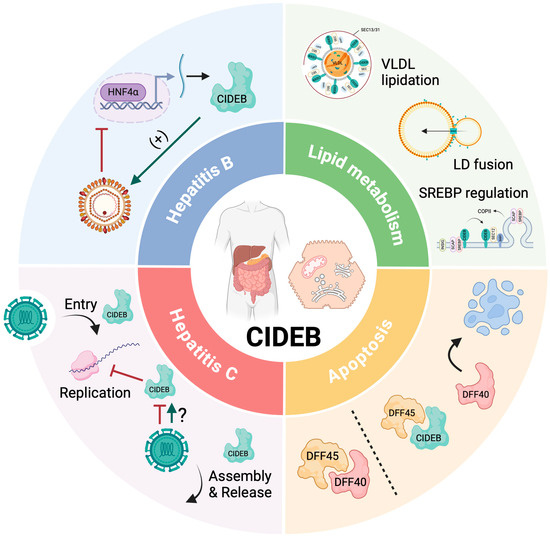
Figure 1.
Overview of CIDEB functions. Lipid metabolism: CIDEB regulates very-low-density lipoprotein (VLDL) lipidation, lipid droplets (LD) fusion, sterol response element-binding protein (SREBP), and chylomicrons. The mechanisms are further illustrated in Figure 2. Apoptosis: In the absence of apoptosis, DFF45 suppresses DFF40. In cases of apoptosis, DFF45 separates from DFF40, so DFF40 releases DNA fragmentation. CIDEB controls the activity of the DFF40/DFF45 complex by sequestration of DFF45. Hepatitis C: CIDEB is involved at different stages of the HCV replication cycle, including entry, replication, assembly, and release. CIDEB acts as a cofactor for HCV entry into hepatocytes and regulates the VLDL pathway components used by HCV for assembly and release, while also reducing viral replication. Hepatitis B: Overexpression of CIDEB enhances HBV production by increasing HBV core promoter activity, while its downregulation in HBV-infected cells and patients is likely due to reduced levels of HNF4α, a transcriptional regulator of CIDEB, which may modulate viral replication. HNF4α, hepatocyte nuclear factor 4 alpha; LD, lipid droplet. This figure was created with BioRender.com (accessed on 30 May 2024).
2. CIDEB: Structure, Expression, and Phylogeny
CIDEB, a cell-death-inducing DNA fragmentation factor-like effector family member, shares overlapping functions with CIDEA and CIDEC. The human CIDEB gene is located on chromosome 14q12 and contains eight exons.
Four transcript variants of CIDEB have been identified, including three long transcripts composed of 219 amino acids (aa), with a molecular mass of 24.7 kDa, and one short transcript of 61 aa, with a molecular mass of 6.3 kDa. Evolutionarily, the N-terminal CAD domain of DFFA was duplicated to form the CIDE-N domain, exhibiting a broad phylogenetic distribution from lower organisms to mammals [22]. The amino acid sequence identity of CIDEB ranges from 42.92% to 84.07% among different species, with a stable genomic structure and conserved intron phase in vertebrates [22,23]. Additionally, the CIDE-N domain has acquired the CIDE-C domain, which is unique to vertebrates [22].
CIDEB protein comprises two domains: the N-terminal (CIDE-N) and the C-terminal (CIDE-C) domains [24]. CIDE-N includes a five-stranded twisted ß sheet and two α-helical structures with a topology of an alpha/ß roll [25]. The CIDE-N domain of CIDEB and its interaction interface are homologous with the CIDEs in the N-terminal regions of DFF40 (DNA fragmentation factor) and DFF45 [24,26]. CIDE interacts through weak homophilic binding in a yin/yang-like orientation with two complementary bipolar surfaces. A higher affinity of the interaction would need other domains [25,27,28]. The CIDE-N domain could regulate the activity of the CIDE-C domain [24].
The CIDE-C domain has apoptotic activity and contains the aa 166–195, which include an extended α-helical structure that localizes CIDEB in the mitochondrial membrane [22]. It enables CIDEB to form homodimers, heterodimers, and maybe oligomers with other family members [28]. However, the high-affinity interaction of CIDEB/CIDEB needs residues upstream or downstream of aa 166–195.
CIDEB mRNA and protein are highly expressed in the liver and less strongly in the Duodenum, small intestine, spleen, kidney, and pancreas [23,29,30,31]. Subcellularly, CIDEB is found in the smooth endoplasmic reticulum (ER), cytoplasmatic lipid droplets (LD), and Golgi apparatus [30,32,33]. A hydrophobic region, aa 166–195, in the CIDE-C domain, integrates CIDEB into the ER membranes and LDs [32].
3. CIDEB in Lipid Metabolism
CIDEB is involved in different aspects of cellular lipid metabolism (Figure 2). CIDEB is pivotal in regulating very-low-density lipoprotein (VLDL) lipidation. VLDL lipidation undergoes two major steps [34]. First, VLDL forms through the co-translational lipidation of nascent apolipoprotein B-100 (ApoB-100) with triglycerides (TGs), phospholipids, and cholesterol esters, followed by fusion with luminal LD [35]. These vesicles exit the ER via COPII (coat protein complex II)-coated transport vesicles and undergo the second, bulk lipidation step, likely in the Golgi apparatus, where neutral lipids are added before secretion [34,36,37,38]. It has been hypothesized that CIDEB supports the direct transport of TG from cytoplasmic LD in pre-VLDL particles by interacting with ApoB analog to its mechanism in LD-contact sites [32,33]. While CIDEB increases LD sizes in hepatocytes, perilipin-2 acts as a negative regulator [33]. CIDEB brings cytosolic LDs and pre-VLDL particles close together, counteracted by perilipin-2 present on the LD surfaces [33]. CIDEB and perilipin-2 regulate hepatocyte lipolysis and re-esterification, contributing to VLDL lipidation [33].
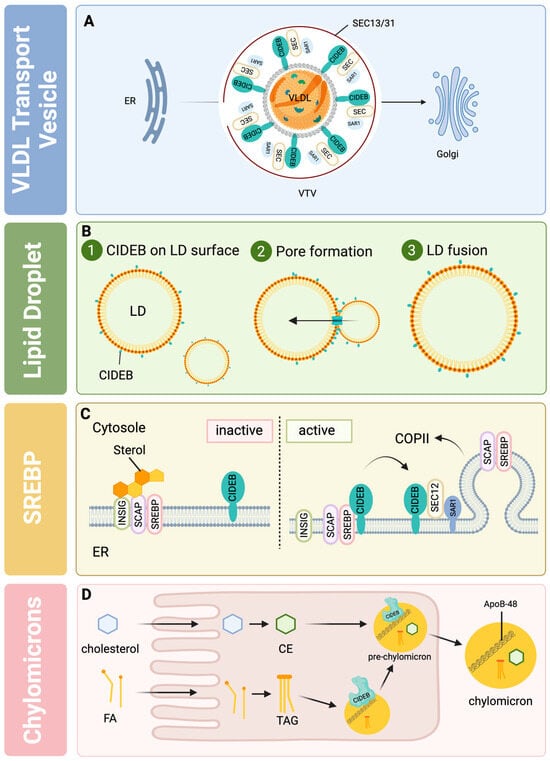
Figure 2.
The role of CIDEB in lipid metabolism: (A) VTV (VLDL transport vesicles), aided by CIDEB, interact with SEC components Sar1 and SEC13/31 to facilitate the transport of VLDLs from the ER to the Golgi apparatus; (B) CIDEB promotes lipid droplet fusion via pore formation to facilitate lipid transfer and increase lipid droplet size; (C) CIDEB enhances the interaction between the SREBP-SCAP complex and COPII components on the ER membrane, facilitating SREBP activation and export in the absence of sterols by disrupting the inhibitory binding of INSIG; (D) CIDEB regulates enterocyte cholesterol and fatty acid uptake and facilitates chylomicron formation by interacting with ApoB-48. CE, cholesterol ester; ER, endoplasmic reticulum; FA, fatty acid; Golgi, Golgi apparatus; LD, lipid droplet; TAG, triglyceride; VTV, VLDL transport vesicle. This figure was created with BioRender.com (accessed on 30 May 2024).
