

An Integrated Pathogenetic Model of Primary Biliary Cholangitis


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Genetics and Epigenetics
Epigenetics
3. The Autophagy Theory
4. The Senescence Theory
5. Apoptosis in PBC
6. The Bicarbonate Umbrella Theory
The miR-506-AE2-sAC Model for PBC
7. Bile Acids
8. Gut Microbiota
9. Environment
9.1. Molecular Mimicry
9.2. The Viral Etiology of PBC
10. Immunology Theory
10.1. Innate Immunity
10.1.1. Monocytes and Macrophages
10.1.2. Biliary Innate Immunity
10.2. Adaptive Immunity
11. The Endothelin Theory
12. Interaction of Pathogenetic Factors in PBC
13. The Integrated Theory
14. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Stemmer, A.; Galili, T.; Kozlovski, T.; Zeevi, Y.; Marcus-Kalish, M.; Benjamini, Y.; Mitelpunkt, A. Current and Potential Approaches for Defining Disease Signatures: A Systematic Review. J. Mol. Neurosci. 2019, 67, 550–558. [Google Scholar] [PubMed]
- Trivella, J.; John, B.V.; Levy, C. Primary biliary cholangitis: Epidemiology, prognosis, and treatment. Hepatol. Commun. 2023, 7, e0179. [Google Scholar] [PubMed]
- Trivedi, P.J.; Hirschfield, G.M. Recent advances in clinical practice: Epidemiology of autoimmune liver diseases. Gut 2021, 70, 1989–2003. [Google Scholar] [PubMed]
- Lv, T.; Chen, S.; Li, M.; Zhang, D.; Kong, Y.; Jia, J. Regional variation and temporal trend of primary biliary cholangitis epidemiology: A systematic review and meta-analysis. J. Gastroenterol. Hepatol. 2021, 36, 1423–1434. [Google Scholar]
- Zeng, N.; Duan, W.; Chen, S.; Wu, S.; Ma, H.; Ou, X.; You, H.; Kong, Y.; Jia, J. Epidemiology and clinical course of primary biliary cholangitis in the Asia-Pacific region: A systematic review and meta-analysis. Hepatol. Int. 2019, 13, 788–799. [Google Scholar]
- Lu, M.; Zhou, Y.; Haller, I.V.; Romanelli, R.J.; VanWormer, J.J.; Rodriguez, C.V.; Anderson, H.; Boscarino, J.A.; Schmidt, M.A.; Daida, Y.G.; et al. Increasing Prevalence of Primary Biliary Cholangitis and Reduced Mortality With Treatment. Clin. Gastroenterol. Hepatol. 2018, 16, 1342–1350.e1. [Google Scholar] [CrossRef]
- Li, H.; Guan, Y.; Han, C.; Zhang, Y.; Liu, Q.; Wei, W.; Ma, Y. The pathogenesis, models and therapeutic advances of primary biliary cholangitis. Biomed. Pharmacother. 2021, 140, 111754. [Google Scholar]
- Prince, M.I.; Chetwynd, A.; Diggle, P.; Jarner, M.; Metcalf, J.V.; James, O.F. The geographical distribution of primary biliary cirrhosis in a well-defined cohort. Hepatology 2001, 34, 1083–1088. [Google Scholar]
- Arbour, L.; Rupps, R.; Field, L.; Ross, P.; Erikson, A.; Henderson, H.; Hill, W.; Yoshida, E. Characteristics of primary biliary cirrhosis in British Columbia’s First Nations population. Can. J. Gastroenterol. 2005, 19, 305–310. [Google Scholar]
- Arbour, L.; Field, L.; Ross, P.; Erikson, A.; Yoshida, E. The mystery of primary biliary cirrhosis in British Columbia’s First Nations people. Int. J. Circumpolar Health 2004, 63, 185–188. [Google Scholar]
- Koulentaki, M.; Mantaka, A.; Sifaki-Pistolla, D.; Thalassinos, E.; Tzanakis, N.; Kouroumalis, E. Geoepidemiology and space-time analysis of Primary biliary cirrhosis in Crete, Greece. Liver Int. 2014, 34, e200–e207. [Google Scholar] [PubMed]
- Roberts, S.B.; Hirschfield, G.M.; Worobetz, L.J.; Vincent, C.; Flemming, J.A.; Cheung, A.; Qumosani, K.; Swain, M.; Grbic, D.; Ko, H.H.; et al. Ethnicity, disease severity, and survival in Canadian patients with primary biliary cholangitis. Hepatology 2022, 76, 303–316. [Google Scholar] [PubMed]
- Lindor, K.D.; Bowlus, C.L.; Boyer, J.; Levy, C.; Mayo, M. Primary Biliary Cholangitis: 2018 Practice Guidance from the American Association for the Study of Liver Diseases. Hepatology 2019, 69, 394–419. [Google Scholar] [PubMed]
- Joplin, R.E.; Wallace, L.L.; Lindsay, J.G.; Palmer, J.M.; Yeaman, S.J.; Neuberger, J.M. The human biliary epithelial cell plasma membrane antigen in primary biliary cirrhosis: Pyruvate dehydrogenase X? Gastroenterology 1997, 113, 1727–1733. [Google Scholar]
- Nguyen, H.H.; Fritzler, M.J.; Swain, M.G. A Review on Biomarkers for the Evaluation of Autoimmune Cholestatic Liver Diseases and Their Overlap Syndromes. Front. Mol. Med. 2022, 2, 914505. [Google Scholar]
- Yamagiwa, S.; Kamimura, H.; Takamura, M.; Aoyagi, Y. Autoantibodies in primary biliary cirrhosis: Recent progress in research on the pathogenetic and clinical significance. World J. Gastroenterol. 2014, 20, 2606–2612. [Google Scholar]
- Joshi, S.; Cauch-Dudek, K.; Heathcote, E.J.; Lindor, K.; Jorgensen, R.; Klein, R. Antimitochondrial antibody profiles: Are they valid prognostic indicators in primary biliary cirrhosis? Am. J. Gastroenterol. 2002, 97, 999–1002. [Google Scholar]
- Elkoshi, Z. Autoimmune diseases refractory to corticosteroids and immunosuppressants. Front. Immunol. 2024, 15, 1447337. [Google Scholar] [CrossRef]
- Ronca, V.; Carbone, M.; Bernuzzi, F.; Malinverno, F.; Mousa, H.S.; Gershwin, M.E.; Invernizzi, P. From pathogenesis to novel therapies in the treatment of primary biliary cholangitis. Expert Rev. Clin. Immunol. 2017, 13, 1121–1131. [Google Scholar] [CrossRef]
- Syed, H.; Penner, T.; Mason, A.L. Linking Human Betaretrovirus with Autoimmunity and Liver Disease in Patients with Primary Biliary Cholangitis. Viruses 2022, 14, 1941. [Google Scholar] [CrossRef]
- Zandanell, S.; Strasser, M.; Feldman, A.; Strebinger, G.; Aigner, G.; Niederseer, D.; Laimer, M.; Mussnig, B.; Paulweber, B.; Datz, C.; et al. Similar clinical outcome of AMA immunoblot-M2-negative compared to immunoblot-positive subjects over six years of follow-up. Postgrad. Med. 2021, 133, 291–298. [Google Scholar]
- Tanaka, A.; Leung, P.S.C.; Gershwin, M.E. Pathogen infections and primary biliary cholangitis. Clin. Exp. Immunol. 2019, 195, 25–34. [Google Scholar]
- Hirschfield, G.M.; Chapman, R.W.; Karlsen, T.H.; Lammert, F.; Lazaridis, K.N.; Mason, A.L. The genetics of complex cholestatic disorders. Gastroenterology 2013, 144, 1357–1374. [Google Scholar]
- Zografos, T.A.; Gatselis, N.; Zachou, K.; Liaskos, C.; Gabeta, S.; Koukoulis, G.K.; Dalekos, G.N. Primary biliary cirrhosis-specific autoantibodies in first degree relatives of Greek primary biliary cirrhosis patients. World J. Gastroenterol. 2012, 18, 4721–4728. [Google Scholar]
- Mantaka, A.; Koulentaki, M.; Chlouverakis, G.; Enele-Melono, J.M.; Darivianaki, A.; Tzardi, M.; Kouroumalis, E.A. Primary biliary cirrhosis in a genetically homogeneous population: Disease associations and familial occurrence rates. BMC Gastroenterol. 2012, 12, 110. [Google Scholar]
- Trivedi, P.J.; Hirschfield, G.M. The Immunogenetics of Autoimmune Cholestasis. Clin. Liver Dis. 2016, 20, 15–31. [Google Scholar]
- Donaldson, P.T.; Baragiotta, A.; Heneghan, M.A.; Floreani, A.; Venturi, C.; Underhill, J.A.; Jones, D.E.; James, O.F.; Bassendine, M.F. HLA class II alleles, genotypes, haplotypes, and amino acids in primary biliary cirrhosis: A large-scale study. Hepatology 2006, 44, 667–674. [Google Scholar]
- Gerussi, A.; Asselta, R.; Invernizzi, P. Genetics of Primary Biliary Cholangitis. Clin. Liver Dis. 2022, 26, 571–582. [Google Scholar]
- Invernizzi, P.; Miozzo, M.; Battezzati, P.M.; Bianchi, I.; Grati, F.R.; Simoni, G.; Selmi, C.; Watnik, M.; Gershwin, M.E.; Podda, M. Frequency of monosomy X in women with primary biliary cirrhosis. Lancet 2004, 363, 533–535. [Google Scholar]
- Qiu, F.; Tang, R.; Zuo, X.; Shi, X.; Wei, Y.; Zheng, X.; Dai, Y.; Gong, Y.; Wang, L.; Xu, P.; et al. A genome-wide association study identifies six novel risk loci for primary biliary cholangitis. Nat. Commun. 2017, 8, 14828. [Google Scholar]
- Nakamura, M.; Nishida, N.; Kawashima, M.; Aiba, Y.; Tanaka, A.; Yasunami, M.; Nakamura, H.; Komori, A.; Nakamuta, M.; Zeniya, M.; et al. Genome-Wide association study identifies TNFSF15 and POU2AF1 as susceptibility loci for primary biliary cirrhosis in the Japanese population. Am. J. Hum. Genet. 2012, 91, 721–728. [Google Scholar]
- Mells, G.F.; Floyd, J.A.; Morley, K.I.; Cordell, H.J.; Franklin, C.S.; Shin, S.Y.; Heneghan, M.A.; Neuberger, J.M.; Donaldson, P.T.; Day, D.B.; et al. Genome-Wide association study identifies 12 new susceptibility loci for primary biliary cirrhosis. Nat. Genet. 2011, 43, 329–332. [Google Scholar]
- Liu, X.; Invernizzi, P.; Lu, Y.; Kosoy, R.; Lu, Y.; Bianchi, I.; Podda, M.; Xu, C.; Xie, G.; Macciardi, F.; et al. Genome-Wide meta-analyses identify three loci associated with primary biliary cirrhosis. Nat. Genet. 2010, 42, 658–660. [Google Scholar]
- Liu, J.Z.; Almarri, M.A.; Gaffney, D.J.; Mells, G.F.; Jostins, L.; Cordell, H.J.; Ducker, S.J.; Day, D.B.; Heneghan, M.A.; Neuberger, J.M.; et al. Dense fine-mapping study identifies new susceptibility loci for primary biliary cirrhosis. Nat. Genet. 2012, 44, 1137–1141. [Google Scholar]
- Juran, B.D.; Hirschfield, G.M.; Invernizzi, P.; Atkinson, E.J.; Li, Y.; Xie, G.; Kosoy, R.; Ransom, M.; Sun, Y.; Bianchi, I.; et al. Immunochip analyses identify a novel risk locus for primary biliary cirrhosis at 13q14, multiple independent associations at four established risk loci and epistasis between 1p31 and 7q32 risk variants. Hum. Mol. Genet. 2012, 21, 5209–5221. [Google Scholar]
- Hirschfield, G.M.; Liu, X.; Han, Y.; Gorlov, I.P.; Lu, Y.; Xu, C.; Lu, Y.; Chen, W.; Juran, B.D.; Coltescu, C.; et al. Variants at IRF5-TNPO3, 17q12-21 and MMEL1 are associated with primary biliary cirrhosis. Nat. Genet. 2010, 42, 655–657. [Google Scholar]
- Gerussi, A.; Carbone, M.; Corpechot, C.; Schramm, C.; Asselta, R.; Invernizzi, P. The genetic architecture of primary biliary cholangitis. Eur. J. Med. Genet. 2021, 64, 104292. [Google Scholar]
- Cordell, H.J.; Fryett, J.J.; Ueno, K.; Darlay, R.; Aiba, Y.; Hitomi, Y.; Kawashima, M.; Nishida, N.; Khor, S.S.; Gervais, O.; et al. An international genome-wide meta-analysis of primary biliary cholangitis: Novel risk loci and candidate drugs. J. Hepatol. 2021, 75, 572–581. [Google Scholar]
- Tam, V.; Patel, N.; Turcotte, M.; Bossé, Y.; Paré, G.; Meyre, D. Benefits and limitations of genome-wide association studies. Nat. Rev. Genet. 2019, 20, 467–484. [Google Scholar]
- Li, Y.; Liu, X.; Wang, Y.; Zhou, Y.; Hu, S.; Yang, H.; Zhong, W.; Zhao, J.; Wang, X.; Chu, H.; et al. Novel HLA-DRB1 alleles contribute risk for disease susceptibility in primary biliary cholangitis. Dig. Liver Dis. 2022, 54, 228–236. [Google Scholar]
- Wang, L.; Li, J.; Wang, C.; Tang, R.; Liang, J.; Gong, Y.; Dai, Y.; Ding, N.; Wu, J.; Dai, N.; et al. Mapping of de novo mutations in primary biliary cholangitis to a disease-specific co-expression network underlying homeostasis and metabolism. J. Genet. Genom. 2022, 49, 145–154. [Google Scholar] [CrossRef]
- Yang, C.Y.; Ma, X.; Tsuneyama, K.; Huang, S.; Takahashi, T.; Chalasani, N.P.; Bowlus, C.L.; Yang, G.X.; Leung, P.S.; Ansari, A.A.; et al. IL-12/Th1 and IL-23/Th17 biliary microenvironment in primary biliary cirrhosis: Implications for therapy. Hepatology 2014, 59, 1944–1953. [Google Scholar]
- Hirschfield, G.M.; Liu, X.; Xu, C.; Lu, Y.; Xie, G.; Lu, Y.; Gu, X.; Walker, E.J.; Jing, K.; Juran, B.D.; et al. Primary biliary cirrhosis associated with HLA, IL12A, and IL12RB2 variants. N. Engl. J. Med. 2009, 360, 2544–2555. [Google Scholar] [CrossRef]
- Hirschfield, G.M.; Gershwin, M.E.; Strauss, R.; Mayo, M.J.; Levy, C.; Zou, B.; Johanns, J.; Nnane, I.P.; Dasgupta, B.; Li, K.; et al. Ustekinumab for patients with primary biliary cholangitis who have an inadequate response to ursodeoxycholic acid: A proof-of-concept study. Hepatology 2016, 64, 189–199. [Google Scholar] [CrossRef]
- Waddington, C.H. The epigenotype. 1942. Int. J. Epidemiol. 2012, 41, 10–13. [Google Scholar]
- Berger, S.L.; Kouzarides, T.; Shiekhattar, R.; Shilatifard, A. An operational definition of epigenetics. Genes Dev. 2009, 23, 781–783. [Google Scholar]
- Peixoto, P.; Cartron, P.F.; Serandour, A.A.; Hervouet, E. From 1957 to Nowadays: A Brief History of Epigenetics. Int. J. Mol. Sci. 2020, 21, 7571. [Google Scholar] [CrossRef]
- Gerussi, A.; Paraboschi, E.M.; Cappadona, C.; Caime, C.; Binatti, E.; Cristoferi, L.; Asselta, R.; Invernizzi, P. The Role of Epigenetics in Primary Biliary Cholangitis. Int. J. Mol. Sci. 2022, 23, 4873. [Google Scholar] [CrossRef]
- Li, Y.; Tang, R.; Ma, X. Epigenetics of Primary Biliary Cholangitis. Adv. Exp. Med. Biol. 2020, 1253, 259–283. [Google Scholar]
