The Intestinal Thread of Fate: How the Microbiota Shapes the Story of Liver Disease

 , , , and
, , , and 
Abstract
1. Introduction
2. Literature Research Methodology
3. Risk Factors and Predisposition for MASLD
3.1. Genetic and Epigenetic Factors
3.1.1. PNPLA3
3.1.2. TM6SF2
3.1.3. GCKR
3.1.4. HSD17B13
3.1.5. Epigenetic and Epitranscriptomic Modifications
3.2. Metabolic Consequences and Other Contributing Factors
3.3. Prevalence of Lean/Non-Obese MASLD and Medication-Induced MASH

{kind=link}
| Category | Specific Factor | Main Pathogenetic Mechanisms | Clinical Effects/Risks | Ref. |
|---|---|---|---|---|
| Genetic | PNPLA3 (I148M, rs738409, G allele) | Impaired lipase activity; accumulation of triglycerides in hepatocytes; lipid droplet remodeling dysfunction | Steatosis, NASH, Fibrosis, Increased risk of HCC | [25,26,27,28,29,30,31,32] |
| PNPLA3 (rs6006460, G>T variant) | Modulates risk in protective direction | Potential protective role against hepatic lipid accumulation | [25,26,27,28,29,30,31,32] | |
| TM6SF2 (E167K, rs5842926, C>T variant) | Reduced VLDL secretion; hepatic lipid accumulation; lower total cholesterol and LDL levels | Progression to advanced hepatic fibrosis, MASLD-HCC | [33,34,35,36] | |
| GCKR (rs1260326, C>T variant) | Enhanced glycolysis and de novo lipogenesis; increased hepatic lipid accumulation | Progression to MASH and fibrosis | [37,38,39] | |
| MBOAT7 polymorphisms | Alters phospholipid remodeling; increases hepatic fat accumulation and inflammation | Associated with fibrosis and progression of MASLD | [25,26] | |
| HSD17B13 (rs72613567, T>TA variant) | Loss-of-function variant; reduced hepatic inflammation and fibrosis, especially in PNPLA3 I148M carriers | Protective against MASLD progression and HCC | [40,41,42] | |
| Epigenetic | DNA methylation | Alters gene expression; promotes pro-inflammatory and pro-fibrotic pathways | Progression from MASLD to MASH, fibrosis, HCC | [43] |
| Histone modifications | Modifies chromatin structure; impacts transcriptional activity of genes related to inflammation and fibrosis | Increased susceptibility to MASLD and progression to advanced liver disease | [43] | |
| Epitranscriptomic | m6A RNA modification | Regulates RNA stability and translation; affects lipid metabolism and oncogenic pathways | Promotes MASLD progression and HCC development | [44] |
| METTL3 (m6A writer) and YTHDF1 (m6A reader) | Modulate gene expression; activate oncogenic signals (e.g., EZH2, IL-6) | Immune dysfunction, enhanced tumorigenesis, risk of HCC | [43,44,45] | |
| Metabolic Syndrome | Central obesity (increased waist circumference) | Visceral adipocyte hypertrophy; increased FFAs; portal delivery of lipids to the liver | Steatosis, mitochondrial dysfunction, ROS production, hepatocyte apoptosis | [46,47,48,49,50] |
| Insulin resistance (IR) | Promotes de novo lipogenesis; impairs fatty acid oxidation; perpetuates hyperglycemia and hyperinsulinemia | Inflammatory and fibrotic liver damage; progression to MASH | [51,52,53,54] | |
| Hypertriglyceridemia | Increases toxic lipid species; generates small dense VLDL and LDL particles with high atherogenic potential | Oxidative stress, inflammation, liver and cardiovascular disease | [55,56,57,58] | |