A specialized transport apparatus exports nascent VLDL from the ER to the Golgi for secretion. COPII packages VLDLs into VLDL transport vesicles (VTV) to exit the ER [36,39] (Figure 2A). By activating its guanine nucleotide exchange factor (Sec12), secretion-associated Ras-related GTPase 1 (Sar1) is activated [40]. Activated Sar1 recruits the inner coat complex Sec23/Sec24 [41] and the outer coat complex Sec13/Sec31 after that to generate COPII-coated vesicles [41] (Figure 2A). ApoB, as a VTV cargo protein, may be too big for normal COPII vesicles [42]. Specialized elements may be required to enable the ER export of VLDLs to handle this cargo. CIDEB is found in VTVs and interacts with the COPII components, Sar1 and Sec24 [43,44], to potentially develop these larger VTVs (Figure 2A) [43]. This is supported by the fact that CIDEB needs the ApoB binding and the LDs association to facilitate VLDL formation [32]. CIDEB interaction with ApoB-100 and its truncations, such as ApoB94, ApoB72, and ApoB48 [43,44], is mediated by aa118–aa165 of CIDEB [32] and is present in overlapping cell regions [33]. The interaction of CIDEB and ApoB-100 could initiate VTV formation [32,43]. The size and type of cargo regulate the formation of different coat complexes, which results in the genesis of vesicles of different sizes [43]. CIDEB knockdown results in deficient VTV building and, instead, VLDL transportation in conventional COPII-coated protein transport vesicles (PTV) [43].
Corresponding to the role of CIDEB in VLDL lipidation, CIDEB-null mice presented increased TG levels in the liver. They showed reduced VLDL secretion with fewer mature VLDL particles in the Golgi apparatus [32,33]. Although the total number of VLDL particles and ApoB was comparable to that in wild-type mice, CIDEB-null mice showed a notable reduction in VLDL size and decreased TG, free fatty acids (FFA), and cholesterol content within these particles and plasma [29,32]. However, plasma levels of ApoB were lower in CIDEB-null mice, possibly because of higher clearance of TG-poor VLDL particles [32].
CIDEB also promotes the increase in number and size of LD [45,46,47,48] (Figure 2A). CIDEB is enriched at LD–LD contacting sites to build up the fusion of LDs [45], and CIDEB regulates LDs fusion activity and storage capacity [45] (Figure 2B). It may function similarly to CIDEC (Figure 2B). CIDEC initiates a pore-like structure between contacted LDs formed by lipid transfer proteins through which lipids are transferred from smaller LDs to larger LDs driven by pressure differences [49]. Xu et al. distinguished two hepatocyte populations according to the size of LDs [45]. Small LD-containing hepatocytes express only CIDEB and no other CIDE proteins. Large LD-containing hepatocytes expressed all three CIDE proteins and exhibited increased lipid exchange activity under the control of CIDEA and fat-specific protein (Fsp-27) [45].
CIDEB-deficient mice have reduced expression levels of the mature form of sterol response element-binding protein SREBP1, SREBP2, and SREBP, cleavage-activating protein (SCAP), and their downstream target genes acetyl-CoA carboxylase, fatty-acid synthase, and stearoyl-CoA desaturase 1 under normal and high-fat, low-fiber diet conditions [29,44,50]. Li et al. reported a 50% reduction of SREBP-2 in the livers of CIDEB-deficient mice [50]. The phenotypes of CIDEB-deficient mice are comparable to those of liver-specific SCAP-deficient mice [50,51]. Su et al. reported no association between enhanced lipid accumulation and decreased SREBP activity, indicating that CIDEB’s effects on VLDL secretion and SREBP activation were distinct.
Under high concentrations of sterol, the SREBP/SCAP complex binds an insulin-induced gene (INSIG) through a sterol-dependent interaction and becomes fixed in the endoplasmic reticulum membranes [52] (Figure 2C). The absence of sterol results in the loss of the interaction between INSIG and SCAP and a structural change in SCAP that exposes a hexapeptide motif to interact with COPII components like Sec proteins [52] (Figure 2C) This could also exhibit the regions that interact with CIDEB; this SCAP domain (aa416–800) is the same, including the hexapeptide motif [44]. CIDEB could enrich the components at ER exit sites and enhance the interaction between the SREBP/SCAP complex and COPII, so the SREBP/SCAP complex can be loaded into COPII vesicles and transported to the Golgi apparatus [44] (Figure 2C). Increased CIDEB expression could make SREBP processing independent of sterol inhibition [44].
Additionally, CIDEB-null mice showed reduced cholesterol uptake, conversion, and biosynthesis [50]. Under a high-calorie diet, lower expression levels of genes regulating cholesterol biosynthesis caused decreased biosynthesis in the liver independent of SREBP expression [50]. Therefore, it was concluded that CIDEB could regulate cholesterol metabolism via the SREBP pathway [50]. CIDEB-null mice had increased cholesterol storage in the liver under a high-cholesterol diet, which could be induced by increased expression levels of low-density lipoprotein receptor in the liver or reduced export through VLDL secretion [50]. This resulted in reduced plasma levels of cholesterol and LDL, decreased lipid accumulation in the heart, and reduced weight.
CIDEB is also part of the lipidation of chylomicrons in the small intestine [30] (Figure 2D). CIDEB-null mice showed increased lipid accumulation in enterocytes, the secretion of smaller chylomicrons, and reduced TG plasma levels in comparison to wild-type mice [30]. ApoB-48 packages intestinal lipids into pre-chylomicrons [53]. The interaction between CIDEB and ApoB-48, which could be facilitated by aa118–165 of CIDEB [30], may regulate the lipidation of chylomicrons, but the underlying mechanism has not been elucidated yet.
CIDEB expression is regulated by transcriptional regulation, protein degradation, and external stimuli. Da et al. identified two promoters of CIDEB, the upstream promotor (Pu) between −3628 and −3345, and the internal promotor (Pi), between −204 and the translation start; Sp1 and Sp3, two transcription factors, regulate both in their basal transcription activity [54]. DNA methylation of CpG sites in the Pu region controls the long transcript of CIDEB [54]. PGC-1α (peroxisome proliferator-activated receptor-gamma coactivator 1-alpha) and hepatocyte nuclear factor 1-alpha (HNF-1α) also transcriptionally regulate CIDEB [54,55,56]. PGC-1α increases VLDL lipidation through CIDEB expression. CIDEB was also downregulated in the adrenals of mice under chronic aspartame exposure [57]. HNF4α regulates the short transcript of CIDEB [54]. A high-fat diet increased CIDEB mRNA expression in the intestine, brain, and heart, while a bile acid diet reduced it [23,30]. The ubiquitin–proteasome system regulates CIDEB via posttranslational degradation [58]. CIDEB protein has a short lifespan [58]. High intracellular lipid levels stabilized CIDEB protein, and decreased intracellular lipid levels induced lower protein stability [58].
Given its multifaceted role in lipid metabolism, CIDEB emerges as a crucial regulator of VLDL lipidation, hepatic lipolysis, and lipid storage. Understanding CIDEB’s interactions and regulatory mechanisms offers valuable insights into therapeutic targets in metabolic disorders.
3.1. CIDEB in Diabetes and Metabolic Syndrome
Metabolic syndrome encompasses a group of metabolic dysregulations, including insulin resistance, dyslipidemia, central obesity, and hypertension. The prevalence of metabolic syndrome is projected to rise due to overnutrition and a lack of activity, leading to obesity [59,60]. CIDEB, expressed in pancreatic beta cells, plays a crucial role in metabolic homeostasis. Its expression is responsive to FFA, particularly palmitate, which enhances CIDEB expression [31]. Knockout (KO) of CIDEB in beta cells inhibits the conversion of FFAs to TGs, resulting in FFA overload and exacerbated lipoapoptosis induced by palmitate supplementation [31].
CIDEB-null mice exhibit enhanced insulin sensitivity and a lean phenotype characterized by reduced body weight, reduced adiposity, and resistance to high-fat-diet-induced obesity [29,32,50]. As observed in CIDEA-null mice, this could be caused by higher energy consumption and reduced lipogenesis in the livers of CIDEB-null mice, leading to increased dietary fat intake and decreased lipid secretion [29,61]. In contrast, human carriers of CIDEB variants do not exhibit anthropometric or lipid-related abnormalities, suggesting species-specific differences in CIDEB function.
3.2. CIDEB in MAFLD
MAFLD has a complex underlying pathophysiology. Overnutrition is the main cause of MAFLD, leading to the growth of adipose depots and inflammatory conditions that encourage insulin resistance. When insulin resistance is present, inappropriate lipolysis of TGs leads to the continuous transport of fatty acids to the liver, which overwhelms its metabolic capacity when combined with elevated de novo lipogenesis. Lipotoxic lipids are created when there is an imbalance in lipid metabolism. These lipids can cause cellular stress, such as oxidative stress and endoplasmic reticulum stress, as well as stimulation of inflammation, apoptosis, tissue regeneration, and fibrogenesis [62,63]. Several poorly understood metabolic, genetic [64,65,66,67], and microbiome-related factors [68,69,70] affect how MAFLD progresses in the liver.