- Lleo, A.; Leung, P.S.C.; Hirschfield, G.M.; Gershwin, E.M. The Pathogenesis of Primary Biliary Cholangitis: A Comprehensive Review. Semin. Liver Dis. 2020, 40, 34–48. [Google Scholar] [CrossRef]
- Selmi, C.; Cavaciocchi, F.; Lleo, A.; Cheroni, C.; De Francesco, R.; Lombardi, S.A.; De Santis, M.; Meda, F.; Raimondo, M.G.; Crotti, C.; et al. Genome-Wide analysis of DNA methylation, copy number variation, and gene expression in monozygotic twins discordant for primary biliary cirrhosis. Front. Immunol. 2014, 5, 128. [Google Scholar]
- Arenas, F.; Hervías, I.; Sáez, E.; Melero, S.; Prieto, J.; Parés, A.; Medina, J.F. Promoter hypermethylation of the AE2/SLC4A2 gene in PBC. JHEP Rep. 2019, 1, 145–153. [Google Scholar] [CrossRef] [PubMed]
- Aran, D.; Toperoff, G.; Rosenberg, M.; Hellman, A. Replication timing-related and gene body-specific methylation of active human genes. Hum. Mol. Genet. 2011, 20, 670–680. [Google Scholar]
- Lleo, A.; Liao, J.; Invernizzi, P.; Zhao, M.; Bernuzzi, F.; Ma, L.; Lanzi, G.; Ansari, A.A.; Coppel, R.L.; Zhang, P.; et al. Immunoglobulin M levels inversely correlate with CD40 ligand promoter methylation in patients with primary biliary cirrhosis. Hepatology 2012, 55, 153–160. [Google Scholar]
- Mitchell, M.M.; Lleo, A.; Zammataro, L.; Mayo, M.J.; Invernizzi, P.; Bach, N.; Shimoda, S.; Gordon, S.; Podda, M.; Gershwin, M.E.; et al. Epigenetic investigation of variably X chromosome inactivated genes in monozygotic female twins discordant for primary biliary cirrhosis. Epigenetics 2011, 6, 95–102. [Google Scholar]
- Jiang, T.; Zhang, H.W.; Wen, Y.P.; Yin, Y.S.; Yang, L.H.; Yang, J.; Lan, T.; Tang, C.W.; Yu, J.K.; Tai, W.L.; et al. 5-Aza-2-deoxycytidine alleviates the progression of primary biliary cholangitis by suppressing the FoxP3 methylation and promoting the Treg/Th17 balance. Int. Immunopharmacol. 2021, 96, 107820. [Google Scholar] [CrossRef]
- Padgett, K.A.; Lan, R.Y.; Leung, P.C.; Lleo, A.; Dawson, K.; Pfeiff, J.; Mao, T.K.; Coppel, R.L.; Ansari, A.A.; Gershwin, M.E. Primary biliary cirrhosis is associated with altered hepatic microRNA expression. J. Autoimmun. 2009, 32, 246–253. [Google Scholar]
- Tomiyama, T.; Yang, G.X.; Zhao, M.; Zhang, W.; Tanaka, H.; Wang, J.; Leung, P.S.; Okazaki, K.; He, X.S.; Lu, Q.; et al. The modulation of co-stimulatory molecules by circulating exosomes in primary biliary cirrhosis. Cell. Mol. Immunol. 2017, 14, 276–284. [Google Scholar]
- Banales, J.M.; Sáez, E.; Uriz, M.; Sarvide, S.; Urribarri, A.D.; Splinter, P.; Tietz Bogert, P.S.; Bujanda, L.; Prieto, J.; Medina, J.F.; et al. Up-Regulation of microRNA 506 leads to decreased Cl-/HCO3- anion exchanger 2 expression in biliary epithelium of patients with primary biliary cirrhosis. Hepatology 2012, 56, 687–697. [Google Scholar] [CrossRef]
- Rodrigues, P.M.; Perugorria, M.J.; Santos-Laso, A.; Bujanda, L.; Beuers, U.; Banales, J.M. Primary biliary cholangitis: A tale of epigenetically-induced secretory failure? J. Hepatol. 2018, 69, 1371–1383. [Google Scholar] [CrossRef]
- Ananthanarayanan, M.; Banales, J.M.; Guerra, M.T.; Spirli, C.; Munoz-Garrido, P.; Mitchell-Richards, K.; Tafur, D.; Saez, E.; Nathanson, M.H. Post-Translational regulation of the type III inositol 1,4,5-trisphosphate receptor by miRNA-506. J. Biol. Chem. 2015, 290, 184–196. [Google Scholar] [PubMed]
- Hirata, K.; Nathanson, M.H. Bile duct epithelia regulate biliary bicarbonate excretion in normal rat liver. Gastroenterology 2001, 121, 396–406. [Google Scholar] [PubMed]
- Minagawa, N.; Nagata, J.; Shibao, K.; Masyuk, A.I.; Gomes, D.A.; Rodrigues, M.A.; Lesage, G.; Akiba, Y.; Kaunitz, J.D.; Ehrlich, B.E.; et al. Cyclic AMP regulates bicarbonate secretion in cholangiocytes through release of ATP into bile. Gastroenterology 2007, 133, 1592–1602. [Google Scholar] [PubMed]
- Shibao, K.; Hirata, K.; Robert, M.E.; Nathanson, M.H. Loss of inositol 1,4,5-trisphosphate receptors from bile duct epithelia is a common event in cholestasis. Gastroenterology 2003, 125, 1175–1187. [Google Scholar]
- Sander, S.; Bullinger, L.; Klapproth, K.; Fiedler, K.; Kestler, H.A.; Barth, T.F.; Möller, P.; Stilgenbauer, S.; Pollack, J.R.; Wirth, T. MYC stimulates EZH2 expression by repression of its negative regulator miR-26a. Blood 2008, 112, 4202–4212. [Google Scholar]
- Sasaki, M.; Ikeda, H.; Sato, Y.; Nakanuma, Y. Decreased expression of Bmi1 is closely associated with cellular senescence in small bile ducts in primary biliary cirrhosis. Am. J. Pathol. 2006, 169, 831–845. [Google Scholar]
- Afonso, M.B.; Rodrigues, P.M.; Simão, A.L.; Gaspar, M.M.; Carvalho, T.; Borralho, P.; Bañales, J.M.; Castro, R.E.; Rodrigues, C.M.P. miRNA-21 ablation protects against liver injury and necroptosis in cholestasis. Cell Death Differ. 2018, 25, 857–872. [Google Scholar]
- Afonso, M.B.; Rodrigues, P.M.; Simão, A.L.; Ofengeim, D.; Carvalho, T.; Amaral, J.D.; Gaspar, M.M.; Cortez-Pinto, H.; Castro, R.E.; Yuan, J.; et al. Activation of necroptosis in human and experimental cholestasis. Cell Death Dis. 2016, 7, e2390. [Google Scholar]
- Nakagawa, R.; Muroyama, R.; Saeki, C.; Goto, K.; Kaise, Y.; Koike, K.; Nakano, M.; Matsubara, Y.; Takano, K.; Ito, S.; et al. miR-425 regulates inflammatory cytokine production in CD4+ T cells via N-Ras upregulation in primary biliary cholangitis. J. Hepatol. 2017, 66, 1223–1230. [Google Scholar]
- Li, X.; Liu, R.; Wang, Y.; Zhu, W.; Zhao, D.; Wang, X.; Yang, H.; Gurley, E.C.; Chen, W.; Hylemon, P.B.; et al. Cholangiocyte-Derived Exosomal lncRNA H19 Promotes Macrophage Activation and Hepatic Inflammation under Cholestatic Conditions. Cells 2020, 9, 190. [Google Scholar] [CrossRef]
- Liu, R.; Li, X.; Zhu, W.; Wang, Y.; Zhao, D.; Wang, X.; Gurley, E.C.; Liang, G.; Chen, W.; Lai, G.; et al. Cholangiocyte-Derived Exosomal Long Noncoding RNA H19 Promotes Hepatic Stellate Cell Activation and Cholestatic Liver Fibrosis. Hepatology 2019, 70, 1317–1335. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Jiao, Z.; Chen, M.; Shen, B.; Shuai, Z. Roles of Non-Coding RNAs in Primary Biliary Cholangitis. Front. Mol. Biosci. 2022, 9, 915993. [Google Scholar]
- Hu, Z.; Huang, Y.; Liu, Y.; Sun, Y.; Zhou, Y.; Gu, M.; Chen, Y.; Xia, R.; Chen, S.; Deng, A.; et al. β-Arrestin 1 modulates functions of autoimmune T cells from primary biliary cirrhosis patients. J. Clin. Immunol. 2011, 31, 346–355. [Google Scholar] [CrossRef] [PubMed]
- Arvaniti, P.; Zachou, K.; Lyberopoulou, A.; Gatselis, N.K.; Brooks, W.H.; Dalekos, G.N.; Renaudineau, Y. Epigenetic Modifications in Generalized Autoimmune Epithelitis: Sjögren’s Syndrome and Primary Biliary Cholangitis. Epigenomes 2019, 3, 15. [Google Scholar] [CrossRef]
- Mizushima, N. A brief history of autophagy from cell biology to physiology and disease. Nat. Cell Biol. 2018, 20, 521–527. [Google Scholar]
- Harnett, M.M.; Pineda, M.A.; Latré de Laté, P.; Eason, R.J.; Besteiro, S.; Harnett, W.; Langsley, G. From Christian de Duve to Yoshinori Ohsumi: More to autophagy than just dining at home. Biomed. J. 2017, 40, 9–22. [Google Scholar]
- Corona Velazquez, A.F.; Jackson, W.T. So Many Roads: The Multifaceted Regulation of Autophagy Induction. Mol. Cell. Biol. 2018, 38, e00303-18. [Google Scholar]
- Sasaki, M.; Nakanuma, Y. Bile Acids and Deregulated Cholangiocyte Autophagy in Primary Biliary Cholangitis. Dig. Dis. 2017, 35, 210–216. [Google Scholar]
- Sasaki, M.; Ikeda, H.; Haga, H.; Manabe, T.; Nakanuma, Y. Frequent cellular senescence in small bile ducts in primary biliary cirrhosis: A possible role in bile duct loss. J. Pathol. 2005, 205, 451–459. [Google Scholar]
- Sasaki, M.; Miyakoshi, M.; Sato, Y.; Nakanuma, Y. A possible involvement of p62/sequestosome-1 in the process of biliary epithelial autophagy and senescence in primary biliary cirrhosis. Liver Int. 2012, 32, 487–499. [Google Scholar]
- Sasaki, M.; Nakanuma, Y. Biliary epithelial apoptosis, autophagy, and senescence in primary biliary cirrhosis. Hepat. Res. Treat. 2010, 2010, 205128. [Google Scholar] [CrossRef] [PubMed]
- Young, A.R.; Narita, M.; Ferreira, M.; Kirschner, K.; Sadaie, M.; Darot, J.F.; Tavaré, S.; Arakawa, S.; Shimizu, S.; Watt, F.M.; et al. Autophagy mediates the mitotic senescence transition. Genes Dev. 2009, 23, 798–803. [Google Scholar] [CrossRef]
- Sasaki, M.; Miyakoshi, M.; Sato, Y.; Nakanuma, Y. Autophagy mediates the process of cellular senescence characterizing bile duct damages in primary biliary cirrhosis. Lab. Investig. 2010, 90, 835–843. [Google Scholar] [CrossRef]
- Sasaki, M.; Ikeda, H.; Nakanuma, Y. Activation of ATM signaling pathway is involved in oxidative stress-induced expression of mito-inhibitory p21WAF1/Cip1 in chronic non-suppurative destructive cholangitis in primary biliary cirrhosis: An immunohistochemical study. J. Autoimmun. 2008, 31, 73–78. [Google Scholar] [CrossRef]
- Sasaki, M.; Ikeda, H.; Yamaguchi, J.; Nakada, S.; Nakanuma, Y. Telomere shortening in the damaged small bile ducts in primary biliary cirrhosis reflects ongoing cellular senescence. Hepatology 2008, 48, 186–195. [Google Scholar] [CrossRef]
- Sasaki, M.; Miyakoshi, M.; Sato, Y.; Nakanuma, Y. Increased expression of mitochondrial proteins associated with autophagy in biliary epithelial lesions in primary biliary cirrhosis. Liver Int. 2013, 33, 312–320. [Google Scholar] [CrossRef]
- Sasaki, M.; Miyakoshi, M.; Sato, Y.; Nakanuma, Y. Modulation of the microenvironment by senescent biliary epithelial cells may be involved in the pathogenesis of primary biliary cirrhosis. J. Hepatol. 2010, 53, 318–325. [Google Scholar] [CrossRef]
- Sasaki, M.; Yoshimura-Miyakoshi, M.; Sato, Y.; Nakanuma, Y. A possible involvement of endoplasmic reticulum stress in biliary epithelial autophagy and senescence in primary biliary cirrhosis. J. Gastroenterol. 2015, 50, 984–995. [Google Scholar] [CrossRef]
- van de Graaf, S.; Beuers, U. Autophagy—Another piece of the puzzle towards understanding primary biliary cirrhosis? Liver Int. 2014, 34, 481–483. [Google Scholar] [CrossRef]
- Münz, C. Antigen processing via autophagy—Not only for MHC class II presentation anymore? Curr. Opin. Immunol. 2010, 22, 89–93. [Google Scholar] [CrossRef]
- Nedjic, J.; Aichinger, M.; Emmerich, J.; Mizushima, N.; Klein, L. Autophagy in thymic epithelium shapes the T-cell repertoire and is essential for tolerance. Nature 2008, 455, 396–400. [Google Scholar] [CrossRef] [PubMed]
- Paludan, C.; Schmid, D.; Landthaler, M.; Vockerodt, M.; Kube, D.; Tuschl, T.; Münz, C. Endogenous MHC class II processing of a viral nuclear antigen after autophagy. Science 2005, 307, 593–596. [Google Scholar] [CrossRef] [PubMed]
- White, E.; Lowe, S.W. Eating to exit: Autophagy-enabled senescence revealed. Genes Dev. 2009, 23, 784–787. [Google Scholar] [CrossRef]
- Xu, Y.; Shen, J.; Ran, Z. Emerging views of mitophagy in immunity and autoimmune diseases. Autophagy 2020, 16, 3–17. [Google Scholar] [CrossRef]
- Allaire, M.; Rautou, P.E.; Codogno, P.; Lotersztajn, S. Autophagy in liver diseases: Time for translation? J. Hepatol. 2019, 70, 985–998. [Google Scholar] [CrossRef]
- Hung, T.M.; Hsiao, C.C.; Lin, C.W.; Lee, P.H. Complex Cell Type-Specific Roles of Autophagy in Liver Fibrosis and Cirrhosis. Pathogens 2020, 9, 225. [Google Scholar] [CrossRef]
- Pantelis, P.; Theocharous, G.; Lagopati, N.; Veroutis, D.; Thanos, D.F.; Lampoglou, G.P.; Pippa, N.; Gatou, M.A.; Tremi, I.; Papaspyropoulos, A.; et al. The Dual Role of Oxidative-Stress-Induced Autophagy in Cellular Senescence: Comprehension and Therapeutic Approaches. Antioxidants 2023, 12, 169. [Google Scholar] [CrossRef]
- Panzitt, K.; Jungwirth, E.; Krones, E.; Lee, J.M.; Pollheimer, M.; Thallinger, G.G.; Kolb-Lenz, D.; Xiao, R.; Thorell, A.; Trauner, M.; et al. FXR-Dependent Rubicon induction impairs autophagy in models of human cholestasis. J. Hepatol. 2020, 72, 1122–1131. [Google Scholar] [CrossRef]
- Hoare, M.; Das, T.; Alexander, G. Ageing, telomeres, senescence, and liver injury. J. Hepatol. 2010, 53, 950–961. [Google Scholar] [CrossRef]
- Sasaki, M.; Nakanuma, Y. Cellular senescence in biliary pathology. Special emphasis on expression of a polycomb group protein EZH2 and a senescent marker p16INK4a in bile ductular tumors and lesions. Histol. Histopathol. 2015, 30, 267–275. [Google Scholar]