| Low HDL-C | Impaired reverse cholesterol transport; pro-inflammatory HDL phenotype with reduced ApoAI and ApoAII levels | Systemic inflammation; hepatic lipid accumulation | [56,57,58] | |
| Impaired fasting glucose/Type 2 Diabetes Mellitus (T2DM) | Chronic hyperinsulinemia; promotes pro-inflammatory state; exacerbates hepatic lipid deposition | Accelerated liver injury, steatosis, fibrosis | [63,64,65] | |
| Arterial hypertension | Activation of RAAS; vascular fibrosis; decreased NO synthesis; impaired hepatic microcirculation | Worsens metabolic, cardiovascular, and hepatic dysfunction | [66,67,68,69,70] | |
| Lean/Non-obese MASLD | Increased visceral adiposity | Excess intra-abdominal fat despite normal BMI; metabolic abnormalities (IR, dyslipidemia) | Hepatic fat accumulation, progression to steatohepatitis and fibrosis | [8,12] |
| Subtle insulin resistance | Mild impairment in insulin action, undetected by conventional markers | Contributes to hepatic lipid accumulation | [50,51] | |
| Dyslipidemia | Elevated triglycerides, low HDL-C | Promotes hepatic steatosis | [53,54,55,56,57] | |
| Specific genetic and epigenetic factors unique to lean MASLD | Genetic variants distinct from classic MASLD | Different pathogenetic pathways; tailored diagnostic and therapeutic approaches | [8,12] | |
| Drug-induced MASH | Tamoxifen | Induces hepatic steatosis via altered lipid metabolism and mitochondrial dysfunction | Drug-induced MASLD/MASH | [46] |
| Corticosteroids | Increases gluconeogenesis and lipogenesis; promotes IR | Hepatic steatosis, progression to MASH | [46] | |
| Antipsychotics (e.g., olanzapine) | Promotes weight gain, IR, and dyslipidemia; direct hepatotoxic effects | MASLD/MASH secondary to pharmacologic agents | [46] | |
| Other Contributing Factors | High-calorie diet | Excess caloric intake; stimulates de novo lipogenesis | Hepatic steatosis, progression to MASLD | [45,46,47,48,49,50,51,52,53,54] |
| Physical inactivity | Reduced energy expenditure; promotes weight gain and IR | Increased risk of MASLD | [45,46,47,48,49,50,51,52,53,54] | |
| Intestinal dysbiosis | Alters gut–liver axis; increases intestinal permeability and endotoxemia | Inflammation, hepatic injury, progression to MASLD | [15] |
4. Pathogenesis of MASLD
5. Intestinal Barriers
6. Gut Microbiota and Microbiome
7. Alterations in Intestinal Barriers in MASLD Pathogenesis
8. Gut Dysbiosis in the Pathogenesis of MASLD
9. Dietary Approach and Gut Microbiota Modulation in the Management of MASLD: Evidence and Perspectives
9.1. Dietary Interventions in MASLD
9.2. Role of Exercise in Modulating the Gut Microbiota
9.3. Antibiotics and Gut Microbiota Modulation in MASLD
9.4. Probiotics, Prebiotics, and Synbiotics in MASLD Management
9.5. Fecal Microbiota Transplantation in MASLD
10. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Eslam, M.; Newsome, P.N.; Sarin, S.K.; Anstee, Q.M.; Targher, G.; Romero-Gomez, M.; Zelber-Sagi, S.; Wai-Sun Wong, V.; Dufour, J.F.; Schattenberg, J.M.; et al. A New Definition for Metabolic Dysfunction-Associated Fatty Liver Disease: An International Expert Consensus Statement. J. Hepatol. 2020, 73, 202–209. [Google Scholar] [CrossRef] [PubMed]