One of the genes involved in the multifactorial MAFLD is CIDEB. CIDEB-null mice showed protection against high-fat-diet-induced liver steatosis, with less accumulation of TGs in the liver and liver inflammation [29,44]. The decreased activation of SREBP may be one of the causes [44]. The mechanism of how CIDEB acts in the pathogenesis of CLD and MAFLD requires further investigation.
3.3. The Protective Role of CIDEB Variants in Liver Disease
Rare variants in the CIDEB gene have garnered attention due to their strong protective effects against CLD, MAFLD, cirrhosis, and viral liver diseases [19]. These variants, encompassing loss-of-function and missense alleles, exhibit a low prevalence of 0.7% overall [19]. Verweij et al. identified 17 independent variants in CIDEB, comprising 12 missense and 5 predicted loss-of-function alleles, which correlated with lower alanine aminotransferase (ALT) levels and reduced odds of liver disease [19]. Notably, each additional alternative-allele copy was associated with decreased ALT levels by −1.24 U per liter and 33% lower odds of liver disease [19].
The protection from CIDEB variants on ALT levels and liver disease risk was particularly pronounced for predicted loss-of-function variants. These variants were linked to even lower ALT levels (−1.57 U per liter) and a 46% reduced risk of liver disease [19]. Interestingly, the disparity in ALT levels between carriers of rare CIDEB variants and noncarriers was more evident in individuals with higher body mass index (BMI), obesity, and type 2 diabetes and carriers of the PNPLA3 (patatin-like phospholipase domain-containing protein 3) I148M variant, a well-known genetic susceptibility factor for MAFLD [64].
Moreover, carriers of rare CIDEB variants exhibited reduced odds of liver diseases, including cirrhosis, along with a lower prevalence of MAFLD [19]. Among obese individuals, the difference in the prevalence of liver disease between carriers and noncarriers of the CIDEB variant was greatest [19] Rare coding variants of CIDEB were also associated with lower odds of hepatocellular carcinoma compared to noncarriers [19].
Among individuals carrying rare coding variants of CIDEB, the nonalcoholic fatty liver disease activity score was lower at biopsy, independent of BMI, diabetes, and PNPLA3 I148M genotype [19]. They also had a lower liver fat percentage; the association was amplified in overweight individuals and the PNPLA3 I148M genotype.
Notably, the associations between CIDEB variants and liver health were consistent across different ancestries, although individuals of non-European descent were comparatively underrepresented in the genetic analysis [19].
In conclusion, the presence of rare coding variants in CIDEB offers a hopeful opportunity to mitigate liver diseases and metabolic disorders, highlighting the importance of further research in this area.
4. CIDEB in Apoptosis
CIDEB, first reported as an activator of apoptosis due to its homology with the CIDE-N domain of DNA fragmentation factor 45 (DFF45), induces DNA fragmentation, nuclear condensation, and membrane blebbing, which are pivotal steps in apoptosis [24,25,31]. Dimerization and localization in mitochondria are crucial for CIDEB-induced apoptosis [28], with the C-terminal domain mediating homodimerization and harboring the effector domain essential for cell death activity [71]. This includes the effector domain necessary for cell death activity [71]. Though mitochondrial localization is primarily observed in the late stages of cell death, endogenous CIDEB is not localized in mitochondria [28,32]. CIDEB dimerization likely enhances local concentration and facilitates mitochondrial targeting [28].
The N-terminal region, akin to DFF45, may modulate the C-terminal region, potentially regulating apoptotic activity [24]. Without apoptosis, DFF45 suppresses DFF40, a nuclease, by interacting with the CIDE-N domains (Figure 1). This way, DFF45 can also inhibit CIDEB and its apoptotic activity [24]. Following apoptosis, the effector caspase cleaves DFF45’s inhibitor domain, causing DFF45 to separate from DFF40, so DFF40 releases DNA fragmentation [72] (Figure 1). Lugovsky et al. demonstrated an interaction between CIDE-N of CIDEB and both CIDE-N of DFF40 and DFF45. CIDEB controls the activity of the DFF40/DFF45 complex in vitro [25] via sequestration of DFF45 [47].
Although it was initially reported that CIDEB-induced apoptosis is caspase-independent, later research linked CIDEB-induced apoptosis to caspase-dependent cell death [71]. This process is mediated by the release of cytochrome c from mitochondria, which activates the cytochrome c/caspase-9/caspase-3 pathway [71].
5. CIDEB in Viral Hepatitis
5.1. CIDEB in Hepatitis C
CIDEB influences many steps of the HCV replication cycle, including entry, replication, assembly, and release (Figure 1). It serves as an essential cofactor for the late-step entry of HCV into hepatocytes, potentially facilitating the membrane fusion between HCV and endosome membranes. However, its absence on plasma or endosomal membranes upon HCV infection suggests the involvement of additional transmembrane proteins in mediating a connection between CIDEB and HCV glycoproteins [46]. Alternatively, the refractoriness of CIDEB-deficient cells to HCV infection may stem from indirect effects on HCV entry via viral or cellular membrane lipid composition modifications [46]. The involvement of CIDEB in HCV replication remains contentious. While some studies have reported that CIDEB does not affect HCV RNA replication [46,73], others have observed that CIDEB overexpression reduces HCV replication, and CIDEB knockdown enhances viral replication and protein release (Figure 1). These conflicting findings suggest a potential antiviral effect of CIDEB, primarily exerted at the translational level [47].
Not all LD need HCV core proteins on their surfaces to produce high-titer HCV [74]. Instead, CIDEB could prevent HCV infection by blocking the development of LD through mechanisms such as SREBP-1 and SREBP-2 depletion [46,75]. Nevertheless, Singaravelu et al. further noted that CIDEB’s antiviral effect against HCV replication is separate from its LD fusogenic role [47], suggesting that the CIDEB’s action against HCV involves different pathways. The reduction of HCV RNA levels by CIDEB is correlated with its proapoptotic activity through a caspase-independent mechanism, but basal expression levels of CIDEB did not promote cell death [47].
The role of CIDEB in HCV assembly and release is better understood (Figure 1). It is a critical regulator of HCV’s VLDL pathway components for assembly and release. HCV uses VLDL pathway components for assembly and release [76,77]; CIDEB has been identified as a critical regulator of this pathway [73]. Prior studies have demonstrated that HNF4α influences HCV assembly and release by regulating downstream components involved in VLDL assembly and secretion [73]. Notably, CIDEB is transcriptionally promoted by HNF4α [54,55,56], which underlines its potential role in this pathway.
The regulation of CIDEB protein levels during HCV infection has been inconsistently described. Conversely, HCV infection effectively decreases CIDEB protein levels [46,58,71] through a post-transcriptional mechanism likely involving translational silencing and accelerated protein degradation [46] through a proteolytic cleavage event in the “competition-displacement model”, independent of cellular proteasomal degradation. This cleavage event is facilitated by the HCV core protein located on the surface of LD [58]. HCV core protein coats LDs extensively, which may displace native LD proteins like CIDEB and present them to proteases that would otherwise be unable to access [58]. Alternatively, the HCV core protein could activate a protease that may cleave CIDEB, but this protease has yet to be identified [58]. Wu et al. [46] proposed that the downregulation of the proapoptotic CIDEB may increase the survival of infected cells, facilitating the development of chronic infections.
On the other hand, HCV infection has been found to upregulate CIDEB expression [47]. Singaravelu et al. proposed that this induction of CIDEB expression could characterize a host response to the pro-viral accumulation of lipids induced by HCV infection consistent with known modulation of lipid metabolism [47]. HCV activates PGC-1a, which regulates VLDL assembly through CIDEB [55], further supporting the connection between HCV-induced CIDEB expression and viral manipulation of lipid metabolism [47]. The variations in those findings could be the result of different models.
Steatosis is a common feature observed in patients infected with HCV [78,79,80], partly induced by increasing lipogenesis. HCV infection leads to increased stability of LD as well as CIDEB [58]. This observation raises the possibility of a link between lipid regulation in infected cells and the modulation of CIDEB [58]. This is supported by the finding that the density profile of VLDL particles secreted from CIDEB KO cells resembles that of particles secreted from HCV-infected cells, as opposed to wild-type uninfected cells. The HCV core protein could exploit this stabilization mechanism to contribute to hepatic steatosis by enhancing the stability of cytoplasmic LD [58,81,82].