- Brain, J.G.; Robertson, H.; Thompson, E.; Humphreys, E.H.; Gardner, A.; Booth, T.A.; Jones, D.E.; Afford, S.C.; von Zglinicki, T.; Burt, A.D.; et al. Biliary epithelial senescence and plasticity in acute cellular rejection. Am. J. Transplant. 2013, 13, 1688–1702. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, M.; Ikeda, H.; Sato, Y.; Nakanuma, Y. Proinflammatory cytokine-induced cellular senescence of biliary epithelial cells is mediated via oxidative stress and activation of ATM pathway: A culture study. Free Radic. Res. 2008, 42, 625–632. [Google Scholar] [PubMed]
- Birch, J.; Gil, J. Senescence and the SASP: Many therapeutic avenues. Genes Dev. 2020, 34, 1565–1576. [Google Scholar] [PubMed]
- Sasaki, M.; Miyakoshi, M.; Sato, Y.; Nakanuma, Y. Chemokine-chemokine receptor CCL2-CCR2 and CX3CL1-CX3CR1 axis may play a role in the aggravated inflammation in primary biliary cirrhosis. Dig. Dis. Sci. 2014, 59, 358–364. [Google Scholar]
- Gorgoulis, V.; Adams, P.D.; Alimonti, A.; Bennett, D.C.; Bischof, O.; Bishop, C.; Campisi, J.; Collado, M.; Evangelou, K.; Ferbeyre, G.; et al. Cellular Senescence: Defining a Path Forward. Cell 2019, 179, 813–827. [Google Scholar] [CrossRef]
- Lunz, J.G., 3rd; Contrucci, S.; Ruppert, K.; Murase, N.; Fung, J.J.; Starzl, T.E.; Demetris, A.J. Replicative senescence of biliary epithelial cells precedes bile duct loss in chronic liver allograft rejection: Increased expression of p21(WAF1/Cip1) as a disease marker and the influence of immunosuppressive drugs. Am. J. Pathol. 2001, 158, 1379–1390. [Google Scholar] [CrossRef]
- Sasaki, M.; Nakanuma, Y. Novel approach to bile duct damage in primary biliary cirrhosis: Participation of cellular senescence and autophagy. Int. J. Hepatol. 2012, 2012, 452143. [Google Scholar]
- Barron-Millar, B.; Ogle, L.; Mells, G.; Flack, S.; Badrock, J.; Sandford, R.; Kirby, J.; Palmer, J.; Jopson, L.; Brain, J.; et al. The Serum Proteome and Ursodeoxycholic Acid Response in Primary Biliary Cholangitis. Hepatology 2021, 74, 3269–3283. [Google Scholar]
- Tabibian, J.H.; O’Hara, S.P.; Splinter, P.L.; Trussoni, C.E.; LaRusso, N.F. Cholangiocyte senescence by way of N-ras activation is a characteristic of primary sclerosing cholangitis. Hepatology 2014, 59, 2263–2275. [Google Scholar]
- Sasaki, M.; Sato, Y.; Nakanuma, Y. Interferon-Induced protein with tetratricopeptide repeats 3 may be a key factor in primary biliary cholangitis. Sci. Rep. 2021, 11, 11413. [Google Scholar]
- O’Hara, S.P.; Splinter, P.L.; Trussoni, C.E.; Pisarello, M.J.; Loarca, L.; Splinter, N.S.; Schutte, B.F.; LaRusso, N.F. ETS Proto-oncogene 1 Transcriptionally Up-regulates the Cholangiocyte Senescence-associated Protein Cyclin-dependent Kinase Inhibitor 2A. J. Biol. Chem. 2017, 292, 4833–4846. [Google Scholar] [PubMed]
- Sasaki, M.; Sato, Y.; Nakanuma, Y. An involvement of Hippo-yes-associated protein pathway in biliary epithelial senescence in primary biliary cholangitis. Clin. Res. Hepatol. Gastroenterol. 2023, 47, 102106. [Google Scholar] [PubMed]
- Sasaki, M.; Sato, Y.; Nakanuma, Y. Increased p16INK4a-expressing senescent bile ductular cells are associated with inadequate response to ursodeoxycholic acid in primary biliary cholangitis. J. Autoimmun. 2020, 107, 102377. [Google Scholar] [PubMed]
- Wijayasiri, P.; Astbury, S.; Kaye, P.; Oakley, F.; Alexander, G.J.; Kendall, T.J.; Aravinthan, A.D. Role of Hepatocyte Senescence in the Activation of Hepatic Stellate Cells and Liver Fibrosis Progression. Cells 2022, 11, 2221. [Google Scholar] [CrossRef]
- Kennedy, L.; Francis, H.; Invernizzi, P.; Venter, J.; Wu, N.; Carbone, M.; Gershwin, M.E.; Bernuzzi, F.; Franchitto, A.; Alvaro, D.; et al. Secretin/Secretin receptor signaling mediates biliary damage and liver fibrosis in early-stage primary biliary cholangitis. FASEB J. 2019, 33, 10269–10279. [Google Scholar]
- Zhou, T.; Wu, N.; Meng, F.; Venter, J.; Giang, T.K.; Francis, H.; Kyritsi, K.; Wu, C.; Franchitto, A.; Alvaro, D.; et al. Knockout of secretin receptor reduces biliary damage and liver fibrosis in Mdr2-/- mice by diminishing senescence of cholangiocytes. Lab. Investig. 2018, 98, 1449–1464. [Google Scholar]
- Sasaki, M.; Miyakoshi, M.; Sato, Y.; Nakanuma, Y. Autophagy may precede cellular senescence of bile ductular cells in ductular reaction in primary biliary cirrhosis. Dig. Dis. Sci. 2012, 57, 660–666. [Google Scholar]
- Overi, D.; Carpino, G.; Cristoferi, L.; Onori, P.; Kennedy, L.; Francis, H.; Zucchini, N.; Rigamonti, C.; Viganò, M.; Floreani, A.; et al. Role of ductular reaction and ductular-canalicular junctions in identifying severe primary biliary cholangitis. JHEP Rep. 2022, 4, 100556. [Google Scholar]
- Lenci, I.; Carnì, P.; Milana, M.; Bicaj, A.; Signorello, A.; Baiocchi, L. Sequence of events leading to primary biliary cholangitis. World J. Gastroenterol. 2023, 29, 5305–5312. [Google Scholar]
- Tsomidis, I.; Notas, G.; Voumvouraki, A.; Samonakis, D.; Koulentaki, M.; Kouroumalis, E. Hepatic Lysosomal Enzyme Activity in Primary Biliary Cholangitis. Fibrosis 2023, 1, 10005. [Google Scholar]
- Chen, Y.; Guo, G.; Guo, S.; Shimoda, S.; Shroyer, K.R.; Tang, Y.; Wu, Y. Intracellular B7-H4 suppresses bile duct epithelial cell apoptosis in human primary biliary cirrhosis. Inflammation 2011, 34, 688–697. [Google Scholar] [PubMed]
- Harada, K.; Furubo, S.; Ozaki, S.; Hiramatsu, K.; Sudo, Y.; Nakanuma, Y. Increased expression of WAF1 in intrahepatic bile ducts in primary biliary cirrhosis relates to apoptosis. J. Hepatol. 2001, 34, 500–506. [Google Scholar] [PubMed]
- Tinmouth, J.; Lee, M.; Wanless, I.R.; Tsui, F.W.; Inman, R.; Heathcote, E.J. Apoptosis of biliary epithelial cells in primary biliary cirrhosis and primary sclerosing cholangitis. Liver 2002, 22, 228–234. [Google Scholar] [PubMed]
- D’Arcy, M.S. Cell death: A review of the major forms of apoptosis, necrosis and autophagy. Cell Biol. Int. 2019, 43, 582–592. [Google Scholar]
- Shojaie, L.; Iorga, A.; Dara, L. Cell Death in Liver Diseases: A Review. Int. J. Mol. Sci. 2020, 21, 9682. [Google Scholar] [CrossRef]
- Harada, K.; Kono, N.; Tsuneyama, K.; Nakanuma, Y. Cell-Kinetic study of proliferating bile ductules in various hepatobiliary diseases. Liver 1998, 18, 277–284. [Google Scholar]
- Harada, K.; Ozaki, S.; Gershwin, M.E.; Nakanuma, Y. Enhanced apoptosis relates to bile duct loss in primary biliary cirrhosis. Hepatology 1997, 26, 1399–1405. [Google Scholar]
- Takeda, K.; Kojima, Y.; Ikejima, K.; Harada, K.; Yamashina, S.; Okumura, K.; Aoyama, T.; Frese, S.; Ikeda, H.; Haynes, N.M.; et al. Death receptor 5 mediated-apoptosis contributes to cholestatic liver disease. Proc. Natl. Acad. Sci. USA 2008, 105, 10895–10900. [Google Scholar]
- Perez, M.J.; Briz, O. Bile-Acid-Induced cell injury and protection. World J. Gastroenterol. 2009, 15, 1677–1689. [Google Scholar]
- Odin, J.A.; Huebert, R.C.; Casciola-Rosen, L.; LaRusso, N.F.; Rosen, A. Bcl-2-Dependent oxidation of pyruvate dehydrogenase-E2, a primary biliary cirrhosis autoantigen, during apoptosis. J. Clin. Investig. 2001, 108, 223–232. [Google Scholar]
- Lleo, A.; Bowlus, C.L.; Yang, G.X.; Invernizzi, P.; Podda, M.; Van de Water, J.; Ansari, A.A.; Coppel, R.L.; Worman, H.J.; Gores, G.J.; et al. Biliary apotopes and anti-mitochondrial antibodies activate innate immune responses in primary biliary cirrhosis. Hepatology 2010, 52, 987–998. [Google Scholar] [PubMed]
- Lleo, A.; Selmi, C.; Invernizzi, P.; Podda, M.; Coppel, R.L.; Mackay, I.R.; Gores, G.J.; Ansari, A.A.; Van de Water, J.; Gershwin, M.E. Apotopes and the biliary specificity of primary biliary cirrhosis. Hepatology 2009, 49, 871–879. [Google Scholar] [PubMed]
- Cubero, F.J.; Peng, J.; Liao, L.; Su, H.; Zhao, G.; Zoubek, M.E.; Macías-Rodríguez, R.; Ruiz-Margain, A.; Reißing, J.; Zimmermann, H.W.; et al. Inactivation of caspase 8 in liver parenchymal cells confers protection against murine obstructive cholestasis. J. Hepatol. 2018, 69, 1326–1334. [Google Scholar] [PubMed]
- Eguchi, A.; Koyama, Y.; Wree, A.; Johnson, C.D.; Nakamura, R.; Povero, D.; Kneiber, D.; Tameda, M.; Contreras, P.; Spada, A.; et al. Emricasan, a pan-caspase inhibitor, improves survival and portal hypertension in a murine model of common bile-duct ligation. J. Mol. Med. 2018, 96, 575–583. [Google Scholar]
- Rong, G.; Zhong, R.; Lleo, A.; Leung, P.S.; Bowlus, C.L.; Yang, G.X.; Yang, C.Y.; Coppel, R.L.; Ansari, A.A.; Cuebas, D.A.; et al. Epithelial cell specificity and apotope recognition by serum autoantibodies in primary biliary cirrhosis. Hepatology 2011, 54, 196–203. [Google Scholar]
- Azad, A.I.; Krishnan, A.; Troop, L.; Li, Y.; Katsumi, T.; Pavelko, K.; Kostallari, E.; Guicciardi, M.E.; Gores, G.J. Targeted Apoptosis of Ductular Reactive Cells Reduces Hepatic Fibrosis in a Mouse Model of Cholestasis. Hepatology 2020, 72, 1013–1028. [Google Scholar]
- Wang, Y.W.; Lin, C.I.; Chen, H.W.; Wu, J.C.; Chuang, Y.H. Apoptotic biliary epithelial cells and gut dysbiosis in the induction of murine primary biliary cholangitis. J. Transl. Autoimmun. 2022, 6, 100182. [Google Scholar]
- Schoemaker, M.H.; Conde de la Rosa, L.; Buist-Homan, M.; Vrenken, T.E.; Havinga, R.; Poelstra, K.; Haisma, H.J.; Jansen, P.L.; Moshage, H. Tauroursodeoxycholic acid protects rat hepatocytes from bile acid-induced apoptosis via activation of survival pathways. Hepatology 2004, 39, 1563–1573. [Google Scholar]
- Manousou, P.; Kolios, G.; Drygiannakis, I.; Koulentaki, M.; Pyrovolaki, K.; Voumvouraki, A.; Notas, G.; Bourikas, L.; Papadaki, H.A.; Kouroumalis, E. CXCR3 axis in patients with primary biliary cirrhosis: A possible novel mechanism of the effect of ursodeoxycholic acid. Clin. Exp. Immunol. 2013, 172, 9–15. [Google Scholar]
- Medina, J.F. Role of the anion exchanger 2 in the pathogenesis and treatment of primary biliary cirrhosis. Dig. Dis. 2011, 29, 103–112. [Google Scholar]
- Prieto, J.; Banales, J.M.; Medina, J.F. Primary biliary cholangitis: Pathogenic mechanisms. Curr. Opin. Gastroenterol. 2021, 37, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Mardones, P.; Medina, J.F.; Elferink, R.P. Activation of cyclic AMP Signaling in Ae2-deficient mouse fibroblasts. J. Biol. Chem. 2008, 283, 12146–12153. [Google Scholar] [CrossRef] [PubMed]
- Hohenester, S.; Wenniger, L.M.; Paulusma, C.C.; van Vliet, S.J.; Jefferson, D.M.; Elferink, R.P.; Beuers, U. A biliary HCO3− umbrella constitutes a protective mechanism against bile acid-induced injury in human cholangiocytes. Hepatology 2012, 55, 173–183. [Google Scholar] [CrossRef]
- Li, H.L.; Verhoeven, A.; Elferink, R.O. The role of soluble adenylyl cyclase in sensing and regulating intracellular pH. Pflug. Arch. 2024, 476, 457–465. [Google Scholar] [CrossRef] [PubMed]
- Arenas, F.; Hervias, I.; Uriz, M.; Joplin, R.; Prieto, J.; Medina, J.F. Combination of ursodeoxycholic acid and glucocorticoids upregulates the AE2 alternate promoter in human liver cells. J. Clin. Investig. 2008, 118, 695–709. [Google Scholar] [CrossRef]
- Banales, J.M.; Arenas, F.; Rodríguez-Ortigosa, C.M.; Sáez, E.; Uriarte, I.; Doctor, R.B.; Prieto, J.; Medina, J.F. Bicarbonate-rich choleresis induced by secretin in normal rat is taurocholate-dependent and involves AE2 anion exchanger. Hepatology 2006, 43, 266–275. [Google Scholar] [CrossRef]
- Uriarte, I.; Banales, J.M.; Sáez, E.; Arenas, F.; Oude Elferink, R.P.; Prieto, J.; Medina, J.F. Bicarbonate secretion of mouse cholangiocytes involves Na(+)-HCO(3)(−) cotransport in addition to Na(+)-independent Cl(−)/HCO(3)(−) exchange. Hepatology 2010, 51, 891–902. [Google Scholar] [CrossRef]
- Chang, J.C.; Go, S.; de Waart, D.R.; Munoz-Garrido, P.; Beuers, U.; Paulusma, C.C.; Oude Elferink, R. Soluble Adenylyl Cyclase Regulates Bile Salt-Induced Apoptosis in Human Cholangiocytes. Hepatology 2016, 64, 522–534. [Google Scholar] [CrossRef]
- Chen, Y.; Cann, M.J.; Litvin, T.N.; Iourgenko, V.; Sinclair, M.L.; Levin, L.R.; Buck, J. Soluble adenylyl cyclase as an evolutionarily conserved bicarbonate sensor. Science 2000, 289, 625–628. [Google Scholar] [CrossRef]
- Beuers, U.; Hohenester, S.; de Buy Wenniger, L.J.; Kremer, A.E.; Jansen, P.L.; Elferink, R.P. The biliary HCO(3)(−) umbrella: A unifying hypothesis on pathogenetic and therapeutic aspects of fibrosing cholangiopathies. Hepatology 2010, 52, 1489–1496. [Google Scholar] [CrossRef]