- Fahed, G.; Aoun, L.; Bou Zerdan, M.; Allam, S.; Bou Zerdan, M.; Bouferraa, Y.; Assi, H.I. Metabolic Syndrome: Updates on Pathophysiology and Management in 2021. Int. J. Mol. Sci. 2022, 23, 786. [Google Scholar] [CrossRef] [PubMed]
- Chalasani, N.; Younossi, Z.; Lavine, J.E.; Charlton, M.; Cusi, K.; Rinella, M.; Harrison, S.A.; Brunt, E.M.; Sanyal, A.J. The Diagnosis and Management of Nonalcoholic Fatty Liver Disease: Practice Guidance from the American Association for the Study of Liver Diseases. Hepatology 2018, 67, 328–357. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.M.; Golabi, P.; Paik, J.M.; Henry, A.; Van Dongen, C.; Henry, L. The Global Epidemiology of Nonalcoholic Fatty Liver Disease (NAFLD) and Nonalcoholic Steatohepatitis (NASH): A Systematic Review. Hepatology 2023, 77, 1335–1347. [Google Scholar] [CrossRef]
- Estes, C.; Anstee, Q.M.; Arias-Loste, M.T.; Bantel, H.; Bellentani, S.; Caballeria, J.; Colombo, M.; Craxi, A.; Crespo, J.; Day, C.P.; et al. Modeling NAFLD Disease Burden in China, France, Germany, Italy, Japan, Spain, United Kingdom, and United States for the Period 2016–2030. J. Hepatol. 2018, 69, 896–904. [Google Scholar] [CrossRef]
- Targher, G.; Corey, K.E.; Byrne, C.D. NAFLD, and Cardiovascular and Cardiac Diseases: Factors Influencing Risk, Prediction and Treatment. Diabetes Metab. 2021, 47, 101215. [Google Scholar] [CrossRef]
- Adams, L.A.; Anstee, Q.M.; Tilg, H.; Targher, G. Non-Alcoholic Fatty Liver Disease and Its Relationship with Cardiovascular Disease and Other Extrahepatic Diseases. Gut 2017, 66, 1138–1153. [Google Scholar] [CrossRef]
- Li, M.; Chen, W.; Deng, Y.; Xie, W. Impacts of Cardiometabolic Risk Factors and Alcohol Consumption on All-Cause Mortality among MASLD and Its Subgroups. Nutr. Metab. Cardiovasc. Dis. 2024, 34, 2085–2094. [Google Scholar] [CrossRef]
- Vetrano, E.; Rinaldi, L.; Mormone, A.; Giorgione, C.; Galiero, R.; Caturano, A.; Nevola, R.; Marfella, R.; Sasso, F.C. Non-Alcoholic Fatty Liver Disease (NAFLD), Type 2 Diabetes, and Non-Viral Hepatocarcinoma: Pathophysiological Mechanisms and New Therapeutic Strategies. Biomedicines 2023, 11, 468. [Google Scholar] [CrossRef]
- Friedman, S.L.; Neuschwander-Tetri, B.A.; Rinella, M.; Sanyal, A.J. Mechanisms of NAFLD Development and Therapeutic Strategies. Nat. Med. 2018, 24, 908–922. [Google Scholar] [CrossRef]
- Byrne, C.D.; Targher, G. NAFLD: A Multisystem Disease. J. Hepatol. 2015, 62, S47–S64. [Google Scholar] [CrossRef] [PubMed]
- Salvatore, T.; Galiero, R.; Caturano, A.; Rinaldi, L.; Criscuolo, L.; Di Martino, A.; Albanese, G.; Vetrano, E.; Catalini, C.; Sardu, C.; et al. Current Knowledge on the Pathophysiology of Lean/Normal-Weight Type 2 Diabetes. Int. J. Mol. Sci. 2022, 24, 658. [Google Scholar] [CrossRef] [PubMed]
- Tilg, H.; Adolph, T.E.; Dudek, M.; Knolle, P. Non-Alcoholic Fatty Liver Disease: The Interplay Between Metabolism, Microbes and Immunity. Nat. Metab. 2021, 3, 1596–1607. [Google Scholar] [CrossRef] [PubMed]