The HCV protein NS5A is essential for both viral assembly and replication [83]. Recent research has clarified its interactions with CIDEB, providing insight into HCV pathogenesis’s complex mechanisms. Specifically, domain I of NS5A interacts with the N terminus of CIDEB [73], and domain III of NS5A interacts with ApoE [84], a crucial component of lipoprotein metabolism. The interaction between NS5A and CIDEB may bring NS5A closer to ApoE, potentially facilitating the formation of infectious HCV particles [73]. The connection of ApoE with HCV particles is linked to the interaction between CIDEB and NS5A [73]. CIDEB silencing disrupts this connection, leading to increased quantities of HCV RNA and core protein, generally linked to non-infectious virus particles. Furthermore, the decreasing rate of identifying NS5A-CIDEB interaction in various species is associated with their different vulnerability to HCV infection [73]. This observation suggests that CIDEB might be critical in determining HCV host tropism [73].
NS2 is a transmembrane polypeptide of HCV that exhibits a distinct orientation within the cell, with its C terminus located in the lumen of the ER and the N terminus in the cytosol [85]. There are different reports on whether CIDEB and NS2 interact. An interaction was observed under basal expression, only in overexpression of CIDEB [73], or not at all [46,86,87]. NS2 aa 135–139 could interact with the CIDEB killing domain, which may prevent CIDEB-induced apoptosis by disrupting its dimerization [71]. Erdtmann et al. further propose that inhibiting pro-apoptotic CIDEB might be a general strategy that viruses use, including HCV, to escape host cell defenses, which may lead to viral persistence in HCV pathogenesis.
Carriers of rare coding variants in CIDEB had lower odds of viral hepatitis-associated liver disease (OR per allele, 0.69; 95% CI, 0.54 to 0.88; p < 0.002) [19].
Overall, CIDEB’s multifaceted involvement in different stages of the HCV lifecycle highlights its potential as a therapeutic target and underscores the intricate interplay between lipid metabolism and viral infection.
5.2. CIDEB in Hepatitis B
CIDEB is involved in the HBV replication cycle by regulating viral replication. Overexpression of CIDEB resulted in enhanced HBV production through increased activity of the HBV core promoter [88]. On the other hand, HBV-producing cells had decreased CIDEB expression [88,89]. Because hepatocyte nuclear factor 4 alpha (HNF4α) levels were lower in HBV-infected cells [90], the downregulation of CIDEB in HBV-infected cells may be attributed to lower levels of hepatocyte HNF4α, a known transcriptional regulator of CIDEB [54,56,90]. HBV-induced reduction in HNF4α expression could consequently lower CIDEB levels, contributing to the modulation of viral replication [88].
Consistent with the reported functions of CIDEB, HBV-producing cells had fewer TGs and smaller sizes of lipid droplets [88]. HBV-infected patients had a lower prevalence of MAFLD and metabolic syndrome [91,92,93]. This may be caused by CIDEB inhibition. More research will be needed to fully understand the implication and significance of CIDEB in HBV.
6. Discussion
CIDEB is a multifaceted protein that plays roles in diverse metabolic pathways. Initially identified as an apoptosis-inducing protein, subsequent studies revealed its critical role in lipid metabolism, including regulating chylomicrons, VLDL, lipid droplets, and SREBP. Most functional studies on CIDEB are based on animal models, highlighting the need for human disease models to further understand its role.
Historically, treatment options for MAFLD were limited to diet and regular exercise due to the scarcity of effective drugs. The advent of resmetirom has revolutionized MAFLD treatment by targeting inflammation and fibrosis, thereby improving liver function and reducing disease progression. Other drugs for MAFLD are also in development.
A study of a common loss-of-function mutation in HSD17B13 and its protection from liver disease prompted the creation of inhibitory small interfering RNAs (siRNAs) against HSD17B13, which are presently being tested in clinical trials (ClinicalTrials.gov numbers, NCT04565717 and NCT04202354). Despite this progress, rare coding variants, which might significantly impact, have received little attention.
Rare germline variants in CIDEB have been found to protect against the development of liver disorders such as cirrhosis, MAFLD, and viral hepatitis. Further research into the mechanisms of the variants is necessary. We have summarized the currently available models (Supplementary Table S1). Although the rare germline variants have a low prevalence, their importance as therapeutic targets must be considered. Preclinical research is underway to develop a new drug using an siRNA approach against CIDEB from Regeneron Pharmaceuticals.
Nonetheless, further research is needed to comprehend the essential function CIDEB plays in lipid metabolism.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/livers4030030/s1, Table S1. Overview of in-vitro and in-vivo models for CIDEB. References [94,95,96,97,98,99,100,101,102,103] are cited in the Supplementary Materials.
Author Contributions
Conceptualization, L.W. and N.H.; resources, N.H. and I.M.S.; writing—original draft preparation, L.W. and N.H.; writing—review and editing, P.T., J.M., A.R.-S. and I.M.S.; visualization, L.W. and N.H.; supervision, N.H. and I.M.S.; project administration, N.H.; funding acquisition, N.H. All authors have read and agreed to the published version of the manuscript.
Funding
This work was funded by the Einstein Center for Regenerative Therapies.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Materials, further inquiries can be directed to the corresponding author.
Conflicts of Interest
The authors declare no conflicts of interest.
References
- Naghavi, M.; Ong, K.L.; Aali, A.; Ababneh, H.S.; Abate, Y.H.; Abbafati, C.; Abbasgholizadeh, R.; Abbasian, M.; Abbasi-Kangevari, M.; Abbastabar, H.; et al. Global Burden of 288 Causes of Death and Life Expectancy Decomposition in 204 Countries and Territories and 811 Subnational Locations, 1990–2021: A Systematic Analysis for the Global Burden of Disease Study 2021. Lancet 2024, 403, 2100–2132. [Google Scholar] [CrossRef] [PubMed]
- Moon, A.M.; Singal, A.G.; Tapper, E.B. Contemporary Epidemiology of Chronic Liver Disease and Cirrhosis. Clin. Gastroenterol. Hepatol. 2020, 18, 2650–2666. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.M.; Wong, G.; Anstee, Q.M.; Henry, L. The Global Burden of Liver Disease. Clin. Gastroenterol. Hepatol. 2023, 21, 1978–1991. [Google Scholar] [CrossRef]
- Eslam, M.; Newsome, P.N.; Sarin, S.K.; Anstee, Q.M.; Targher, G.; Romero-Gomez, M.; Zelber-Sagi, S.; Wai-Sun Wong, V.; Dufour, J.-F.; Schattenberg, J.M.; et al. A New Definition for Metabolic Dysfunction-Associated Fatty Liver Disease: An International Expert Consensus Statement. J. Hepatol. 2020, 73, 202–209. [Google Scholar] [CrossRef] [PubMed]
- EASL–EASD–EASO Clinical Practice Guidelines for the Management of Non-Alcoholic Fatty Liver Disease. J. Hepatol. 2016, 64, 1388–1402. [CrossRef] [PubMed]
- Loomba, R.; Sanyal, A.J. The Global NAFLD Epidemic. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 686–690. [Google Scholar] [CrossRef]
- Younossi, Z.M.; Golabi, P.; Paik, J.M.; Henry, A.; Van Dongen, C.; Henry, L. The Global Epidemiology of Nonalcoholic Fatty Liver Disease (NAFLD) and Nonalcoholic Steatohepatitis (NASH): A Systematic Review. Hepatology 2023, 77, 1335–1347. [Google Scholar] [CrossRef]
- Wong, M.C.S.; Huang, J.; Wang, J.; Chan, P.S.F.; Lok, V.; Chen, X.; Leung, C.; Wang, H.H.X.; Lao, X.Q.; Zheng, Z.-J. Global, Regional and Time-Trend Prevalence of Central Obesity: A Systematic Review and Meta-Analysis of 13.2 Million Subjects. Eur. J. Epidemiol. 2020, 35, 673–683. [Google Scholar] [CrossRef]
- Koh, J.H.; Ng, C.H.; Nah, B.; Tan, D.J.H.; Loomba, R.; Huang, D.Q. NASH Is the Leading Cause of Hepatocellular Carcinoma in Liver Transplant Candidates. Clin. Gastroenterol. Hepatol. 2024, 22, 197–199.e3. [Google Scholar] [CrossRef]
- Younossi, Z.; Stepanova, M.; Ong, J.P.; Jacobson, I.M.; Bugianesi, E.; Duseja, A.; Eguchi, Y.; Wong, V.W.; Negro, F.; Yilmaz, Y.; et al. Nonalcoholic Steatohepatitis Is the Fastest Growing Cause of Hepatocellular Carcinoma in Liver Transplant Candidates. Clin. Gastroenterol. Hepatol. 2018, 17, 748–755.e3. [Google Scholar] [CrossRef]
- Anstee, Q.M.; Day, C.P. The Genetics of NAFLD. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 645–655. [Google Scholar] [CrossRef] [PubMed]
- Semmler, G.; Datz, C.; Reiberger, T.; Trauner, M. Diet and Exercise in NAFLD/NASH: Beyond the Obvious. Liver Int. 2021, 41, 2249–2268. [Google Scholar] [CrossRef]
- FDA Approves First Treatment for Patients with Liver Scarring Due to Fatty Liver Disease. FDA. Available online: https://www.fda.gov/news-events/press-announcements/fda-approves-first-treatment-patients-liver-scarring-due-fatty-liver-disease (accessed on 20 May 2024).