- Chang, J.C.; Beuers, U.; Oude Elferink, R.P. The Emerging Role of Soluble Adenylyl Cyclase in Primary Biliary Cholangitis. Dig. Dis. 2017, 35, 217–223. [Google Scholar] [PubMed]
- Kennedy, L.; Carpino, G.; Owen, T.; Ceci, L.; Kundu, D.; Meadows, V.; Kyritsi, K.; Franchitto, A.; Onori, P.; Isidan, A.; et al. Secretin alleviates biliary and liver injury during late-stage primary biliary cholangitis via restoration of secretory processes. J. Hepatol. 2023, 78, 99–113. [Google Scholar] [CrossRef] [PubMed]
- Medina, J.F.; Martínez-Ansó, E.; Vazquez, J.J.; Prieto, J. Decreased anion exchanger 2 immunoreactivity in the liver of patients with primary biliary cirrhosis. Hepatology 1997, 25, 12–17. [Google Scholar]
- Prieto, J.; Qian, C.; García, N.; Díez, J.; Medina, J.F. Abnormal expression of anion exchanger genes in primary biliary cirrhosis. Gastroenterology 1993, 105, 572–578. [Google Scholar]
- Melero, S.; Spirlì, C.; Zsembery, A.; Medina, J.F.; Joplin, R.E.; Duner, E.; Zuin, M.; Neuberger, J.M.; Prieto, J.; Strazzabosco, M. Defective regulation of cholangiocyte Cl−/HCO3(−) and Na+/H+ exchanger activities in primary biliary cirrhosis. Hepatology 2002, 35, 1513–1521. [Google Scholar]
- Hisamoto, S.; Shimoda, S.; Harada, K.; Iwasaka, S.; Onohara, S.; Chong, Y.; Nakamura, M.; Bekki, Y.; Yoshizumi, T.; Ikegami, T.; et al. Hydrophobic bile acids suppress expression of AE2 in biliary epithelial cells and induce bile duct inflammation in primary biliary cholangitis. J. Autoimmun. 2016, 75, 150–160. [Google Scholar]
- Prieto, J.; García, N.; Martí-Climent, J.M.; Peñuelas, I.; Richter, J.A.; Medina, J.F. Assessment of biliary bicarbonate secretion in humans by positron emission tomography. Gastroenterology 1999, 117, 167–172. [Google Scholar]
- She, X.; Rohl, C.A.; Castle, J.C.; Kulkarni, A.V.; Johnson, J.M.; Chen, R. Definition, conservation and epigenetics of housekeeping and tissue-enriched genes. BMC Genom. 2009, 10, 269. [Google Scholar]
- Salas, J.T.; Banales, J.M.; Sarvide, S.; Recalde, S.; Ferrer, A.; Uriarte, I.; Oude Elferink, R.P.; Prieto, J.; Medina, J.F. Ae2a,b-deficient mice develop antimitochondrial antibodies and other features resembling primary biliary cirrhosis. Gastroenterology 2008, 134, 1482–1493. [Google Scholar] [CrossRef]
- Concepcion, A.R.; Salas, J.T.; Sarvide, S.; Sáez, E.; Ferrer, A.; López, M.; Portu, A.; Banales, J.M.; Hervás-Stubbs, S.; Oude Elferink, R.P.; et al. Anion exchanger 2 is critical for CD8(+) T cells to maintain pHi homeostasis and modulate immune responses. Eur. J. Immunol. 2014, 44, 1341–1351. [Google Scholar]
- Concepcion, A.R.; Salas, J.T.; Sáez, E.; Sarvide, S.; Ferrer, A.; Portu, A.; Uriarte, I.; Hervás-Stubbs, S.; Oude Elferink, R.P.; Prieto, J.; et al. CD8+ T cells undergo activation and programmed death-1 repression in the liver of aged Ae2a,b-/- mice favoring autoimmune cholangitis. Oncotarget 2015, 6, 28588–28606. [Google Scholar] [CrossRef] [PubMed]
- van Niekerk, J.; Kersten, R.; Beuers, U. Role of Bile Acids and the Biliary HCO3− Umbrella in the Pathogenesis of Primary Biliary Cholangitis. Clin. Liver Dis. 2018, 22, 457–479. [Google Scholar] [PubMed]
- Floreani, A.; Gabbia, D.; De Martin, S. Primary biliary cholangitis: Primary autoimmune disease or primary secretory defect. Expert Rev. Gastroenterol. Hepatol. 2023, 17, 863–870. [Google Scholar] [PubMed]
- Ma, D.; Ma, J.; Zhao, C.; Tai, W. Reasons why women are more likely to develop primary biliary cholangitis. Heliyon 2024, 10, e25634. [Google Scholar]
- Erice, O.; Munoz-Garrido, P.; Vaquero, J.; Perugorria, M.J.; Fernandez-Barrena, M.G.; Saez, E.; Santos-Laso, A.; Arbelaiz, A.; Jimenez-Agüero, R.; Fernandez-Irigoyen, J.; et al. MicroRNA-506 promotes primary biliary cholangitis-like features in cholangiocytes and immune activation. Hepatology 2018, 67, 1420–1440. [Google Scholar]
- Chang, J.C.; Go, S.; Verhoeven, A.J.; Beuers, U.; Oude Elferink, R.P.J. Role of the bicarbonate-responsive soluble adenylyl cyclase in cholangiocyte apoptosis in primary biliary cholangitis; a new hypothesis. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 1232–1239. [Google Scholar]
- Mayo, M.J. Mechanisms and molecules: What are the treatment targets for primary biliary cholangitis? Hepatology 2022, 76, 518–531. [Google Scholar]
- Gulamhusein, A.F.; Hirschfield, G.M. Primary biliary cholangitis: Pathogenesis and therapeutic opportunities. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 93–110. [Google Scholar]
- Guo, Z.; He, K.; Pang, K.; Yang, D.; Lyu, C.; Xu, H.; Wu, D. Exploring Advanced Therapies for Primary Biliary Cholangitis: Insights from the Gut Microbiota-Bile Acid-Immunity Network. Int. J. Mol. Sci. 2024, 25, 4321. [Google Scholar] [CrossRef]
- Shi, Q.; Yuan, X.; Zeng, Y.; Wang, J.; Zhang, Y.; Xue, C.; Li, L. Crosstalk between Gut Microbiota and Bile Acids in Cholestatic Liver Disease. Nutrients 2023, 15, 2411. [Google Scholar] [CrossRef]
- Fiorucci, S.; Biagioli, M.; Zampella, A.; Distrutti, E. Bile Acids Activated Receptors Regulate Innate Immunity. Front. Immunol. 2018, 9, 1853. [Google Scholar]
- Gong, Z.; Zhou, J.; Zhao, S.; Tian, C.; Wang, P.; Xu, C.; Chen, Y.; Cai, W.; Wu, J. Chenodeoxycholic acid activates NLRP3 inflammasome and contributes to cholestatic liver fibrosis. Oncotarget 2016, 7, 83951–83963. [Google Scholar] [PubMed]
- Jansen, P.L.; Ghallab, A.; Vartak, N.; Reif, R.; Schaap, F.G.; Hampe, J.; Hengstler, J.G. The ascending pathophysiology of cholestatic liver disease. Hepatology 2017, 65, 722–738. [Google Scholar] [PubMed]
- Li, Y.; Tang, R.; Leung, P.S.C.; Gershwin, M.E.; Ma, X. Bile acids and intestinal microbiota in autoimmune cholestatic liver diseases. Autoimmun. Rev. 2017, 16, 885–896. [Google Scholar]
- Malhi, H.; Kaufman, R.J. Endoplasmic reticulum stress in liver disease. J. Hepatol. 2011, 54, 795–809. [Google Scholar]
- Sokol, R.J.; Dahl, R.; Devereaux, M.W.; Yerushalmi, B.; Kobak, G.E.; Gumpricht, E. Human hepatic mitochondria generate reactive oxygen species and undergo the permeability transition in response to hydrophobic bile acids. J. Pediatr. Gastroenterol. Nutr. 2005, 41, 235–243. [Google Scholar]
- Ljubuncic, P.; Abu-Salach, O.; Bomzon, A. Ursodeoxycholic acid and superoxide anion. World J. Gastroenterol. 2005, 11, 4875–4878. [Google Scholar]
- Ljubuncic, P.; Tanne, Z.; Bomzon, A. Ursodeoxycholic acid suppresses extent of lipid peroxidation in diseased liver in experimental cholestatic liver disease. Dig. Dis. Sci. 2000, 45, 1921–1928. [Google Scholar]
- Serviddio, G.; Pereda, J.; Pallardó, F.V.; Carretero, J.; Borras, C.; Cutrin, J.; Vendemiale, G.; Poli, G.; Viña, J.; Sastre, J. Ursodeoxycholic acid protects against secondary biliary cirrhosis in rats by preventing mitochondrial oxidative stress. Hepatology 2004, 39, 711–720. [Google Scholar]
- Muñoz, S.J.; Heubi, J.E.; Balistreri, W.F.; Maddrey, W.C. Vitamin E deficiency in primary biliary cirrhosis: Gastrointestinal malabsorption, frequency and relationship to other lipid-soluble vitamins. Hepatology 1989, 9, 525–531. [Google Scholar]
- Aboutwerat, A.; Pemberton, P.W.; Smith, A.; Burrows, P.C.; McMahon, R.F.; Jain, S.K.; Warnes, T.W. Oxidant stress is a significant feature of primary biliary cirrhosis. Biochim. Biophys. Acta 2003, 1637, 142–150. [Google Scholar] [CrossRef]
- Notas, G.; Miliaraki, N.; Kampa, M.; Dimoulios, F.; Matrella, E.; Hatzidakis, A.; Castanas, E.; Kouroumalis, E. Patients with primary biliary cirrhosis have increased serum total antioxidant capacity measured with the crocin bleaching assay. World J. Gastroenterol. 2005, 11, 4194–4198. [Google Scholar] [CrossRef] [PubMed]
- Celli, A.; Que, F.G.; Gores, G.J.; LaRusso, N.F. Glutathione depletion is associated with decreased Bcl-2 expression and increased apoptosis in cholangiocytes. Am. J. Physiol. 1998, 275, G749–G757. [Google Scholar] [CrossRef]
- Bleier, J.I.; Katz, S.C.; Chaudhry, U.I.; Pillarisetty, V.G.; Kingham, T.P., 3rd; Shah, A.B.; Raab, J.R.; DeMatteo, R.P. Biliary obstruction selectively expands and activates liver myeloid dendritic cells. J. Immunol. 2006, 176, 7189–7195. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, K.M.; Allen, K.M.; Rockwell, C.E.; Towery, K.; Luyendyk, J.P.; Copple, B.L. IL-17A synergistically enhances bile acid-induced inflammation during obstructive cholestasis. Am. J. Pathol. 2013, 183, 1498–1507. [Google Scholar] [CrossRef] [PubMed]
- Hang, S.; Paik, D.; Yao, L.; Kim, E.; Trinath, J.; Lu, J.; Ha, S.; Nelson, B.N.; Kelly, S.P.; Wu, L.; et al. Bile acid metabolites control TH17 and Treg cell differentiation. Nature 2019, 576, 143–148. [Google Scholar] [CrossRef]
- Paik, D.; Yao, L.; Zhang, Y.; Bae, S.; D’Agostino, G.D.; Zhang, M.; Kim, E.; Franzosa, E.A.; Avila-Pacheco, J.; Bisanz, J.E.; et al. Human gut bacteria produce ΤH17-modulating bile acid metabolites. Nature 2022, 603, 907–912. [Google Scholar] [CrossRef]
- Chen, W.; Wei, Y.; Xiong, A.; Li, Y.; Guan, H.; Wang, Q.; Miao, Q.; Bian, Z.; Xiao, X.; Lian, M.; et al. Comprehensive Analysis of Serum and Fecal Bile Acid Profiles and Interaction with Gut Microbiota in Primary Biliary Cholangitis. Clin. Rev. Allergy Immunol. 2020, 58, 25–38. [Google Scholar] [CrossRef]
- Luo, X.; You, X. Genetic predisposition of the gastrointestinal microbiome and primary biliary cholangitis: A bi-directional, two-sample Mendelian randomization analysis. Front. Endocrinol. 2023, 14, 1225742. [Google Scholar] [CrossRef]
- Cui, Y.; Guo, Y.; Kong, Y.; Zhang, G. Association between gut microbiota and autoimmune cholestatic liver disease, a Mendelian randomization study. Front. Microbiol. 2024, 15, 1348027. [Google Scholar] [CrossRef]
- Pabst, O.; Hornef, M.W.; Schaap, F.G.; Cerovic, V.; Clavel, T.; Bruns, T. Gut-liver axis: Barriers and functional circuits. Nat. Rev. Gastroenterol. Hepatol. 2023, 20, 447–461. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.D.; Zhao, Z.B.; Ma, W.T.; Liu, Q.Z.; Gao, C.Y.; Li, L.; Wang, J.; Tsuneyama, K.; Liu, B.; Zhang, W.; et al. Gut microbiota translocation promotes autoimmune cholangitis. J. Autoimmun. 2018, 95, 47–57. [Google Scholar] [CrossRef] [PubMed]
- Feld, J.J.; Meddings, J.; Heathcote, E.J. Abnormal intestinal permeability in primary biliary cirrhosis. Dig. Dis. Sci. 2006, 51, 1607–1613. [Google Scholar] [CrossRef]
- Haruta, I.; Hashimoto, E.; Kato, Y.; Kikuchi, K.; Kato, H.; Yagi, J.; Uchiyama, T.; Kobayash, M.; Shiratori, K. Lipoteichoic acid may affect the pathogenesis of bile duct damage in primary biliary cirrhosis. Autoimmunity 2006, 39, 129–135. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Zhao, S.; Zhou, G.; Liang, L.; Guo, X.; Mao, P.; Zhou, X.; Wang, H.; Nan, Y.; Xu, D.; et al. Altered biliary epithelial cell and monocyte responses to lipopolysaccharide as a TLR ligand in patients with primary biliary cirrhosis. Scand. J. Gastroenterol. 2011, 46, 485–494. [Google Scholar] [CrossRef]
- Zhang, L.; Yang, L.; Chu, H. Targeting Gut Microbiota for the Treatment of Primary Biliary Cholangitis: From Bench to Bedside. J. Clin. Transl. Hepatol. 2023, 11, 958–966. [Google Scholar] [CrossRef]
- Terziroli Beretta-Piccoli, B.; Mieli-Vergani, G.; Vergani, D.; Vierling, J.M.; Adams, D.; Alpini, G.; Banales, J.M.; Beuers, U.; Björnsson, E.; Bowlus, C.; et al. The challenges of primary biliary cholangitis: What is new and what needs to be done. J. Autoimmun. 2019, 105, 102328. [Google Scholar] [CrossRef]
- Wagner, M.; Fickert, P. Drug Therapies for Chronic Cholestatic Liver Diseases. Annu. Rev. Pharmacol. Toxicol. 2020, 60, 503–527. [Google Scholar] [CrossRef]
- Chen, M.L.; Takeda, K.; Sundrud, M.S. Emerging roles of bile acids in mucosal immunity and inflammation. Mucosal Immunol. 2019, 12, 851–861. [Google Scholar] [CrossRef]
- Joyce, S.A.; Gahan, C.G. Bile Acid Modifications at the Microbe-Host Interface: Potential for Nutraceutical and Pharmaceutical Interventions in Host Health. Annu. Rev. Food Sci. Technol. 2016, 7, 313–333. [Google Scholar] [CrossRef]
- Tang, R.; Wei, Y.; Li, Y.; Chen, W.; Chen, H.; Wang, Q.; Yang, F.; Miao, Q.; Xiao, X.; Zhang, H.; et al. Gut microbial profile is altered in primary biliary cholangitis and partially restored after UDCA therapy. Gut 2018, 67, 534–541. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, M.; Moriya, K.; Nakayama, J.; Inoue, T.; Momoda, R.; Kawaratani, H.; Namisaki, T.; Sato, S.; Douhara, A.; Kaji, K.; et al. Gut dysbiosis associated with clinical prognosis of patients with primary biliary cholangitis. Hepatol. Res. 2020, 50, 840–852. [Google Scholar] [PubMed]