- Schnabl, B.; Brenner, D.A. Interactions Between the Intestinal Microbiome and Liver Diseases. Gastroenterology 2014, 146, 1513–1524. [Google Scholar] [CrossRef]
- Albillos, A.; De Gottardi, A.; Rescigno, M. The Gut-Liver Axis in Liver Disease: Pathophysiological Basis for Therapy. J. Hepatol. 2020, 72, 558–577. [Google Scholar] [CrossRef]
- Sharpton, S.R.; Schnabl, B.; Knight, R.; Loomba, R. Current Concepts, Opportunities, and Challenges of Gut Microbiome-Based Personalized Medicine in Nonalcoholic Fatty Liver Disease. Cell Metab. 2021, 33, 21–32. [Google Scholar] [CrossRef]
- Miele, L.; Marrone, G.; Lauritano, C.; Cefalo, C.; Gasbarrini, A.; Day, C.; Grieco, A. Gut-Liver Axis and Microbiota in NAFLD: Insight Pathophysiology for Novel Therapeutic Target. Curr. Pharm. Des. 2013, 19, 5314–5324. [Google Scholar] [CrossRef]
- Aron-Wisnewsky, J.; Gaborit, B.; Dutour, A.; Clement, K. Gut Microbiota and Non-Alcoholic Fatty Liver Disease: New Insights. Clin. Microbiol. Infect. 2013, 19, 338–348. [Google Scholar] [CrossRef]
- Miele, L.; Valenza, V.; La Torre, G.; Montalto, M.; Cammarota, G.; Ricci, R.; Mascianà, R.; Forgione, A.; Gabrieli, M.L.; Perotti, G.; et al. Increased Intestinal Permeability and Tight Junction Alterations in Nonalcoholic Fatty Liver Disease. Hepatology 2009, 49, 1877–1887. [Google Scholar] [CrossRef]
- Zampino, R.; Marrone, A.; Rinaldi, L.; Guerrera, B.; Nevola, R.; Boemio, A.; Iuliano, N.; Giordano, M.; Passariello, N.; Sasso, F.C.; et al. Endotoxinemia Contributes to Steatosis, Insulin Resistance and Atherosclerosis in Chronic Hepatitis C: The Role of Pro-Inflammatory Cytokines and Oxidative Stress. Infection 2018, 46, 793–799. [Google Scholar] [CrossRef]
- Borrelli, A.; Bonelli, P.; Tuccillo, F.M.; Goldfine, I.D.; Evans, J.L.; Buonaguro, F.M.; Mancini, A. Role of Gut Microbiota and Oxidative Stress in the Progression of Non-Alcoholic Fatty Liver Disease to Hepatocarcinoma: Current and Innovative Therapeutic Approaches. Redox Biol. 2018, 15, 467–479. [Google Scholar] [CrossRef] [PubMed]
- Lombardi, R.; Petta, S.; Pisano, G.; Dongiovanni, P.; Rinaldi, L.; Adinolfi, L.E.; Acierno, C.; Valenti, L.; Boemi, R.; Spatola, F.; et al. FibroScan Identifies Patients with Nonalcoholic Fatty Liver Disease and Cardiovascular Damage. Clin. Gastroenterol. Hepatol. 2020, 18, 517–519. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Roberts, A.B.; Buffa, J.A.; Levison, B.S.; Zhu, W.; Org, E.; Gu, X.; Huang, Y.; Zamanian-Daryoush, M.; Culley, M.K.; et al. Non-Lethal Inhibition of Gut Microbial Trimethylamine Production for the Treatment of Atherosclerosis. Cell 2015, 163, 1585–1595. [Google Scholar] [CrossRef] [PubMed]
- Martin-Grau, M.; Monleón, D. The Role of Microbiota-Related Co-Metabolites in MASLD Progression: A Narrative Review. Curr. Issues Mol. Biol. 2024, 46, 6377–6389. [Google Scholar] [CrossRef]
- Trépo, E.; Valenti, L. Update on NAFLD Genetics: From New Variants to the Clinic. J. Hepatol. 2020, 72, 1196–1209. [Google Scholar] [CrossRef]
- Targher, G.; Corey, K.E.; Byrne, C.D.; Roden, M. The Complex Link between NAFLD and Type 2 Diabetes Mellitus—Mechanisms and Treatments. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 599–612. [Google Scholar] [CrossRef]