- Armstrong, M.J.; Gaunt, P.; Aithal, G.P.; Barton, D.; Hull, D.; Parker, R.; Hazlehurst, J.M.; Guo, K.; Abouda, G.; Aldersley, M.A.; et al. Liraglutide Safety and Efficacy in Patients with Non-Alcoholic Steatohepatitis (LEAN): A Multicentre, Double-Blind, Randomised, Placebo-Controlled Phase 2 Study. Lancet 2016, 387, 679–690. [Google Scholar] [CrossRef] [PubMed]
- Newsome, P.N.; Buchholtz, K.; Cusi, K.; Linder, M.; Okanoue, T.; Ratziu, V.; Sanyal, A.J.; Sejling, A.-S.; Harrison, S.A. A Placebo-Controlled Trial of Subcutaneous Semaglutide in Nonalcoholic Steatohepatitis. N. Engl. J. Med. 2021, 384, 1113–1124. [Google Scholar] [CrossRef]
- Aithal, G.P.; Thomas, J.A.; Kaye, P.V.; Lawson, A.; Ryder, S.D.; Spendlove, I.; Austin, A.S.; Freeman, J.G.; Morgan, L.; Webber, J. Randomized, Placebo-Controlled Trial of Pioglitazone in Nondiabetic Subjects with Nonalcoholic Steatohepatitis. Gastroenterology 2008, 135, 1176–1184. [Google Scholar] [CrossRef] [PubMed]
- Tahara, A.; Kurosaki, E.; Yokono, M.; Yamajuku, D.; Kihara, R.; Hayashizaki, Y.; Takasu, T.; Imamura, M.; Li, Q.; Tomiyama, H.; et al. Effects of SGLT2 Selective Inhibitor Ipragliflozin on Hyperglycemia, Hyperlipidemia, Hepatic Steatosis, Oxidative Stress, Inflammation, and Obesity in Type 2 Diabetic Mice. Eur. J. Pharmacol. 2013, 715, 246–255. [Google Scholar] [CrossRef] [PubMed]
- Emdin, C.A.; Haas, M.E.; Khera, A.V.; Aragam, K.; Chaffin, M.; Klarin, D.; Hindy, G.; Jiang, L.; Wei, W.-Q.; Feng, Q.; et al. A Missense Variant in Mitochondrial Amidoxime Reducing Component 1 Gene and Protection against Liver Disease. PLoS Genet. 2020, 16, e1008629. [Google Scholar] [CrossRef]
- Verweij, N.; Haas, M.E.; Nielsen, J.B.; Sosina, O.A.; Kim, M.; Akbari, P.; De, T.; Hindy, G.; Bovijn, J.; Persaud, T.; et al. Germline Mutations in CIDEB and Protection against Liver Disease. N. Engl. J. Med. 2022, 387, 332–344. [Google Scholar] [CrossRef]
- Abul-Husn, N.S.; Cheng, X.; Li, A.H.; Xin, Y.; Schurmann, C.; Stevis, P.; Liu, Y.; Kozlitina, J.; Stender, S.; Wood, G.C.; et al. A Protein-Truncating HSD17B13 Variant and Protection from Chronic Liver Disease. N. Engl. J. Med. 2018, 378, 1096–1106. [Google Scholar] [CrossRef]
- Motomura, T.; Amirneni, S.; Diaz-Aragon, R.; Faccioli, L.A.P.; Malizio, M.R.; Coard, M.C.; Kocas-Kilicarslan, Z.N.; Frau, C.; Haep, N.; Ostrowska, A.; et al. Is HSD17B13 Genetic Variant a Protector for Liver Dysfunction? Future Perspective as a Potential Therapeutic Target. J. Pers. Med. 2021, 11, 619. [Google Scholar] [CrossRef]
- Wu, C.; Zhang, Y.; Sun, Z.; Li, P. Molecular Evolution of Cide Family Proteins: Novel Domain Formation in Early Vertebrates and the Subsequent Divergence. BMC Evol. Biol. 2008, 8, 159. [Google Scholar] [CrossRef]
- Li, Q.-L.; Zheng, H.; Luo, Z.; Wu, L.-X.; Xu, P.-C.; Guo, J.-C.; Song, Y.-F.; Tan, X.-Y. Characterization and Expression Analysis of Seven Lipid Metabolism-Related Genes in Yellow Catfish Pelteobagrus Fulvidraco Fed High Fat and Bile Acid Diet. Gene 2024, 894, 147972. [Google Scholar] [CrossRef]
- Inohara, N.; Koseki, T.; Chen, S.; Wu, X.; Núñez, G. CIDE, a Novel Family of Cell Death Activators with Homology to the 45 kDa Subunit of the DNA Fragmentation Factor. EMBO J. 1998, 17, 2526–2533. [Google Scholar] [CrossRef]
- Lugovskoy, A.A.; Zhou, P.; Chou, J.J.; McCarty, J.S.; Li, P.; Wagner, G. Solution Structure of the CIDE-N Domain of CIDE-B and a Model for CIDE-N/CIDE-N Interactions in the DNA Fragmentation Pathway of Apoptosis. Cell 1999, 99, 747–755. [Google Scholar] [CrossRef]
- Zou, H.; Henzel, W.J.; Liu, X.; Lutschg, A.; Wang, X. Apaf-1, a Human Protein Homologous to C. Elegans CED-4, Participates in Cytochrome c–Dependent Activation of Caspase-3. Cell 1997, 90, 405–413. [Google Scholar] [CrossRef] [PubMed]
- McCarty, J.S.; Toh, S.Y.; Li, P. Study of DFF45 in Its Role of Chaperone and Inhibitor: Two Independent Inhibitory Domains of DFF40 Nuclease Activity. Biochem. Biophys. Res. Commun. 1999, 264, 176–180. [Google Scholar] [CrossRef]
- Chen, Z.; Guo, K.; Toh, S.Y.; Zhou, Z.; Li, P. Mitochondria Localization and Dimerization Are Required for CIDE-B to Induce Apoptosis. J. Biol. Chem. 2000, 275, 22619–22622. [Google Scholar] [CrossRef] [PubMed]
- Li, J.Z.; Ye, J.; Xue, B.; Qi, J.; Zhang, J.; Zhou, Z.; Li, Q.; Wen, Z.; Li, P. Cideb Regulates Diet-Induced Obesity, Liver Steatosis, and Insulin Sensitivity by Controlling Lipogenesis and Fatty Acid Oxidation. Diabetes 2007, 56, 2523–2532. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.-J.; Wang, C.; Yuan, Y.; Wang, H.; Wu, J.; Liu, F.; Li, L.; Gao, X.; Zhao, Y.-L.; Hu, P.-Z.; et al. Cideb Facilitates the Lipidation of Chylomicrons in the Small Intestine. J. Lipid Res. 2014, 55, 1279–1287. [Google Scholar] [CrossRef]
- Li, H.; Song, Y.; Zhang, L.J.; Li, F.F.; Gu, Y.; Zhang, J.; Dong, W.P.; Xue, L.; Zhang, L.Y.; Liu, F.; et al. Cell Death-Inducing DFF45-like Effector b (Cideb) Is Present in Pancreatic Beta-Cells and Involved in Palmitate Induced Beta-Cell Apoptosis. Diabetes/Metab. Res. Rev. 2012, 28, 145–155. [Google Scholar] [CrossRef]
- Ye, J.; Li, J.Z.; Liu, Y.; Li, X.; Yang, T.; Ma, X.; Li, Q.; Yao, Z.; Li, P. Cideb, an ER- and Lipid Droplet-Associated Protein, Mediates VLDL Lipidation and Maturation by Interacting with Apolipoprotein B. Cell Metab. 2009, 9, 177–190. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Ye, J.; Zhou, L.; Gu, W.; Fisher, E.A.; Li, P. eroxisome Proliferator-Activated Receptor-γ Coactivator-1α (PGC-1α) Stimulates VLDL Assembly through Activ VLDL Lipidation. J. Lipid Res. 2012, 53, 1877–1889. [Google Scholar] [CrossRef]
- Olofsson, S.-O.; Stillemark-Billton, P.; Asp, L. Intracellular Assembly of VLDL: Two Major Steps in Separate Cell Compartments. Trends Cardiovasc. Med. 2000, 10, 338–345. [Google Scholar] [CrossRef]