- Kitahata, S.; Yamamoto, Y.; Yoshida, O.; Tokumoto, Y.; Kawamura, T.; Furukawa, S.; Kumagi, T.; Hirooka, M.; Takeshita, E.; Abe, M.; et al. Ileal mucosa-associated microbiota overgrowth associated with pathogenesis of primary biliary cholangitis. Sci. Rep. 2021, 11, 19705. [Google Scholar] [CrossRef]
- Jiang, H.; Yu, Y.; Hu, X.; Du, B.; Shao, Y.; Wang, F.; Chen, L.; Yan, R.; Li, L.; Lv, L. The fecal microbiota of patients with primary biliary cholangitis (PBC) causes PBC-like liver lesions in mice and exacerbates liver damage in a mouse model of PBC. Gut Microbes 2024, 16, 2383353. [Google Scholar] [PubMed]
- Wu, L.; Zhou, J.; Zhou, A.; Lei, Y.; Tang, L.; Hu, S.; Wang, S.; Xiao, X.; Chen, Q.; Tu, D.; et al. Lactobacillus acidophilus ameliorates cholestatic liver injury through inhibiting bile acid synthesis and promoting bile acid excretion. Gut Microbes 2024, 16, 2390176. [Google Scholar]
- Arpaia, N.; Campbell, C.; Fan, X.; Dikiy, S.; van der Veeken, J.; deRoos, P.; Liu, H.; Cross, J.R.; Pfeffer, K.; Coffer, P.J.; et al. Metabolites produced by commensal bacteria promote peripheral regulatory T-cell generation. Nature 2013, 504, 451–455. [Google Scholar]
- Chen, J.; Vitetta, L. The Role of Butyrate in Attenuating Pathobiont-Induced Hyperinflammation. Immune Netw. 2020, 20, e15. [Google Scholar]
- Schulthess, J.; Pandey, S.; Capitani, M.; Rue-Albrecht, K.C.; Arnold, I.; Franchini, F.; Chomka, A.; Ilott, N.E.; Johnston, D.G.W.; Pires, E.; et al. The Short Chain Fatty Acid Butyrate Imprints an Antimicrobial Program in Macrophages. Immunity 2019, 50, 432–445.e7. [Google Scholar]
- Wang, H.B.; Wang, P.Y.; Wang, X.; Wan, Y.L.; Liu, Y.C. Butyrate enhances intestinal epithelial barrier function via up-regulation of tight junction protein Claudin-1 transcription. Dig. Dis. Sci. 2012, 57, 3126–3135. [Google Scholar] [CrossRef]
- Li, B.; Zhang, J.; Chen, Y.; Wang, Q.; Yan, L.; Wang, R.; Wei, Y.; You, Z.; Li, Y.; Miao, Q.; et al. Alterations in microbiota and their metabolites are associated with beneficial effects of bile acid sequestrant on icteric primary biliary Cholangitis. Gut Microbes 2021, 13, 1946366. [Google Scholar] [CrossRef]
- Martinez-Gili, L.; Pechlivanis, A.; McDonald, J.A.K.; Begum, S.; Badrock, J.; Dyson, J.K.; Jones, R.; Hirschfield, G.; Ryder, S.D.; Sandford, R.; et al. Bacterial and metabolic phenotypes associated with inadequate response to ursodeoxycholic acid treatment in primary biliary cholangitis. Gut Microbes 2023, 15, 2208501. [Google Scholar] [CrossRef] [PubMed]
- Lammert, C.; Shin, A.; Xu, H.; Hemmerich, C.; O’Connell, T.M.; Chalasani, N. Short-Chain fatty acid and fecal microbiota profiles are linked to fibrosis in primary biliary cholangitis. FEMS Microbiol. Lett. 2021, 368, fnab038. [Google Scholar] [CrossRef] [PubMed]
- Kouroumalis, E. Environmental agents involved in the cause of primary biliary cirrhosis. Dis. Markers 2010, 29, 329–336. [Google Scholar] [CrossRef]
- Rieger, R.; Leung, P.S.; Jeddeloh, M.R.; Kurth, M.J.; Nantz, M.H.; Lam, K.S.; Barsky, D.; Ansari, A.A.; Coppel, R.L.; Mackay, I.R.; et al. Identification of 2-nonynoic acid, a cosmetic component, as a potential trigger of primary biliary cirrhosis. J. Autoimmun. 2006, 27, 7–16. [Google Scholar] [CrossRef]
- Tanaka, A.; Leung, P.S.; Gershwin, M.E. Environmental basis of primary biliary cholangitis. Exp. Biol. Med. 2018, 243, 184–189. [Google Scholar] [CrossRef]
- Mattner, J.; Savage, P.B.; Leung, P.; Oertelt, S.S.; Wang, V.; Trivedi, O.; Scanlon, S.T.; Pendem, K.; Teyton, L.; Hart, J.; et al. Liver autoimmunity triggered by microbial activation of natural killer T cells. Cell Host Microbe 2008, 3, 304–315. [Google Scholar]
- Wang, J.J.; Yang, G.X.; Zhang, W.C.; Lu, L.; Tsuneyama, K.; Kronenberg, M.; Véla, J.L.; Lopez-Hoyos, M.; He, X.S.; Ridgway, W.M.; et al. Escherichia coli infection induces autoimmune cholangitis and anti-mitochondrial antibodies in non-obese diabetic (NOD).B6 (Idd10/Idd18) mice. Clin. Exp. Immunol. 2014, 175, 192–201. [Google Scholar] [CrossRef]
- Katsumi, T.; Sato, H.; Murakami, R.; Hanatani, T.; Uchiyama, F.; Suzuki, F.; Maki, K.; Hoshikawa, K.; Haga, H.; Saito, T.; et al. Identification of microbial antigens in liver tissues involved in the pathogenesis of primary biliary cholangitis using 16S rRNA metagenome analysis. PLoS ONE 2024, 19, e0308912. [Google Scholar]
- Kim, K.A.; Kim, Y.S.; Park, S.H.; Chung, W.J.; Choi, D.H.; Jang, E.S.; Jeong, S.H. Environmental risk factors and comorbidities of primary biliary cholangitis in Korea: A case-control study. Korean J. Intern. Med. 2021, 36, 313–321. [Google Scholar]
- Matsumoto, K.; Ohfuji, S.; Abe, M.; Komori, A.; Takahashi, A.; Fujii, H.; Kawata, K.; Noritake, H.; Tadokoro, T.; Honda, A.; et al. Environmental factors, medical and family history, and comorbidities associated with primary biliary cholangitis in Japan: A multicenter case-control study. J. Gastroenterol. 2022, 57, 19–29. [Google Scholar] [CrossRef]
- Mantaka, A.; Koulentaki, M.; Samonakis, D.; Sifaki-Pistolla, D.; Voumvouraki, A.; Tzardi, M.; Kouroumalis, E. Association of smoking with liver fibrosis and mortality in primary biliary cholangitis. Eur. J. Gastroenterol. Hepatol. 2018, 30, 1461–1469. [Google Scholar] [PubMed]
- Wijarnpreecha, K.; Werlang, M.; Panjawatanan, P.; Pungpapong, S.; Lukens, F.J.; Harnois, D.M.; Ungprasert, P. Smoking & risk of advanced liver fibrosis among patients with primary biliary cholangitis: A systematic review & meta-analysis. Indian J. Med. Res. 2021, 154, 806–812. [Google Scholar]
- Dyson, J.K.; Blain, A.; Foster Shirley, M.D.; Hudson, M.; Rushton, S.; Jeffreys Jones, D.E. Geo-Epidemiology and environmental co-variate mapping of primary biliary cholangitis and primary sclerosing cholangitis. JHEP Rep. 2020, 3, 100202. [Google Scholar]
- Yang, Y.; Choi, J.; Chen, Y.; Invernizzi, P.; Yang, G.; Zhang, W.; Shao, T.H.; Jordan, F.; Nemeria, N.S.; Coppel, R.L.; et al. E. coli and the etiology of human PBC: Antimitochondrial antibodies and spreading determinants. Hepatology 2022, 75, 266–279. [Google Scholar] [PubMed]
- Rojas, M.; Restrepo-Jiménez, P.; Monsalve, D.M.; Pacheco, Y.; Acosta-Ampudia, Y.; Ramírez-Santana, C.; Leung, P.S.C.; Ansari, A.A.; Gershwin, M.E.; Anaya, J.M. Molecular mimicry and autoimmunity. J. Autoimmun. 2018, 95, 100–123. [Google Scholar] [PubMed]
- Leung, P.S.; Wang, J.; Naiyanetr, P.; Kenny, T.P.; Lam, K.S.; Kurth, M.J.; Gershwin, M.E. Environment and primary biliary cirrhosis: Electrophilic drugs and the induction of AMA. J. Autoimmun. 2013, 41, 79–86. [Google Scholar] [CrossRef]
- Bae, H.R.; Hodge, D.L.; Yang, G.X.; Leung, P.S.C.; Chodisetti, S.B.; Valencia, J.C.; Sanford, M.; Fenimore, J.M.; Rahman, Z.S.M.; Tsuneyama, K.; et al. The interplay of type I and type II interferons in murine autoimmune cholangitis as a basis for sex-biased autoimmunity. Hepatology 2018, 67, 1408–1419. [Google Scholar]
- Bae, H.R.; Leung, P.S.; Tsuneyama, K.; Valencia, J.C.; Hodge, D.L.; Kim, S.; Back, T.; Karwan, M.; Merchant, A.S.; Baba, N.; et al. Chronic expression of interferon-gamma leads to murine autoimmune cholangitis with a female predominance. Hepatology 2016, 64, 1189–1201. [Google Scholar]
- Mason, A.L. The evidence supports a viral aetiology for primary biliary cirrhosis. J. Hepatol. 2011, 54, 1312–1314. [Google Scholar] [CrossRef]
- Sutton, I.; Neuberger, J. Primary biliary cirrhosis: Seeking the silent partner of autoimmunity. Gut 2002, 50, 743–746. [Google Scholar]
- Xu, L.; Shen, Z.; Guo, L.; Fodera, B.; Keogh, A.; Joplin, R.; O’Donnell, B.; Aitken, J.; Carman, W.; Neuberger, J.; et al. Does a betaretrovirus infection trigger primary biliary cirrhosis? Proc. Natl. Acad. Sci. USA 2003, 100, 8454–8459. [Google Scholar] [PubMed]
- Mason, A.L. Is PBC a viral infectious disease? Best Pract. Res. Clin. Gastroenterol. 2018, 34, 27–39. [Google Scholar] [PubMed]
- Turvey, S.L.; Saxinger, L.; Mason, A.L. Apples to Apples? A Comparison of Real-World Tolerability of Antiretrovirals in Patients with Human Immunodeficiency Virus Infection and Patients with Primary Biliary Cholangitis. Viruses 2022, 14, 516. [Google Scholar] [CrossRef]
- Goubran, M.; Wang, W.; Indik, S.; Faschinger, A.; Wasilenko, S.T.; Bintner, J.; Carpenter, E.J.; Zhang, G.; Nuin, P.; Macintyre, G.; et al. Isolation of a Human Betaretrovirus from Patients with Primary Biliary Cholangitis. Viruses 2022, 14, 886. [Google Scholar] [CrossRef]
- Wang, W.; Indik, S.; Wasilenko, S.T.; Faschinger, A.; Carpenter, E.J.; Tian, Z.; Zhang, Y.; Wong, G.K.; Mason, A.L. Frequent proviral integration of the human betaretrovirus in biliary epithelium of patients with autoimmune and idiopathic liver disease. Aliment. Pharmacol. Ther. 2015, 41, 393–405. [Google Scholar] [CrossRef]
- Montano-Loza, A.J.; Hansen, B.E.; Corpechot, C.; Roccarina, D.; Thorburn, D.; Trivedi, P.; Hirschfield, G.; McDowell, P.; Poupon, R.; Dumortier, J.; et al. Factors Associated With Recurrence of Primary Biliary Cholangitis After Liver Transplantation and Effects on Graft and Patient Survival. Gastroenterology 2019, 156, 96–107.e1. [Google Scholar]
- Lytvyak, E.; Niazi, M.; Pai, R.; He, D.; Zhang, G.; Hübscher, S.G.; Mason, A.L. Combination antiretroviral therapy improves recurrent primary biliary cholangitis following liver transplantation. Liver Int. 2021, 41, 1879–1883. [Google Scholar]
- Krenkel, O.; Tacke, F. Liver macrophages in tissue homeostasis and disease. Nat. Rev. Immunol. 2017, 17, 306–321. [Google Scholar] [CrossRef]
- Zimmermann, H.W.; Seidler, S.; Nattermann, J.; Gassler, N.; Hellerbrand, C.; Zernecke, A.; Tischendorf, J.J.; Luedde, T.; Weiskirchen, R.; Trautwein, C.; et al. Functional contribution of elevated circulating and hepatic non-classical CD14CD16 monocytes to inflammation and human liver fibrosis. PLoS ONE 2010, 5, e11049. [Google Scholar]
- Ma, W.T.; Gao, F.; Gu, K.; Chen, D.K. The Role of Monocytes and Macrophages in Autoimmune Diseases: A Comprehensive Review. Front. Immunol. 2019, 10, 1140. [Google Scholar]
- Peng, A.; Ke, P.; Zhao, R.; Lu, X.; Zhang, C.; Huang, X.; Tian, G.; Huang, J.; Wang, J.; Invernizzi, P.; et al. Elevated circulating CD14lowCD16+ monocyte subset in primary biliary cirrhosis correlates with liver injury and promotes Th1 polarization. Clin. Exp. Med. 2016, 16, 511–521. [Google Scholar] [CrossRef] [PubMed]
- Shimoda, S.; Harada, K.; Niiro, H.; Taketomi, A.; Maehara, Y.; Tsuneyama, K.; Kikuchi, K.; Nakanuma, Y.; Mackay, I.R.; Gershwin, M.E.; et al. CX3CL1 (fractalkine): A signpost for biliary inflammation in primary biliary cirrhosis. Hepatology 2010, 51, 567–575. [Google Scholar] [CrossRef] [PubMed]
- Mao, T.K.; Lian, Z.X.; Selmi, C.; Ichiki, Y.; Ashwood, P.; Ansari, A.A.; Coppel, R.L.; Shimoda, S.; Ishibashi, H.; Gershwin, M.E. Altered monocyte responses to defined TLR ligands in patients with primary biliary cirrhosis. Hepatology 2005, 42, 802–808. [Google Scholar] [CrossRef] [PubMed]
- Honda, Y.; Yamagiwa, S.; Matsuda, Y.; Takamura, M.; Ichida, T.; Aoyagi, Y. Altered expression of TLR homolog RP105 on monocytes hypersensitive to LPS in patients with primary biliary cirrhosis. J. Hepatol. 2007, 47, 404–411. [Google Scholar] [CrossRef]
- Tsuneyama, K.; Harada, K.; Yasoshima, M.; Hiramatsu, K.; Mackay, C.R.; Mackay, I.R.; Gershwin, M.E.; Nakanuma, Y. Monocyte chemotactic protein-1, -2, and -3 are distinctively expressed in portal tracts and granulomata in primary biliary cirrhosis: Implications for pathogenesis. J. Pathol. 2001, 193, 102–109. [Google Scholar] [CrossRef]
- Reuveni, D.; Gore, Y.; Leung, P.S.C.; Lichter, Y.; Moshkovits, I.; Kaminitz, A.; Brazowski, E.; Lefebvre, E.; Vig, P.; Varol, C.; et al. The Critical Role of Chemokine (C-C Motif) Receptor 2-Positive Monocytes in Autoimmune Cholangitis. Front. Immunol. 2018, 9, 1852. [Google Scholar] [CrossRef]
- Ronca, V.; Chen, Q.B.; Lygoura, V.; Ben-Mustapha, I.; Shums, Z.; Trifa, M.; Carbone, M.; Mancuso, C.; Milani, C.; Bernuzzi, F.; et al. Autoantibodies in patients with interleukin 12 receptor beta 1 deficiency. J. Dig. Dis. 2019, 20, 363–370. [Google Scholar] [CrossRef]