- Wetterau, J.R.; Aggerbeck, L.P.; Bouma, M.-E.; Eisenberg, C.; Munck, A.; Hermier, M.; Schmitz, J.; Gay, G.; Rader, D.J.; Gregg, R.E. Absence of Microsomal Triglyceride Transfer Protein in Individuals with Abetalipoproteinemia. Science 1992, 258, 999–1001. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, S.; Siddiqi, S.A. Intracellular Trafficking and Secretion of Very Low Density Lipoproteins. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 1079–1086. [Google Scholar] [CrossRef]
- Yamaguchi, J.; Gamble, M.V.; Conlon, D.; Liang, J.; Ginsberg, H.N. The Conversion of ApoB100 Low Density Lipoprotein/High Density Lipoprotein Particles to ApoB100 Very Low Density Lipoproteins in Response to Oleic Acid Occurs in the Endoplasmic Reticulum and Not in the Golgi in McA RH7777 Cells. J. Biol. Chem. 2003, 278, 42643–42651. [Google Scholar] [CrossRef] [PubMed]
- Gusarova, V.; Brodsky, J.L.; Fisher, E.A. Apolipoprotein B100 Exit from the Endoplasmic Reticulum (ER) Is COPII-Dependent, and Its Lipidation to Very Low Density Lipoprotein Occurs Post-ER. J. Biol. Chem. 2003, 278, 48051–48058. [Google Scholar] [CrossRef] [PubMed]
- Siddiqi, S.A. VLDL Exits from the Endoplasmic Reticulum in a Specialized Vesicle, the VLDL Transport Vesicle, in Rat Primary Hepatocytes. Biochem. J. 2008, 413, 333–342. [Google Scholar] [CrossRef]
- Yuan, L.; Kenny, S.J.; Hemmati, J.; Xu, K.; Schekman, R. TANGO1 and SEC12 Are Copackaged with Procollagen I to Facilitate the Generation of Large COPII Carriers. Proc. Natl. Acad. Sci. USA 2018, 115, E12255–E12264. [Google Scholar] [CrossRef]
- Zanetti, G.; Pahuja, K.B.; Studer, S.; Shim, S.; Schekman, R. COPII and the Regulation of Protein Sorting in Mammals. Nat. Cell Biol. 2012, 14, 20–28. [Google Scholar] [CrossRef]
- Fromme, J.C.; Schekman, R. COPII-Coated Vesicles: Flexible Enough for Large Cargo? Curr. Opin. Cell Biol. 2005, 17, 345–352. [Google Scholar] [CrossRef]
- Tiwari, S.; Siddiqi, S.; Siddiqi, S.A. CideB Protein Is Required for the Biogenesis of Very Low Density Lipoprotein (VLDL) Transport Vesicle. J. Biol. Chem. 2013, 288, 5157–5165. [Google Scholar] [CrossRef]
- Su, L.; Zhou, L.; Chen, F.-J.; Wang, H.; Qian, H.; Sheng, Y.; Zhu, Y.; Yu, H.; Gong, X.; Cai, L.; et al. Cideb Controls Sterol-Regulated ER Export of SREBP/SCAP by Promoting Cargo Loading at ER Exit Sites. EMBO J. 2019, 38, e100156. [Google Scholar] [CrossRef]
- Xu, W.; Wu, L.; Yu, M.; Chen, F.-J.; Arshad, M.; Xia, X.; Ren, H.; Yu, J.; Xu, L.; Xu, D.; et al. Differential Roles of Cell Death-Inducing DNA Fragmentation Factor-α-like Effector (CIDE) Proteins in Promoting Lipid Droplet Fusion and olecular Cloning, Genomic Organization, Chromosome. J. Biol. Chem. 2016, 291, 4282–4293. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Lee, E.M.; Hammack, C.; Robotham, J.M.; Basu, M.; Lang, J.; Brinton, M.A.; Tang, H. Cell Death-Inducing DFFA-Like Effector b Is Required for Hepatitis C Virus Entry into Hepatocytes. J. Virol. 2014, 88, 8433–8444. [Google Scholar] [CrossRef] [PubMed]
- Singaravelu, R.; Delcorde, J.; Lyn, R.K.; Steenbergen, R.H.; Jones, D.M.; Tyrrell, D.L.; Russell, R.S.; Pezacki, J.P. Investigating the Antiviral Role of Cell Death-Inducing DFF45-like Effector B in HCV Replication. FEBS J. 2014, 281, 3751–3765. [Google Scholar] [CrossRef]
- Singaravelu, R.; Lyn, R.K.; Srinivasan, P.; Delcorde, J.; Steenbergen, R.H.; Tyrrell, D.L.; Pezacki, J.P. Human Serum Activates CIDEB-Mediated Lipid Droplet Enlargement in Hepatoma Cells. Biochem. Biophys. Res. Commun. 2013, 441, 447–452. [Google Scholar] [CrossRef] [PubMed]
- Gong, J.; Sun, Z.; Wu, L.; Xu, W.; Schieber, N.; Xu, D.; Shui, G.; Yang, H.; Parton, R.G.; Li, P. Fsp27 Promotes Lipid Droplet Growth by Lipid Exchange and Transfer at Lipid Droplet Contact Sites. J. Cell Biol. 2011, 195, 953–963. [Google Scholar] [CrossRef] [PubMed]
- Li, J.Z.; Lei, Y.; Wang, Y.; Zhang, Y.; Ye, J.; Xia, X.; Pan, X.; Li, P. Control of Cholesterol Biosynthesis, Uptake and Storage in Hepatocytes by Cideb. Biochim. Biophys. Acta 2010, 1801, 577–586. [Google Scholar] [CrossRef]
- Matsuda, M.; Korn, B.S.; Hammer, R.E.; Moon, Y.-A.; Komuro, R.; Horton, J.D.; Goldstein, J.L.; Brown, M.S.; Shimomura, I. SREBP Cleavage-Activating Protein (SCAP) Is Required for Increased Lipid Synthesis in Liver Induced by Cholesterol Deprivation and Insulin Elevation. Genes Dev. 2001, 15, 1206–1216. [Google Scholar] [CrossRef] [PubMed]
- Chandrasekaran, P.; Weiskirchen, R. The Role of SCAP/SREBP as Central Regulators of Lipid Metabolism in Hepatic Steatosis. Int. J. Mol. Sci. 2024, 25, 1109. [Google Scholar] [CrossRef]
- Ko, C.-W.; Qu, J.; Black, D.D.; Tso, P. Regulation of Intestinal Lipid Metabolism: Current Concepts and Relevance to Disease. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 169–183. [Google Scholar] [CrossRef]
- Da, L.; Li, D.; Yokoyama, K.K.; Li, T.; Zhao, M. Dual Promoters Control the Cell-Specific Expression of the Human Cell Death-Inducing DFF45-like Effector B Gene. Biochem. J. 2006, 393 Pt 3, 779–788. [Google Scholar] [CrossRef][Green Version]
- Chen, Z.; Norris, J.Y.; Finck, B.N. Peroxisome Proliferator-Activated Receptor-γ Coactivator-1α (PGC-1α) Stimulates VLDL Assembly through Activation of Cell Death-Inducing DFFA-like Effector B (CideB). J. Biol. Chem. 2010, 285, 25996. [Google Scholar] [CrossRef]
- Daigo, K.; Kawamura, T.; Ohta, Y.; Ohashi, R.; Katayose, S.; Tanaka, T.; Aburatani, H.; Naito, M.; Kodama, T.; Ihara, S.; et al. Proteomic Analysis of Native Hepatocyte Nuclear Factor-4α (HNF4α) Isoforms, Phosphorylation Status, and Interactive Cofactors. J. Biol. Chem. 2011, 286, 674–686. [Google Scholar] [CrossRef] [PubMed]
- Collison, K.S.; Inglis, A.; Shibin, S.; Saleh, S.; Andres, B.; Ubungen, R.; Thiam, J.; Mata, P.; Al-Mohanna, F.A. Effect of Developmental NMDAR Antagonism with CGP 39551 on Aspartame-Induced Hypothalamic and Adrenal Gene Expression. PLoS ONE 2018, 13, e0194416. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.M.; Alsagheir, A.; Wu, X.; Hammack, C.; McLauchlan, J.; Watanabe, N.; Wakita, T.; Kneteman, N.M.; Douglas, D.N.; Tang, H. Hepatitis C Virus-Induced Degradation of Cell Death-Inducing DFFA-Like Effector B Leads to Hepatic Lipid Dysregulation. J. Virol. 2016, 90, 4174–4185. [Google Scholar] [CrossRef]