- Sasatomi, K.; Noguchi, K.; Sakisaka, S.; Sata, M.; Tanikawa, K. Abnormal accumulation of endotoxin in biliary epithelial cells in primary biliary cirrhosis and primary sclerosing cholangitis. J. Hepatol. 1998, 29, 409–416. [Google Scholar] [CrossRef]
- Alabraba, E.B.; Lai, V.; Boon, L.; Wigmore, S.J.; Adams, D.H.; Afford, S.C. Coculture of human liver macrophages and cholangiocytes leads to CD40-dependent apoptosis and cytokine secretion. Hepatology 2008, 47, 552–562. [Google Scholar] [CrossRef]
- Mulcahy, V.; Liaskou, E.; Martin, J.E.; Kotagiri, P.; Badrock, J.; Jones, R.L.; Rushbrook, S.M.; Ryder, S.D.; Thorburn, D.; Taylor-Robinson, S.D.; et al. Regulation of immune responses in primary biliary cholangitis: A transcriptomic analysis of peripheral immune cells. Hepatol. Commun. 2023, 7, e0110. [Google Scholar] [CrossRef]
- Sato, K.; Hall, C.; Glaser, S.; Francis, H.; Meng, F.; Alpini, G. Pathogenesis of Kupffer Cells in Cholestatic Liver Injury. Am. J. Pathol. 2016, 186, 2238–2247. [Google Scholar] [PubMed]
- Fu, H.Y.; Xu, J.M.; Ai, X.; Dang, F.T.; Tan, X.; Yu, H.Y.; Feng, J.; Yang, W.X.; Ma, H.T.; Tu, R.F.; et al. The Clostridium Metabolite P-Cresol Sulfate Relieves Inflammation of Primary Biliary Cholangitis by Regulating Kupffer Cells. Cells 2022, 11, 3782. [Google Scholar] [CrossRef] [PubMed]
- Harada, K.; Nakanuma, Y. Cholangiopathy with respect to biliary innate immunity. Int. J. Hepatol. 2012, 2012, 793569. [Google Scholar] [PubMed]
- Szabo, G.; Dolganiuc, A.; Mandrekar, P. Pattern recognition receptors: A contemporary view on liver diseases. Hepatology 2006, 44, 287–298. [Google Scholar] [CrossRef]
- Chen, X.M.; O’Hara, S.P.; Nelson, J.B.; Splinter, P.L.; Small, A.J.; Tietz, P.S.; Limper, A.H.; LaRusso, N.F. Multiple TLRs are expressed in human cholangiocytes and mediate host epithelial defense responses to Cryptosporidium parvum via activation of NF-kappaB. J. Immunol. 2005, 175, 7447–7456. [Google Scholar]
- Harada, K.; Isse, K.; Nakanuma, Y. Interferon gamma accelerates NF-kappaB activation of biliary epithelial cells induced by Toll-like receptor and ligand interaction. J. Clin. Pathol. 2006, 59, 184–190. [Google Scholar] [CrossRef]
- Harada, K.; Nakanuma, Y. Biliary innate immunity: Function and modulation. Mediat. Inflamm. 2010, 2010, 373878. [Google Scholar]
- Harada, K.; Ohba, K.; Ozaki, S.; Isse, K.; Hirayama, T.; Wada, A.; Nakanuma, Y. Peptide antibiotic human beta-defensin-1 and -2 contribute to antimicrobial defense of the intrahepatic biliary tree. Hepatology 2004, 40, 925–932. [Google Scholar]
- Isse, K.; Harada, K.; Zen, Y.; Kamihira, T.; Shimoda, S.; Harada, M.; Nakanuma, Y. Fractalkine and CX3CR1 are involved in the recruitment of intraepithelial lymphocytes of intrahepatic bile ducts. Hepatology 2005, 41, 506–516. [Google Scholar]
- Shimoda, S.; Harada, K.; Niiro, H.; Yoshizumi, T.; Soejima, Y.; Taketomi, A.; Maehara, Y.; Tsuneyama, K.; Nakamura, M.; Komori, A.; et al. Biliary epithelial cells and primary biliary cirrhosis: The role of liver-infiltrating mononuclear cells. Hepatology 2008, 47, 958–965. [Google Scholar]
- Chuang, Y.H.; Lian, Z.X.; Tsuneyama, K.; Chiang, B.L.; Ansari, A.A.; Coppel, R.L.; Gershwin, M.E. Increased killing activity and decreased cytokine production in NK cells in patients with primary biliary cirrhosis. J. Autoimmun. 2006, 26, 232–240. [Google Scholar] [PubMed]
- Ronca, V.; Mancuso, C.; Milani, C.; Carbone, M.; Oo, Y.H.; Invernizzi, P. Immune system and cholangiocytes: A puzzling affair in primary biliary cholangitis. J. Leucoc. Biol. 2020, 108, 659–671. [Google Scholar]
- Jiang, X.; Lian, M.; Li, Y.; Zhang, W.; Wang, Q.; Wei, Y.; Zhang, J.; Chen, W.; Xiao, X.; Miao, Q.; et al. The immunobiology of mucosal-associated invariant T cell (MAIT) function in primary biliary cholangitis: Regulation by cholic acid-induced Interleukin-7. J. Autoimmun. 2018, 90, 64–75. [Google Scholar] [CrossRef]
- Banales, J.M.; Huebert, R.C.; Karlsen, T.; Strazzabosco, M.; LaRusso, N.F.; Gores, G.J. Cholangiocyte pathobiology. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 269–281. [Google Scholar] [PubMed]
- Lleo, A.; Maroni, L.; Glaser, S.; Alpini, G.; Marzioni, M. Role of cholangiocytes in primary biliary cirrhosis. Semin. Liver Dis. 2014, 34, 273–284. [Google Scholar]
- Harada, K.; Sato, Y.; Itatsu, K.; Isse, K.; Ikeda, H.; Yasoshima, M.; Zen, Y.; Matsui, A.; Nakanuma, Y. Innate immune response to double-stranded RNA in biliary epithelial cells is associated with the pathogenesis of biliary atresia. Hepatology 2007, 46, 1146–1154. [Google Scholar]
- O’Hara, S.P.; Tabibian, J.H.; Splinter, P.L.; LaRusso, N.F. The dynamic biliary epithelia: Molecules, pathways, and disease. J. Hepatol. 2013, 58, 575–582. [Google Scholar]
- Tabibian, J.H.; O’Hara, S.P.; Lindor, K.D. Primary sclerosing cholangitis and the microbiota: Current knowledge and perspectives on etiopathogenesis and emerging therapies. Scand. J. Gastroenterol. 2014, 49, 901–908. [Google Scholar]
- O’Hara, S.P.; Splinter, P.L.; Trussoni, C.E.; Gajdos, G.B.; Lineswala, P.N.; LaRusso, N.F. Cholangiocyte N-Ras protein mediates lipopolysaccharide-induced interleukin 6 secretion and proliferation. J. Biol. Chem. 2011, 286, 30352–30360. [Google Scholar]
- Schrumpf, E.; Tan, C.; Karlsen, T.H.; Sponheim, J.; Björkström, N.K.; Sundnes, O.; Alfsnes, K.; Kaser, A.; Jefferson, D.M.; Ueno, Y.; et al. The biliary epithelium presents antigens to and activates natural killer T cells. Hepatology 2015, 62, 1249–1259. [Google Scholar]
- Rong, G.H.; Yang, G.X.; Ando, Y.; Zhang, W.; He, X.S.; Leung, P.S.; Coppel, R.L.; Ansari, A.A.; Zhong, R.; Gershwin, M.E. Human intrahepatic biliary epithelial cells engulf blebs from their apoptotic peers. Clin. Exp. Immunol. 2013, 172, 95–103. [Google Scholar] [CrossRef] [PubMed]
- He, X.S.; Gershwin, M.E.; Ansari, A.A. Checkpoint-Based immunotherapy for autoimmune diseases—Opportunities and challenges. J. Autoimmun. 2017, 79, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Shi, T.Y.; Zhang, F.C. Role of autoimmunity in primary biliary cirrhosis. World J. Gastroenterol. 2012, 18, 7141–7148. [Google Scholar] [CrossRef] [PubMed]
- Shuai, Z.; Wang, J.; Badamagunta, M.; Choi, J.; Yang, G.; Zhang, W.; Kenny, T.P.; Guggenheim, K.; Kurth, M.J.; Ansari, A.A.; et al. The fingerprint of antimitochondrial antibodies and the etiology of primary biliary cholangitis. Hepatology 2017, 65, 1670–1682. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, W.; Leung, P.S.; Bowlus, C.L.; Dhaliwal, S.; Coppel, R.L.; Ansari, A.A.; Yang, G.X.; Wang, J.; Kenny, T.P.; et al. Ongoing activation of autoantigen-specific B cells in primary biliary cirrhosis. Hepatology 2014, 60, 1708–1716. [Google Scholar] [CrossRef]
- Wang, L.; Wang, F.S.; Chang, C.; Gershwin, M.E. Breach of tolerance: Primary biliary cirrhosis. Semin. Liver Dis. 2014, 34, 297–317. [Google Scholar] [CrossRef]
- Chung, B.K.; Guevel, B.T.; Reynolds, G.M.; Gupta Udatha, D.B.; Henriksen, E.K.; Stamataki, Z.; Hirschfield, G.M.; Karlsen, T.H.; Liaskou, E. Phenotyping and auto-antibody production by liver-infiltrating B cells in primary sclerosing cholangitis and primary biliary cholangitis. J. Autoimmun. 2017, 77, 45–54. [Google Scholar] [CrossRef]
- Sakaguchi, S. Naturally arising CD4+ regulatory t cells for immunologic self-tolerance and negative control of immune responses. Annu. Rev. Immunol. 2004, 22, 531–562. [Google Scholar] [CrossRef]
- Harada, K.; Isse, K.; Sato, Y.; Ozaki, S.; Nakanuma, Y. Endotoxin tolerance in human intrahepatic biliary epithelial cells is induced by upregulation of IRAK-M. Liver Int. 2006, 26, 935–942. [Google Scholar] [CrossRef]
- Lan, R.Y.; Cheng, C.; Lian, Z.X.; Tsuneyama, K.; Yang, G.X.; Moritoki, Y.; Chuang, Y.H.; Nakamura, T.; Saito, S.; Shimoda, S.; et al. Liver-Targeted and peripheral blood alterations of regulatory T cells in primary biliary cirrhosis. Hepatology 2006, 43, 729–737. [Google Scholar] [CrossRef]
- Liaskou, E.; Patel, S.R.; Webb, G.; Bagkou Dimakou, D.; Akiror, S.; Krishna, M.; Mells, G.; Jones, D.E.; Bowman, S.J.; Barone, F.; et al. Increased sensitivity of Treg cells from patients with PBC to low dose IL-12 drives their differentiation into IFN-γ secreting cells. J. Autoimmun. 2018, 94, 143–155. [Google Scholar] [PubMed]
- Trivedi, P.J.; Cullen, S. Etiopathogenesis of primary biliary cirrhosis: An overview of recent developments. Hepatol. Int. 2013, 7, 28–47. [Google Scholar] [PubMed]
- Gulamhusein, A.F.; Juran, B.D.; Lazaridis, K.N. Genome-Wide Association Studies in Primary Biliary Cirrhosis. Semin. Liver Dis. 2015, 35, 392–401. [Google Scholar]
- Ichiki, Y.; Selmi, C.; Shimoda, S.; Ishibashi, H.; Gordon, S.C.; Gershwin, M.E. Mitochondrial antigens as targets of cellular and humoral auto-immunity in primary biliary cirrhosis. Clin. Rev. Allergy Immunol. 2005, 28, 83–91. [Google Scholar] [CrossRef]
- Benson, G.D.; Kikuchi, K.; Miyakawa, H.; Tanaka, A.; Watnik, M.R.; Gershwin, M.E. Serial analysis of antimitochondrial antibody in patients with primary biliary cirrhosis. Clin. Dev. Immunol. 2004, 11, 129–133. [Google Scholar] [CrossRef]
- Moteki, S.; Leung, P.S.; Dickson, E.R.; Van Thiel, D.H.; Galperin, C.; Buch, T.; Alarcon-Segovia, D.; Kershenobich, D.; Kawano, K.; Coppel, R.L.; et al. Epitope mapping and reactivity of autoantibodies to the E2 component of 2-oxoglutarate dehydrogenase complex in primary biliary cirrhosis using recombinant 2-oxoglutarate dehydrogenase complex. Hepatology 1996, 23, 436–444. [Google Scholar]
- Allina, J.; Hu, B.; Sullivan, D.M.; Fiel, M.I.; Thung, S.N.; Bronk, S.F.; Huebert, R.C.; van de Water, J.; LaRusso, N.F.; Gershwin, M.E.; et al. T cell targeting and phagocytosis of apoptotic biliary epithelial cells in primary biliary cirrhosis. J. Autoimmun. 2006, 27, 232–241. [Google Scholar] [CrossRef]
- Kaplan, M.M.; Gershwin, M.E. Primary biliary cirrhosis. N. Engl. J. Med. 2005, 353, 1261–1273. [Google Scholar] [CrossRef]
- Leung, P.S.; Coppel, R.L.; Gershwin, M.E. Etiology of primary biliary cirrhosis: The search for the culprit. Semin. Liver Dis. 2005, 25, 327–336. [Google Scholar]
- Oertelt, S.; Rieger, R.; Selmi, C.; Invernizzi, P.; Ansari, A.A.; Coppel, R.L.; Podda, M.; Leung, P.S.; Gershwin, M.E. A sensitive bead assay for antimitochondrial antibodies: Chipping away at AMA-negative primary biliary cirrhosis. Hepatology 2007, 45, 659–665. [Google Scholar] [CrossRef]
- Amano, K.; Leung, P.S.; Rieger, R.; Quan, C.; Wang, X.; Marik, J.; Suen, Y.F.; Kurth, M.J.; Nantz, M.H.; Ansari, A.A.; et al. Chemical xenobiotics and mitochondrial autoantigens in primary biliary cirrhosis: Identification of antibodies against a common environmental, cosmetic, and food additive, 2-octynoic acid. J. Immunol. 2005, 174, 5874–5883. [Google Scholar] [PubMed]
- Wakabayashi, K.; Lian, Z.X.; Leung, P.S.; Moritoki, Y.; Tsuneyama, K.; Kurth, M.J.; Lam, K.S.; Yoshida, K.; Yang, G.X.; Hibi, T.; et al. Loss of tolerance in C57BL/6 mice to the autoantigen E2 subunit of pyruvate dehydrogenase by a xenobiotic with ensuing biliary ductular disease. Hepatology 2008, 48, 531–540. [Google Scholar] [PubMed]
- Wakabayashi, K.; Yoshida, K.; Leung, P.S.; Moritoki, Y.; Yang, G.X.; Tsuneyama, K.; Lian, Z.X.; Hibi, T.; Ansari, A.A.; Wicker, L.S.; et al. Induction of autoimmune cholangitis in non-obese diabetic (NOD).1101 mice following a chemical xenobiotic immunization. Clin. Exp. Immunol. 2009, 155, 577–586. [Google Scholar] [PubMed]
- Dahlqvist, G.; Gaouar, F.; Carrat, F.; Meurisse, S.; Chazouillères, O.; Poupon, R.; Johanet, C.; Corpechot, C.; French Network of Immunology Laboratories. Large-Scale characterization study of patients with antimitochondrial antibodies but nonestablished primary biliary cholangitis. Hepatology 2017, 65, 152–163. [Google Scholar] [CrossRef]
- Koulentaki, M.; Moscandrea, J.; Dimoulios, P.; Chatzicostas, C.; Kouroumalis, E.A. Survival of anti-mitochondrial antibody-positive and -negative primary biliary cirrhosis patients on ursodeoxycholic acid treatment. Dig. Dis. Sci. 2004, 49, 1190–1195. [Google Scholar]
- Kita, H.; Lian, Z.X.; Van de Water, J.; He, X.S.; Matsumura, S.; Kaplan, M.; Luketic, V.; Coppel, R.L.; Ansari, A.A.; Gershwin, M.E. Identification of HLA-A2-restricted CD8(+) cytotoxic T cell responses in primary biliary cirrhosis: T cell activation is augmented by immune complexes cross-presented by dendritic cells. J. Exp. Med. 2002, 195, 113–123. [Google Scholar]