- Aguilar, M.; Bhuket, T.; Torres, S.; Liu, B.; Wong, R.J. Prevalence of the Metabolic Syndrome in the United States, 2003–2012. JAMA 2015, 313, 1973–1974. [Google Scholar] [CrossRef] [PubMed]
- Noubiap, J.J.; Nansseu, J.R.; Lontchi-Yimagou, E.; Nkeck, J.R.; Nyaga, U.F.; Ngouo, A.T.; Tounouga, D.N.; Tianyi, F.-L.; Foka, A.J.; Ndoadoumgue, A.L.; et al. Geographic Distribution of Metabolic Syndrome and Its Components in the General Adult Population: A Meta-Analysis of Global Data from 28 Million Individuals. Diabetes Res. Clin. Pract. 2022, 188, 109924. [Google Scholar] [CrossRef]
- Zhou, Z.; Yon Toh, S.; Chen, Z.; Guo, K.; Peng Ng, C.; Ponniah, S.; Lin, S.-C.; Hong, W.; Li, P. Cidea-Deficient Mice Have Lean Phenotype and Are Resistant to Obesity. Nat. Genet. 2003, 35, 49–56. [Google Scholar] [CrossRef]
- Friedman, S.L.; Neuschwander-Tetri, B.A.; Rinella, M.; Sanyal, A.J. Mechanisms of NAFLD Development and Therapeutic Strategies. Nat. Med. 2018, 24, 908–922. [Google Scholar] [CrossRef] [PubMed]
- Sanyal, A.J. Past, Present and Future Perspectives in Nonalcoholic Fatty Liver Disease. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 377–386. [Google Scholar] [CrossRef] [PubMed]
- Romeo, S.; Kozlitina, J.; Xing, C.; Pertsemlidis, A.; Cox, D.; Pennacchio, L.A.; Boerwinkle, E.; Cohen, J.C.; Hobbs, H.H. Genetic Variation in PNPLA3 Confers Susceptibility to Nonalcoholic Fatty Liver Disease. Nat. Genet. 2008, 40, 1461–1465. [Google Scholar] [CrossRef]
- Meffert, P.J.; Repp, K.D.; Völzke, H.; Weiss, F.U.; Homuth, G.; Kühn, J.P.; Lerch, M.M.; Aghdassi, A.A. The PNPLA3 SNP Rs738409:G Allele Is Associated with Increased Liver Disease-Associated Mortality but Reduced Overall Mortality in a Population-Based Cohort. J. Hepatol. 2018, 68, 858–860. [Google Scholar] [CrossRef]
- Stender, S.; Kozlitina, J.; Nordestgaard, B.G.; Tybjærg-Hansen, A.; Hobbs, H.H.; Cohen, J.C. Adiposity Amplifies the Genetic Risk of Fatty Liver Disease Conferred by Multiple Loci. Nat. Genet. 2017, 49, 842–847. [Google Scholar] [CrossRef]
- Eslam, M.; George, J. Genetic Contributions to NAFLD: Leveraging Shared Genetics to Uncover Systems Biology. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 40–52. [Google Scholar] [CrossRef]
- Aron-Wisnewsky, J.; Warmbrunn, M.V.; Nieuwdorp, M.; Clément, K. Nonalcoholic Fatty Liver Disease: Modulating Gut Microbiota to Improve Severity? Gastroenterology 2020, 158, 1881–1898. [Google Scholar] [CrossRef]
- Caussy, C.; Tripathi, A.; Humphrey, G.; Bassirian, S.; Singh, S.; Faulkner, C.; Bettencourt, R.; Rizo, E.; Richards, L.; Xu, Z.Z.; et al. A Gut Microbiome Signature for Cirrhosis Due to Nonalcoholic Fatty Liver Disease. Nat. Commun. 2019, 10, 1406. [Google Scholar] [CrossRef] [PubMed]
- Loomba, R.; Seguritan, V.; Li, W.; Long, T.; Klitgord, N.; Bhatt, A.; Dulai, P.S.; Caussy, C.; Bettencourt, R.; Highlander, S.K.; et al. Gut Microbiome Based Metagenomic Signature for Non-Invasive Detection of Advanced Fibrosis in Human Nonalcoholic Fatty Liver Disease. Cell Metab. 2017, 25, 1054–1062.e5. [Google Scholar] [CrossRef]
- Erdtmann, L.; Franck, N.; Lerat, H.; Le Seyec, J.; Gilot, D.; Cannie, I.; Gripon, P.; Hibner, U.; Guguen-Guillouzo, C. The Hepatitis C Virus NS2 Protein Is an Inhibitor of CIDE-B-Induced Apoptosis. J. Biol. Chem. 2003, 278, 18256–18264. [Google Scholar] [CrossRef]
- Ha, H.J.; Park, H.H. Molecular Basis of Apoptotic DNA Fragmentation by DFF40. Cell Death Dis. 2022, 13, 198. [Google Scholar] [CrossRef] [PubMed]
- Cai, H.; Yao, W.; Li, L.; Li, X.; Hu, L.; Mai, R.; Peng, T. Cell-Death-Inducing DFFA-like Effector B Contributes to the Assembly of Hepatitis C Virus (HCV) Particles and Interacts with HCV NS5A. Sci. Rep. 2016, 6, 27778. [Google Scholar] [CrossRef]
- Boson, B.; Granio, O.; Bartenschlager, R.; Cosset, F.-L. A Concerted Action of Hepatitis C Virus P7 and Nonstructural Protein 2 Regulates Core Localization at the Endoplasmic Reticulum and Virus Assembly. PLoS Pathog. 2011, 7, e1002144. [Google Scholar] [CrossRef]
- Li, Q.; Pène, V.; Krishnamurthy, S.; Cha, H.; Liang, T.J. Hepatitis C Virus Infection Activates a Novel Innate Pathway Involving IKKα in Lipogenesis and Viral Assembly. Nat. Med. 2013, 19, 722–729. [Google Scholar] [CrossRef]
- Bassendine, M.F.; Sheridan, D.A.; Felmlee, D.J.; Bridge, S.H.; Toms, G.L.; Neely, R.D.G. HCV and the Hepatic Lipid Pathway as a Potential Treatment Target. J. Hepatol. 2011, 55, 1428–1440. [Google Scholar] [CrossRef][Green Version]
- Bassendine, M.F.; Sheridan, D.A.; Bridge, S.H.; Felmlee, D.J.; Neely, R.D.G. Lipids and HCV. Semin. Immunopathol. 2013, 35, 87–100. [Google Scholar] [CrossRef]
- Lambert, J.E.; Bain, V.G.; Ryan, E.A.; Thomson, A.B.R.; Clandinin, M.T. Elevated Lipogenesis and Diminished Cholesterol Synthesis in Patients with Hepatitis C Viral Infection Compared to Healthy Humans. Hepatology 2013, 57, 1697–1704. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Hood, B.L.; Chadwick, S.L.; Liu, S.; Watkins, S.C.; Luo, G.; Conrads, T.P.; Wang, T. Fatty Acid Synthase Is Upregulated during HCV Infection and Regulates HCV Entry and Production. Hepatology 2008, 48, 1396–1403. [Google Scholar] [CrossRef] [PubMed]
- Lerat, H.; Honda, M.; Beard, M.R.; Loesch, K.; Sun, J.; Yang, Y.; Okuda, M.; Gosert, R.; Xiao, S.-Y.; Weinman, S.A.; et al. Steatosis and Liver Cancer in Transgenic Mice Expressing the Structural and Nonstructural Proteins of Hepatitis C Virus. Gastroenterology 2002, 122, 352–365. [Google Scholar] [CrossRef]
- Harris, C.; Herker, E.; Farese, R.V.; Ott, M. Hepatitis C Virus Core Protein Decreases Lipid Droplet Turnover. J. Biol. Chem. 2011, 286, 42615–42625. [Google Scholar] [CrossRef]