- Hsu, W.; Zhang, W.; Tsuneyama, K.; Moritoki, Y.; Ridgway, W.M.; Ansari, A.A.; Coppel, R.L.; Lian, Z.X.; Mackay, I.; Gershwin, M.E. Differential mechanisms in the pathogenesis of autoimmune cholangitis versus inflammatory bowel disease in interleukin-2Ralpha(-/-) mice. Hepatology 2009, 49, 133–140. [Google Scholar]
- Wakabayashi, K.; Lian, Z.X.; Moritoki, Y.; Lan, R.Y.; Tsuneyama, K.; Chuang, Y.H.; Yang, G.X.; Ridgway, W.; Ueno, Y.; Ansari, A.A.; et al. IL-2 receptor alpha(-/-) mice and the development of primary biliary cirrhosis. Hepatology 2006, 44, 1240–1249. [Google Scholar]
- Aoki, C.A.; Roifman, C.M.; Lian, Z.X.; Bowlus, C.L.; Norman, G.L.; Shoenfeld, Y.; Mackay, I.R.; Gershwin, M.E. IL-2 receptor alpha deficiency and features of primary biliary cirrhosis. J. Autoimmun. 2006, 27, 50–53. [Google Scholar]
- Lan, R.Y.; Salunga, T.L.; Tsuneyama, K.; Lian, Z.X.; Yang, G.X.; Hsu, W.; Moritoki, Y.; Ansari, A.A.; Kemper, C.; Price, J.; et al. Hepatic IL-17 responses in human and murine primary biliary cirrhosis. J. Autoimmun. 2009, 32, 43–51. [Google Scholar]
- Jia, H.; Chen, J.; Zhang, X.; Bi, K.; Zhou, H.; Liu, T.; Xu, J.; Diao, H. IL-17A produced by invariant natural killer T cells and CD3+ CD56+ αGalcer-CD1d tetramer-T cells promote liver fibrosis in patients with primary biliary cholangitis. J. Leukoc. Biol. 2022, 112, 1079–1087. [Google Scholar] [CrossRef] [PubMed]
- Chan, C.W.; Chen, H.W.; Wang, Y.W.; Lin, C.I.; Chuang, Y.H. IL-21, not IL-17A, exacerbates murine primary biliary cholangitis. Clin. Exp. Immunol. 2024, 215, 137–147. [Google Scholar] [CrossRef]
- Oertelt, S.; Lian, Z.X.; Cheng, C.M.; Chuang, Y.H.; Padgett, K.A.; He, X.S.; Ridgway, W.M.; Ansari, A.A.; Coppel, R.L.; Li, M.O.; et al. Anti-Mitochondrial antibodies and primary biliary cirrhosis in TGF-beta receptor II dominant-negative mice. J. Immunol. 2006, 177, 1655–1660. [Google Scholar] [CrossRef]
- Gorelik, L.; Flavell, R.A. Abrogation of TGFbeta signaling in T cells leads to spontaneous T cell differentiation and autoimmune disease. Immunity 2000, 12, 171–181. [Google Scholar] [CrossRef]
- Ueno, Y.; Ambrosini, Y.M.; Moritoki, Y.; Ridgway, W.M.; Gershwin, M.E. Murine models of autoimmune cholangitis. Curr. Opin. Gastroenterol. 2010, 26, 274–279. [Google Scholar] [CrossRef]
- Yang, G.X.; Lian, Z.X.; Chuang, Y.H.; Moritoki, Y.; Lan, R.Y.; Wakabayashi, K.; Ansari, A.A.; Flavell, R.A.; Ridgway, W.M.; Coppel, R.L.; et al. Adoptive transfer of CD8(+) T cells from transforming growth factor beta receptor type II (dominant negative form) induces autoimmune cholangitis in mice. Hepatology 2008, 47, 1974–1982. [Google Scholar] [CrossRef]
- Marie, J.C.; Letterio, J.J.; Gavin, M.; Rudensky, A.Y. TGF-beta1 maintains suppressor function and Foxp3 expression in CD4+CD25+ regulatory T cells. J. Exp. Med. 2005, 201, 1061–1067. [Google Scholar] [CrossRef]
- Li, M.O.; Wan, Y.Y.; Sanjabi, S.; Robertson, A.K.; Flavell, R.A. Transforming growth factor-beta regulation of immune responses. Annu. Rev. Immunol. 2006, 24, 99–146. [Google Scholar] [CrossRef]
- Fontenot, J.D.; Gavin, M.A.; Rudensky, A.Y. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat. Immunol. 2003, 4, 330–336. [Google Scholar] [CrossRef]
- Hori, S.; Nomura, T.; Sakaguchi, S. Control of regulatory T cell development by the transcription factor Foxp3. Science 2003, 299, 1057–1061. [Google Scholar] [CrossRef]
- Bettelli, E.; Carrier, Y.; Gao, W.; Korn, T.; Strom, T.B.; Oukka, M.; Weiner, H.L.; Kuchroo, V.K. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature 2006, 441, 235–238. [Google Scholar] [CrossRef] [PubMed]
- Mangan, P.R.; Harrington, L.E.; O’Quinn, D.B.; Helms, W.S.; Bullard, D.C.; Elson, C.O.; Hatton, R.D.; Wahl, S.M.; Schoeb, T.R.; Weaver, C.T. Transforming growth factor-beta induces development of the T(H)17 lineage. Nature 2006, 441, 231–234. [Google Scholar] [CrossRef] [PubMed]
- Veldhoen, M.; Hocking, R.J.; Atkins, C.J.; Locksley, R.M.; Stockinger, B. TGFbeta in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity 2006, 24, 179–189. [Google Scholar] [CrossRef] [PubMed]
- Voumvouraki, A.; Koulentaki, M.; Tzardi, M.; Sfakianaki, O.; Manousou, P.; Notas, G.; Kouroumalis, E. Increased ΤGF-β3 in primary biliary cirrhosis: An abnormality related to pathogenesis? World J. Gastroenterol. 2010, 16, 5057–5064. [Google Scholar] [CrossRef]
- Pyzik, M.; Piccirillo, C.A. TGF-beta1 modulates Foxp3 expression and regulatory activity in distinct CD4+ T cell subsets. J. Leukoc. Biol. 2007, 82, 335–346. [Google Scholar] [CrossRef]
- Zhang, W.; Sharma, R.; Ju, S.T.; He, X.S.; Tao, Y.; Tsuneyama, K.; Tian, Z.; Lian, Z.X.; Fu, S.M.; Gershwin, M.E. Deficiency in regulatory T cells results in development of antimitochondrial antibodies and autoimmune cholangitis. Hepatology 2009, 49, 545–552. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, P.; Gao, X.; Qian, W.; Xu, K. rAAV2-TGF-β(3) decreases collagen synthesis and deposition in the liver of experimental hepatic fibrosis rat. Dig. Dis. Sci. 2010, 55, 2821–2830. [Google Scholar] [CrossRef]
- Chuang, Y.H.; Lian, Z.X.; Cheng, C.M.; Lan, R.Y.; Yang, G.X.; Moritoki, Y.; Chiang, B.L.; Ansari, A.A.; Tsuneyama, K.; Coppel, R.L.; et al. Increased levels of chemokine receptor CXCR3 and chemokines IP-10 and MIG in patients with primary biliary cirrhosis and their first degree relatives. J. Autoimmun. 2005, 25, 126–132. [Google Scholar] [CrossRef]
- Oo, Y.H.; Banz, V.; Kavanagh, D.; Liaskou, E.; Withers, D.R.; Humphreys, E.; Reynolds, G.M.; Lee-Turner, L.; Kalia, N.; Hubscher, S.G.; et al. CXCR3-Dependent recruitment and CCR6-mediated positioning of Th-17 cells in the inflamed liver. J. Hepatol. 2012, 57, 1044–1051. [Google Scholar] [CrossRef]
- Santodomingo-Garzon, T.; Han, J.; Le, T.; Yang, Y.; Swain, M.G. Natural killer T cells regulate the homing of chemokine CXC receptor 3-positive regulatory T cells to the liver in mice. Hepatology 2009, 49, 1267–1276. [Google Scholar] [CrossRef]
- Hintermann, E.; Bayer, M.; Pfeilschifter, J.M.; Luster, A.D.; Christen, U. CXCL10 promotes liver fibrosis by prevention of NK cell mediated hepatic stellate cell inactivation. J. Autoimmun. 2010, 35, 424–435. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.D.; Ma, W.T.; Liu, Q.Z.; Zhao, Z.B.; Liu, M.Z.; Tsuneyama, K.; Gao, J.M.; Ridgway, W.M.; Ansari, A.A.; Gershwin, M.E.; et al. Chemokine receptor CXCR3 deficiency exacerbates murine autoimmune cholangitis by promoting pathogenic CD8+ T cell activation. J. Autoimmun. 2017, 78, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Cai, X.; Tacke, F.; Guillot, A.; Liu, H. Cholangiokines: Undervalued modulators in the hepatic microenvironment. Front. Immunol. 2023, 14, 1192840. [Google Scholar] [CrossRef]
- Oikawa, T.; Takahashi, H.; Ishikawa, T.; Hokari, A.; Otsuki, N.; Azuma, M.; Zeniya, M.; Tajiri, H. Intrahepatic expression of the co-stimulatory molecules programmed death-1, and its ligands in autoimmune liver disease. Pathol. Int. 2007, 57, 485–492. [Google Scholar] [CrossRef]
- Mataki, N.; Kikuchi, K.; Kawai, T.; Higashiyama, M.; Okada, Y.; Kurihara, C.; Hokari, R.; Kawaguchi, A.; Nagao, S.; Kondo, T.; et al. Expression of PD-1, PD-L1, and PD-L2 in the liver in autoimmune liver diseases. Am. J. Gastroenterol. 2007, 102, 302–312. [Google Scholar] [CrossRef]
- Zhang, S.; Tao, X.; Wang, L.; Chen, H.; Zhao, L.; Sun, J.; Bian, S.; Chen, Z.; Shao, T.; Yang, Y.; et al. Downregulation of Programmed Death-1 Pathway Promoting CD8 + T Cell Cytotoxicity in Primary Biliary Cholangitis. Dig. Dis. Sci. 2022, 67, 2981–2993. [Google Scholar] [CrossRef]
- Anderson, A.C.; Joller, N.; Kuchroo, V.K. Lag-3, Tim-3, and TIGIT: Co-inhibitory Receptors with Specialized Functions in Immune Regulation. Immunity 2016, 44, 989–1004. [Google Scholar] [CrossRef]
- Deng, C.; Li, W.; Fei, Y.; Wang, L.; Chen, Y.; Zeng, X.; Zhang, F.; Li, Y. Imbalance of the CD226/TIGIT Immune Checkpoint Is Involved in the Pathogenesis of Primary Biliary Cholangitis. Front. Immunol. 2020, 11, 1619. [Google Scholar] [CrossRef]
- Bhave, P.; Pham, A.; Gordon, A.; Moore, M. Safe administration of anti-PD-1 immunotherapy in a patient with pre-existing primary biliary cholangitis. Immunotherapy 2020, 12, 445–450. [Google Scholar] [CrossRef]
- Yang, Y.; He, X.; Rojas, M.; Leung, P.S.C.; Gao, L. Mechanism-based target therapy in primary biliary cholangitis: Opportunities before liver cirrhosis? Front. Immunol. 2023, 14, 1184252. [Google Scholar] [CrossRef]
- Davenport, A.P.; Hyndman, K.A.; Dhaun, N.; Southan, C.; Kohan, D.E.; Pollock, J.S.; Pollock, D.M.; Webb, D.J.; Maguire, J.J. Endothelin. Pharmacol. Rev. 2016, 68, 357–418. [Google Scholar] [PubMed]
- Schneider, M.P.; Boesen, E.I.; Pollock, D.M. Contrasting actions of endothelin ET(A) and ET(B) receptors in cardiovascular disease. Annu. Rev. Pharmacol. Toxicol. 2007, 47, 731–759. [Google Scholar] [CrossRef] [PubMed]
- Luo, B.; Tang, L.; Wang, Z.; Zhang, J.; Ling, Y.; Feng, W.; Sun, J.Z.; Stockard, C.R.; Frost, A.R.; Chen, Y.F.; et al. Cholangiocyte endothelin 1 and transforming growth factor beta1 production in rat experimental hepatopulmonary syndrome. Gastroenterology 2005, 129, 682–695. [Google Scholar] [PubMed]
- Yadav, N.; Jaber, F.L.; Sharma, Y.; Gupta, P.; Viswanathan, P.; Gupta, S. Efficient Reconstitution of Hepatic Microvasculature by Endothelin Receptor Antagonism in Liver Sinusoidal Endothelial Cells. Hum. Gene Ther. 2019, 30, 365–377. [Google Scholar]
- Zhang, L.; Luo, B.; Dang, Y.W.; He, R.Q.; Chen, G.; Peng, Z.G.; Feng, Z.B. The clinical significance of endothelin receptor type B in hepatocellular carcinoma and its potential molecular mechanism. Exp. Mol. Pathol. 2019, 107, 141–157. [Google Scholar] [CrossRef]
- Cho, J.J.; Hocher, B.; Herbst, H.; Jia, J.D.; Ruehl, M.; Hahn, E.G.; Riecken, E.O.; Schuppan, D. An oral endothelin-A receptor antagonist blocks collagen synthesis and deposition in advanced rat liver fibrosis. Gastroenterology 2000, 118, 1169–1178. [Google Scholar]
- Ling, L.; Kuc, R.E.; Maguire, J.J.; Davie, N.J.; Webb, D.J.; Gibbs, P.; Alexander, G.J.; Davenport, A.P. Comparison of endothelin receptors in normal versus cirrhotic human liver and in the liver from endothelial cell-specific ETB knockout mice. Life Sci. 2012, 91, 716–722. [Google Scholar]
- Kojima, H.; Sakurai, S.; Kuriyama, S.; Yoshiji, H.; Imazu, H.; Uemura, M.; Nakatani, Y.; Yamao, J.; Fukui, H. Endothelin-1 plays a major role in portal hypertension of biliary cirrhotic rats through endothelin receptor subtype B together with subtype A in vivo. J. Hepatol. 2001, 34, 805–811. [Google Scholar] [CrossRef]
- Zipprich, A.; Gittinger, F.; Winkler, M.; Dollinger, M.M.; Ripoll, C. Effect of ET-A blockade on portal pressure and hepatic arterial perfusion in patients with cirrhosis: A proof of concept study. Liver Int. 2021, 41, 554–561. [Google Scholar]
- Ryanto, G.R.T.; Yorifuji, K.; Ikeda, K.; Emoto, N. Chondroitin sulfate mediates liver responses to injury induced by dual endothelin receptor inhibition. Can. J. Physiol. Pharmacol. 2020, 98, 618–624. [Google Scholar]
- Owen, T.; Carpino, G.; Chen, L.; Kundu, D.; Wills, P.; Ekser, B.; Onori, P.; Gaudio, E.; Alpini, G.; Francis, H.; et al. Endothelin Receptor-A Inhibition Decreases Ductular Reaction, Liver Fibrosis, and Angiogenesis in a Model of Cholangitis. Cell. Mol. Gastroenterol. Hepatol. 2023, 16, 513–540. [Google Scholar]
- Dimoulios, P.; Kolios, G.; Notas, G.; Matrella, E.; Xidakis, C.; Koulentaki, M.; Tsagarakis, N.; Kouroumalis, A.; Kouroumalis, E. Ursodeoxycholic acid reduces increased circulating endothelin 2 in primary biliary cirrhosis. Aliment. Pharmacol. Ther. 2005, 21, 227–234. [Google Scholar]
- Kouroumalis, E.; Notas, G. Primary biliary cirrhosis: From bench to bedside. World J. Gastrointest. Pharmacol. Ther. 2015, 6, 32–58. [Google Scholar]
- Tsuneyama, K.; Harada, K.; Kono, N.; Hiramatsu, K.; Zen, Y.; Sudo, Y.; Gershwin, M.E.; Ikemoto, M.; Arai, H.; Nakanuma, Y. Scavenger cells with gram-positive bacterial lipoteichoic acid infiltrate around the damaged interlobular bile ducts of primary biliary cirrhosis. J. Hepatol. 2001, 35, 156–163. [Google Scholar]
- Speciale, L.; Roda, K.; Saresella, M.; Taramelli, D.; Ferrante, P. Different endothelins stimulate cytokine production by peritoneal macrophages and microglial cell line. Immunology 1998, 93, 109–114. [Google Scholar]