- Camus, G.; Schweiger, M.; Herker, E.; Harris, C.; Kondratowicz, A.S.; Tsou, C.-L.; Farese, R.V.; Herath, K.; Previs, S.F.; Roddy, T.P.; et al. The Hepatitis C Virus Core Protein Inhibits Adipose Triglyceride Lipase (ATGL)-Mediated Lipid Mobilization and Enhances the ATGL Interaction with Comparative Gene Identification 58 (CGI-58) and Lipid Droplets. J. Biol. Chem. 2014, 289, 35770–35780. [Google Scholar] [CrossRef]
- Kandangwa, M.; Liu, Q. HCV-2a NS5A Downregulates Viral Translation Predominantly through Domain I. Biochem. Biophys. Res. Commun. 2020, 529, 77–84. [Google Scholar] [CrossRef]
- Benga, W.J.A.; Krieger, S.E.; Dimitrova, M.; Zeisel, M.B.; Parnot, M.; Lupberger, J.; Hildt, E.; Luo, G.; Mclauchlan, J.; Baumert, T.F.; et al. Apolipoprotein E Interacts with Hepatitis C Virus Nonstructural Protein 5A and Determines Assembly of Infectious Particles. Hepatology 2010, 51, 43. [Google Scholar] [CrossRef] [PubMed]
- Santolini, E.; Pacini, L.; Fipaldini, C.; Migliaccio, G.; Monica, N. The NS2 Protein of Hepatitis C Virus Is a Transmembrane Polypeptide. J. Virol. 1995, 69, 7461–7471. [Google Scholar] [CrossRef] [PubMed]
- Phan, T.; Beran, R.K.F.; Peters, C.; Lorenz, I.C.; Lindenbach, B.D. Hepatitis C Virus NS2 Protein Contributes to Virus Particle Assembly via Opposing Epistatic Interactions with the E1-E2 Glycoprotein and NS3-NS4A Enzyme Complexes. J. Virol. 2009, 83, 8379–8395. [Google Scholar] [CrossRef]
- Stapleford, K.A.; Lindenbach, B.D. Hepatitis C Virus NS2 Coordinates Virus Particle Assembly through Physical Interactions with the E1-E2 Glycoprotein and NS3-NS4A Enzyme Complexes. J. Virol. 2011, 85, 1706–1717. [Google Scholar] [CrossRef]
- Yasumoto, J.; Kasai, H.; Yoshimura, K.; Otoguro, T.; Watashi, K.; Wakita, T.; Yamashita, A.; Tanaka, T.; Takeda, S.; Moriishi, K. Hepatitis B Virus Prevents Excessive Viral Production via Reduction of Cell Death-Inducing DFF45-Like Effectors. J Gen Virol. 2017, 98, 1762–1773. [Google Scholar] [CrossRef]
- Nissim, O.; Melis, M.; Diaz, G.; Kleiner, D.E.; Tice, A.; Fantola, G.; Zamboni, F.; Mishra, L.; Farci, P. Liver Regeneration Signature in Hepatitis B Virus (HBV)-Associated Acute Liver Failure Identified by Gene Expression Profiling. PLoS ONE 2012, 7, e49611. [Google Scholar] [CrossRef]
- Wu, Q.; Liu, H.-O.; Liu, Y.-D.; Liu, W.-S.; Pan, D.; Zhang, W.-J.; Yang, L.; Fu, Q.; Xu, J.-J.; Gu, J.-X. Decreased Expression of Hepatocyte Nuclear Factor 4α (Hnf4α)/microRNA-122 (miR-122) Axis in Hepatitis B Virus-Associated Hepatocellular Carcinoma Enhances Potential Oncogenic GALNT10 Protein Activity. J. Biol. Chem. 2015, 290, 1170–1185. [Google Scholar] [CrossRef]
- Yi, S.; Ren, G.; Zhu, Y.; Cong, Q. Correlation Analysis of Hepatic Steatosis and Hepatitis B Virus: A Cross-Sectional Study. Virol. J. 2024, 21, 22. [Google Scholar] [CrossRef] [PubMed]
- Wong, V.W.-S.; Wong, G.L.-H.; Chu, W.C.-W.; Chim, A.M.-L.; Ong, A.; Yeung, D.K.-W.; Yiu, K.K.-L.; Chu, S.H.-T.; Chan, H.-Y.; Woo, J.; et al. Hepatitis B Virus Infection and Fatty Liver in the General Population. J. Hepatol. 2012, 56, 533–540. [Google Scholar] [CrossRef]
- Jinjuvadia, R.; Liangpunsakul, S. Association between Metabolic Syndrome and Its Individual Components with Viral Hepatitis B. Am. J. Med. Sci. 2014, 347, 23–27. [Google Scholar] [CrossRef] [PubMed]
- Hou, B.; Qin, L.; Huang, L. Liver Cancer Cells as the Model for Developing Liver-Targeted RNAi Therapeutics. Biochem. Biophys. Res. Commun. 2023, 644, 85–94. [Google Scholar] [CrossRef]
- Chen, Q.; Fang, W.; Shen, Y.; Xu, D.; Chen, Q.; Cui, K.; Mai, K.; Ai, Q. Suppression of Cideb under Endoplasmic Reticulum Stress Exacerbated Hepatic Inflammation by Inducing Hepatic Steatosis and Oxidative Stress. Free Radic. Biol. Med. 2022, 185, 67–75. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Li, A.; Yang, Z.Q. Molecular Cloning, Genomic Organization, Chromosome Mapping, Tissues Expression Pattern and Identification of a Novel Splicing Variant of Porcine CIDEb Gene. Biochem. Biophys. Res. Commun. 2016, 478, 486–493. [Google Scholar] [CrossRef]
- Qiu, Y.; Yang, X.; Ma, X.; Xiong, Y.; Tian, Z.; Fan, Q.; Wang, L.; Jiang, Z. CIDE Gene Expression in Adipose Tissue, Liver, and Skeletal Muscle from Obese and Lean Pigs. J. Zhejiang Univ. Sci. B 2017, 18, 492–500. [Google Scholar] [CrossRef] [PubMed]
- Ye, P.; Cheah, I.K.; Halliwell, B. High Fat Diets and Pathology in the Guinea Pig. Atherosclerosis or Liver Damage? Biochim. Biophys. Acta (BBA)—Mol. Basis Dis. 2013, 1832, 355–364. [Google Scholar] [CrossRef]
- Fu, J.-F.; Fang, Y.-L.; Liang, L.; Wang, C.-L.; Hong, F.; Dong, G.-P. A Rabbit Model of Pediatric Nonalcoholic Steatohepatitis: The Role of Adiponectin. World J. Gastroenterol. 2009, 15, 912–918. [Google Scholar] [CrossRef]
- Zhang, L.; Wu, X.; Liao, S.; Li, Y.; Zhang, Z.; Chang, Q.; Xiao, R.; Liang, B. Tree Shrew (Tupaia Belangeri Chinensis), a Novel Non-Obese Animal Model of Non-Alcoholic Fatty Liver Disease. Biol. Open 2016, 5, 1545–1552. [Google Scholar] [CrossRef]
- Hammoudeh, S.M.; Hammoudeh, A.M.; Bhamidimarri, P.M.; Mahboub, B.; Halwani, R.; Hamid, Q.; Rahmani, M.; Hamoudi, R. Insight into Molecular Mechanisms Underlying Hepatic Dysfunction in Severe COVID-19 Patients Using Systems Biology. World J. Gastroenterol. 2021, 27, 2850–2870. [Google Scholar] [CrossRef]
- Ng, S.W.K.; Rouhani, F.J.; Brunner, S.F.; Brzozowska, N.; Aitken, S.J.; Yang, M.; Abascal, F.; Moore, L.; Nikitopoulou, E.; Chappell, L.; et al. Convergent Somatic Mutations in Metabolism Genes in Chronic Liver Disease. Nature 2021, 598, 473–478. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Wang, H.; Zhao, J.; Yuan, Y.; Wang, C.; Li, J.; Zhang, L.; Zhang, L.; Li, Q.; Ye, J. Expression of CIDE Proteins in Clear Cell Renal Cell Carcinoma and Their Prognostic Significance. Mol. Cell. Biochem. 2013, 378, 145–151. [Google Scholar] [CrossRef] [PubMed][Green Version]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).