- Grimshaw, M.J.; Wilson, J.L.; Balkwill, F.R. Endothelin-2 is a macrophage chemoattractant: Implications for macrophage distribution in tumors. Eur. J. Immunol. 2002, 32, 2393–2400. [Google Scholar]
- Colucci, G.; Schaffner, F.; Paronetto, F. In situ characterization of the cell-surface antigens of the mononuclear cell infiltrate and bile duct epithelium in primary biliary cirrhosis. Clin. Immunol. Immunopathol. 1986, 41, 35–42. [Google Scholar] [CrossRef]
- Tobe, K.; Tsuchiya, T.; Itoshima, T.; Nagashima, H.; Kobayashi, T. Electron microscopy of fat-storing cells in liver diseases with special reference to cilia and cytoplasmic cholesterol crystals. Arch. Histol. Jpn. 1985, 48, 435–441. [Google Scholar]
- Rockey, D. The cellular pathogenesis of portal hypertension: Stellate cell contractility, endothelin, and nitric oxide. Hepatology 1997, 25, 2–5. [Google Scholar]
- Sfakianaki, O.; Tzardi, M.; Voumvouraki, A.; Afgoustaki, A.; Koulentaki, M.; Kouroumalis, E. Presence of disease specific autoantibodies against liver sinusoidal cells in primary biliary cirrhosis. World J. Hepatol. 2013, 5, 568–576. [Google Scholar]
- Mantaka, A.; Goulielmos, G.N.; Koulentaki, M.; Tsagournis, O.; Voumvouraki, A.; Kouroumalis, E.A. Polymorphisms of genes related to endothelial cells are associated with primary biliary cirrhosis patients of Cretan origin. Hum. Immunol. 2012, 73, 829–835. [Google Scholar]
- Ludwig, J.; Batts, K.P.; MacCarty, R.L. Ischemic cholangitis in hepatic allografts. Mayo Clin. Proc. 1992, 67, 519–526. [Google Scholar]
- Matsunaga, Y.; Terada, T. Peribiliary capillary plexus around interlobular bile ducts in various chronic liver diseases: An immunohistochemical and morphometric study. Pathol. Int. 1999, 49, 869–873. [Google Scholar]
- Strazzabosco, M.; Fabris, L. Functional anatomy of normal bile ducts. Anat. Rec. 2008, 291, 653–660. [Google Scholar]
- Washington, K.; Clavien, P.A.; Killenberg, P. Peribiliary vascular plexus in primary sclerosing cholangitis and primary biliary cirrhosis. Hum. Pathol. 1997, 28, 791–795. [Google Scholar]
- Koda, W.; Harada, K.; Tsuneyama, K.; Kono, N.; Sasaki, M.; Matsui, O.; Nakanuma, Y. Evidence of the participation of peribiliary mast cells in regulation of the peribiliary vascular plexus along the intrahepatic biliary tree. Lab. Investig. 2000, 80, 1007–1017. [Google Scholar]
- Ayres, R.C.; Neuberger, J.M.; Shaw, J.; Joplin, R.; Adams, D.H. Intercellular adhesion molecule-1 and MHC antigens on human intrahepatic bile duct cells: Effect of pro-inflammatory cytokines. Gut 1993, 34, 1245–1249. [Google Scholar]
- Epstein, O.; Thomas, H.C.; Sherlock, S. Primary biliary cirrhosis is a dry gland syndrome with features of chronic graft-versus-host disease. Lancet 1980, 315, 1166–1168. [Google Scholar]
- Howell, C.D.; Li, J.; Chen, W. Role of intercellular adhesion molecule-1 and lymphocyte function-associated antigen-1 during nonsuppurative destructive cholangitis in a mouse graft-versus-host disease model. Hepatology 1999, 29, 766–776. [Google Scholar]
- Kimura, T.; Suzuki, K.; Inada, S.; Hayashi, A.; Isobe, M.; Matsuzaki, Y.; Tanaka, N.; Osuga, T.; Fujiwara, M. Monoclonal antibody against lymphocyte function-associated antigen 1 inhibits the formation of primary biliary cirrhosis-like lesions induced by murine graft-versus-host reaction. Hepatology 1996, 24, 888–894. [Google Scholar]
- Wakae, T.; Takatsuka, H.; Seto, Y.; Iwata, N.; Mori, A.; Okada, M.; Fujimori, Y.; Okamoto, T.; Kakishita, E.; Hara, H. Similarity between hepatic graft-versus-host disease and primary biliary cirrhosis. Hematology 2002, 7, 305–310. [Google Scholar]
- Beschorner, W.E.; Shinn, C.A.; Hess, A.D.; Suresch, D.L.; Santos, G.W. Immune-Related injury to endothelium associated with acute graft-versus-host disease in the rat. Transplant. Proc. 1989, 21, 3025–3027. [Google Scholar]
- Hiroyasu, S.; Shiraishi, M.; Kusano, T.; Muto, Y. Involvement of endothelin in graft-versus-host disease after rat small bowel transplantation. Transpl. Int. 1997, 10, 121–124. [Google Scholar]
- Xu, G.; Gong, Y.; Lu, F.; Wang, B.; Yang, Z.; Chen, L.; Min, J.; Cheng, C.; Jiang, T. Endothelin receptor B enhances liver injury and pro-inflammatory responses by increasing G-protein-coupled receptor kinase-2 expression in primary biliary cholangitis. Sci. Rep. 2022, 12, 19772. [Google Scholar]
- Reshetnyak, V.I.; Maev, I.V. New insights into the pathogenesis of primary biliary cholangitis asymptomatic stage. World J. Gastroenterol. 2023, 29, 5292–5304. [Google Scholar]
- Trivedi, P.J.; Hirschfield, G.M.; Adams, D.H.; Vierling, J.M. Immunopathogenesis of Primary Biliary Cholangitis, Primary Sclerosing Cholangitis and Autoimmune Hepatitis: Themes and Concepts. Gastroenterology 2024, 166, 995–1019. [Google Scholar]
- Zhao, Y.; Wei, S.; Chen, L.; Zhou, X.; Ma, X. Primary biliary cholangitis: Molecular pathogenesis perspectives and therapeutic potential of natural products. Front. Immunol. 2023, 14, 1164202. [Google Scholar]
- Kwon, Y.; Kim, J.W.; Jeoung, J.A.; Kim, M.S.; Kang, C. Autophagy Is Pro-Senescence When Seen in Close-Up, but Anti-Senescence in Long-Shot. Mol. Cells 2017, 40, 607–612. [Google Scholar]
- Chapman, J.; Fielder, E.; Passos, J.F. Mitochondrial dysfunction and cell senescence: Deciphering a complex relationship. FEBS Lett. 2019, 593, 1566–1579. [Google Scholar]
- Abate, M.; Festa, A.; Falco, M.; Lombardi, A.; Luce, A.; Grimaldi, A.; Zappavigna, S.; Sperlongano, P.; Irace, C.; Caraglia, M.; et al. Mitochondria as playmakers of apoptosis, autophagy and senescence. Semin. Cell Dev. Biol. 2020, 98, 139–153. [Google Scholar]
- Zhang, C.; Zhao, Y.; Yu, M.; Qin, J.; Ye, B.; Wang, Q. Mitochondrial Dysfunction and Chronic Liver Disease. Curr. Issues Mol. Biol. 2022, 44, 3156–3165. [Google Scholar] [CrossRef]
- Gossard, A.A.; Lindor, K.D. Current and promising therapy for primary biliary cholangitis. Expert Opin. Pharmacother. 2019, 20, 1161–1167. [Google Scholar]
- Mueller, M.; Thorell, A.; Claudel, T.; Jha, P.; Koefeler, H.; Lackner, C.; Hoesel, B.; Fauler, G.; Stojakovic, T.; Einarsson, C.; et al. Ursodeoxycholic acid exerts farnesoid X receptor-antagonistic effects on bile acid and lipid metabolism in morbid obesity. J. Hepatol. 2015, 62, 1398–1404. [Google Scholar]
- Khambu, B.; Li, T.; Yan, S.; Yu, C.; Chen, X.; Goheen, M.; Li, Y.; Lin, J.; Cummings, O.W.; Lee, Y.A.; et al. Hepatic Autophagy Deficiency Compromises Farnesoid X Receptor Functionality and Causes Cholestatic Injury. Hepatology 2019, 69, 2196–2213. [Google Scholar]
- Sasaki, M.; Sato, Y.; Nakanuma, Y. An impaired biliary bicarbonate umbrella may be involved in dysregulated autophagy in primary biliary cholangitis. Lab. Investig. 2018, 98, 745–754. [Google Scholar]
- Nelyudova, A.; Aksenov, N.; Pospelov, V.; Pospelova, T. By blocking apoptosis, Bcl-2 in p38-dependent manner promotes cell cycle arrest and accelerated senescence after DNA damage and serum withdrawal. Cell Cycle 2007, 6, 2171–2177. [Google Scholar]
- Chang, J.; Wang, Y.; Shao, L.; Laberge, R.M.; Demaria, M.; Campisi, J.; Janakiraman, K.; Sharpless, N.E.; Ding, S.; Feng, W.; et al. Clearance of senescent cells by ABT263 rejuvenates aged hematopoietic stem cells in mice. Nat. Med. 2016, 22, 78–83. [Google Scholar]
- Sanders, Y.Y.; Liu, H.; Zhang, X.; Hecker, L.; Bernard, K.; Desai, L.; Liu, G.; Thannickal, V.J. Histone modifications in senescence-associated resistance to apoptosis by oxidative stress. Redox Biol. 2013, 1, 8–16. [Google Scholar]
- Yosef, R.; Pilpel, N.; Tokarsky-Amiel, R.; Biran, A.; Ovadya, Y.; Cohen, S.; Vadai, E.; Dassa, L.; Shahar, E.; Condiotti, R.; et al. Directed elimination of senescent cells by inhibition of BCL-W and BCL-XL. Nat. Commun. 2016, 7, 11190. [Google Scholar]
- Chen, Q.M.; Liu, J.; Merrett, J.B. Apoptosis or senescence-like growth arrest: Influence of cell-cycle position, p53, p21 and bax in H2O2 response of normal human fibroblasts. Biochem. J. 2000, 347, 543–551. [Google Scholar]
- Seluanov, A.; Gorbunova, V.; Falcovitz, A.; Sigal, A.; Milyavsky, M.; Zurer, I.; Shohat, G.; Goldfinger, N.; Rotter, V. Change of the death pathway in senescent human fibroblasts in response to DNA damage is caused by an inability to stabilize p53. Mol. Cell. Biol. 2001, 21, 1552–1564. [Google Scholar] [CrossRef]
- Gartel, A.L.; Tyner, A.L. The role of the cyclin-dependent kinase inhibitor p21 in apoptosis. Mol. Cancer Ther. 2002, 1, 639–649. [Google Scholar]
- Xia, M.; Knezevic, D.; Vassilev, L.T. p21 does not protect cancer cells from apoptosis induced by nongenotoxic p53 activation. Oncogene 2011, 30, 346–355. [Google Scholar]
- Yosef, R.; Pilpel, N.; Papismadov, N.; Gal, H.; Ovadya, Y.; Vadai, E.; Miller, S.; Porat, Z.; Ben-Dor, S.; Krizhanovsky, V. p21 maintains senescent cell viability under persistent DNA damage response by restraining JNK and caspase signaling. EMBO J. 2017, 36, 2280–2295. [Google Scholar]
- Gorbunova, V.; Seluanov, A.; Pereira-Smith, O.M. Evidence that high telomerase activity may induce a senescent-like growth arrest in human fibroblasts. J. Biol. Chem. 2003, 278, 7692–7698. [Google Scholar] [CrossRef]
- Catz, S.D.; Johnson, J.L. Transcriptional regulation of bcl-2 by nuclear factor kappa B and its significance in prostate cancer. Oncogene 2001, 20, 7342–7351. [Google Scholar]
- Bogert, P.S.; O’Hara, S.P.; LaRusso, N.F. Cellular senescence in the cholangiopathies. Curr. Opin. Gastroenterol. 2022, 38, 121–127. [Google Scholar]
- O’Hara, S.P.; Splinter, P.L.; Trussoni, C.E.; Guicciardi, M.E.; Splinter, N.P.; Al Suraih, M.S.; Nasser-Ghodsi, N.; Stollenwerk, D.; Gores, G.J.; LaRusso, N.F. The transcription factor ETS1 promotes apoptosis resistance of senescent cholangiocytes by epigenetically up-regulating the apoptosis suppressor BCL2L1. J. Biol. Chem. 2019, 294, 18698–18713. [Google Scholar]
- Guicciardi, M.E.; Trussoni, C.E.; Krishnan, A.; Bronk, S.F.; Lorenzo Pisarello, M.J.; O’Hara, S.P.; Splinter, P.L.; Gao, Y.; Vig, P.; Revzin, A.; et al. Macrophages contribute to the pathogenesis of sclerosing cholangitis in mice. J. Hepatol. 2018, 69, 676–686. [Google Scholar]
- Cadamuro, M.; Girardi, N.; Gores, G.J.; Strazzabosco, M.; Fabris, L. The Emerging Role of Macrophages in Chronic Cholangiopathies Featuring Biliary Fibrosis: An Attractive Therapeutic Target for Orphan Diseases. Front. Med. 2020, 7, 115. [Google Scholar] [CrossRef]
- Katsumi, T.; Guicciardi, M.E.; Azad, A.; Bronk, S.F.; Krishnan, A.; Gores, G.J. Activated cholangiocytes release macrophage-polarizing extracellular vesicles bearing the DAMP S100A11. Am. J. Physiol. Cell Physiol. 2019, 317, C788–C799. [Google Scholar] [CrossRef] [PubMed]
- Xia, C.; Braunstein, Z.; Toomey, A.C.; Zhong, J.; Rao, X. S100 Proteins As an Important Regulator of Macrophage Inflammation. Front. Immunol. 2018, 8, 1908. [Google Scholar]
- Inamura, K.; Tsuji, H.; Nakamoto, Y.; Suzuki, M.; Kaneko, S. Transgenic mice aberrantly expressing pyruvate dehydrogenase complex E2 component on biliary epithelial cells do not show primary biliary cirrhosis. Clin. Exp. Immunol. 2006, 145, 93–100. [Google Scholar] [CrossRef]
- Butler, P.; Hamilton-Miller, J.; Baum, H.; Burroughs, A.K. Detection of M2 antibodies in patients with recurrent urinary tract infection using an ELISA and purified PBC specific antigens. Evidence for a molecular mimicry mechanism in the pathogenesis of primary biliary cirrhosis? Biochem. Mol. Biol. Int. 1995, 35, 473–485. [Google Scholar]
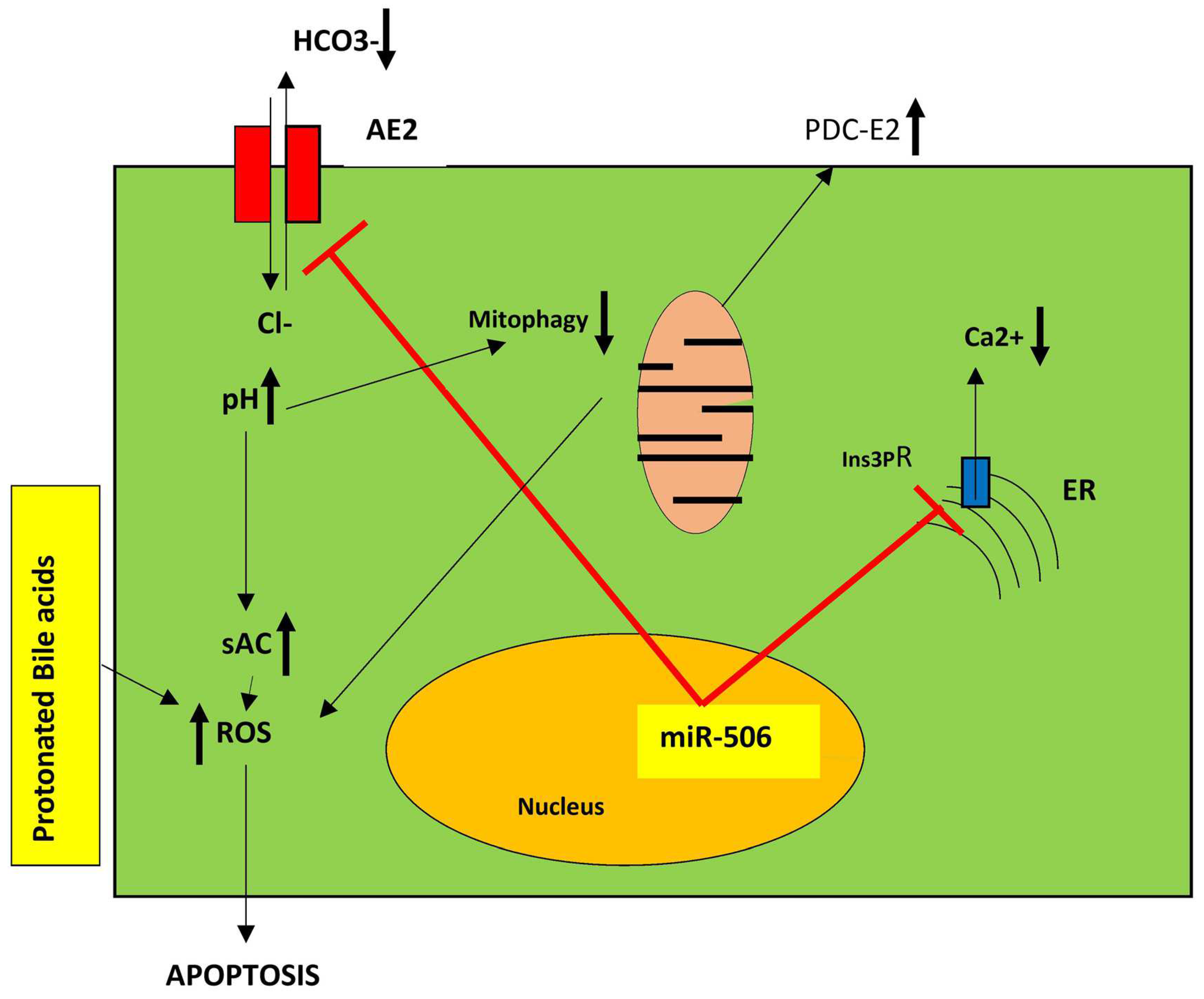
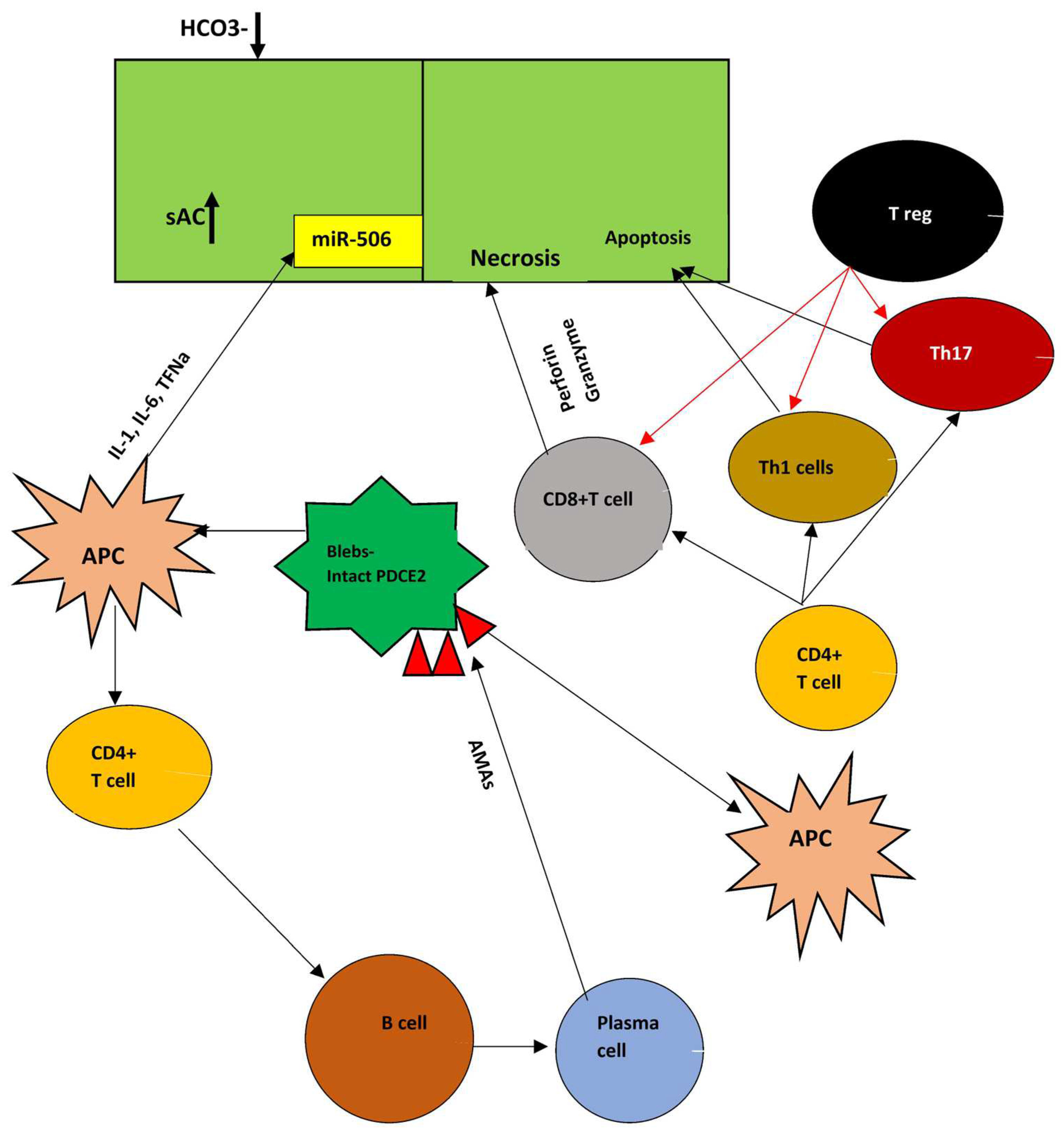

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kouroumalis, E.; Tsomidis, I.; Voumvouraki, A. An Integrated Pathogenetic Model of Primary Biliary Cholangitis. Livers 2025, 5, 15. https://doi.org/10.3390/livers5020015
Kouroumalis E, Tsomidis I, Voumvouraki A. An Integrated Pathogenetic Model of Primary Biliary Cholangitis. Livers. 2025; 5(2):15. https://doi.org/10.3390/livers5020015
Chicago/Turabian StyleKouroumalis, Elias, Ioannis Tsomidis, and Argyro Voumvouraki. 2025. "An Integrated Pathogenetic Model of Primary Biliary Cholangitis" Livers 5, no. 2: 15. https://doi.org/10.3390/livers5020015
APA StyleKouroumalis, E., Tsomidis, I., & Voumvouraki, A. (2025). An Integrated Pathogenetic Model of Primary Biliary Cholangitis. Livers, 5(2), 15. https://doi.org/10.3390/livers5020015