- Sookoian, S. PNPLA3, the Triacylglycerol Synthesis/Hydrolysis/Storage Dilemma, and Nonalcoholic Fatty Liver Disease. World J. Gastroenterol. 2012, 18, 6018. [Google Scholar] [CrossRef]
- Pingitore, P.; Romeo, S. The Role of PNPLA3 in Health and Disease. Biochim. Biophys. Acta BBA—Mol. Cell Biol. Lipids 2019, 1864, 900–906. [Google Scholar] [CrossRef]
- Qadri, S.; Lallukka-Brück, S.; Luukkonen, P.K.; Zhou, Y.; Gastaldelli, A.; Orho-Melander, M.; Sammalkorpi, H.; Juuti, A.; Penttilä, A.K.; Perttilä, J.; et al. The PNPLA3-I148M Variant Increases Polyunsaturated Triglycerides in Human Adipose Tissue. Liver Int. 2020, 40, 2128–2138. [Google Scholar] [CrossRef]
- Wang, Y.; Kory, N.; BasuRay, S.; Cohen, J.C.; Hobbs, H.H. PNPLA3, CGI-58, and Inhibition of Hepatic Triglyceride Hydrolysis in Mice. Hepatology 2019, 69, 2427–2441. [Google Scholar] [CrossRef]
- Liu, Y.L.; Patman, G.L.; Leathart, J.B.S.; Piguet, A.C.; Burt, A.D.; Dufour, J.F.; Day, C.P.; Daly, A.K.; Reeves, H.L.; Anstee, Q.M. Carriage of the PNPLA3 rs738409 C>G Polymorphism Confers an Increased Risk of Non-Alcoholic Fatty Liver Disease-Associated Hepatocellular Carcinoma. J. Hepatol. 2014, 61, 75–81. [Google Scholar] [CrossRef] [PubMed]
- Romeo, S.; Kozlitina, J.; Xing, C.; Pertsemlidis, A.; Cox, D.; Pennacchio, L.A.; Boerwinkle, E.; Cohen, J.C.; Hobbs, H.H. Genetic Variation in PNPLA3 Confers Susceptibility to Nonalcoholic Fatty Liver Disease. Nat. Genet. 2008, 40, 1461–1465. [Google Scholar] [CrossRef] [PubMed]
- Sharma, D.; Mandal, P. NAFLD: Genetics and Its Clinical Implications. Clin. Res. Hepatol. Gastroenterol. 2022, 46, 102003. [Google Scholar] [CrossRef] [PubMed]
- Newberry, E.P.; Hall, Z.; Xie, Y.; Molitor, E.A.; Bayguinov, P.O.; Strout, G.W.; Fitzpatrick, J.A.J.; Brunt, E.M.; Griffin, J.L.; Davidson, N.O. Liver-Specific Deletion of Mouse Tm6sf2 Promotes Steatosis, Fibrosis, and Hepatocellular Cancer. Hepatology 2021, 74, 1203–1219. [Google Scholar] [CrossRef]
- O’Hare, E.A.; Yang, R.; Yerges-Armstrong, L.M.; Sreenivasan, U.; McFarland, R.; Leitch, C.C.; Wilson, M.H.; Narina, S.; Gorden, A.; Ryan, K.A.; et al. TM6SF2 rs58542926 Impacts Lipid Processing in Liver and Small Intestine. Hepatology 2017, 65, 1526–1542. [Google Scholar] [CrossRef]
- Liu, Y.L.; Reeves, H.L.; Burt, A.D.; Tiniakos, D.; McPherson, S.; Leathart, J.B.S.; Allison, M.E.; Alexander, G.J.; Piguet, A.C.; Anty, R.; et al. TM6SF2 rs58542926 Influences Hepatic Fibrosis Progression in Patients with Non-Alcoholic Fatty Liver Disease. Nat. Commun. 2014, 5, 4309. [Google Scholar] [CrossRef]
- Peter, A.; Stefan, N.; Cegan, A.; Walenta, M.; Wagner, S.; Königsrainer, A.; Machicao, F.; Schick, F.; Häring, H.U.; Schleicher, E. Hepatic Glucokinase Expression Is Associated with Lipogenesis and Fatty Liver in Humans. J. Clin. Endocrinol. Metab. 2011, 96, E1126–E1130. [Google Scholar] [CrossRef]
- Meroni, M.; Longo, M.; Tria, G.; Dongiovanni, P. Genetics Is of the Essence to Face NAFLD. Biomedicines 2021, 9, 1359. [Google Scholar] [CrossRef]
- Santoro, N.; Zhang, C.K.; Zhao, H.; Pakstis, A.J.; Kim, G.; Kursawe, R.; Dykas, D.J.; Bale, A.E.; Giannini, C.; Pierpont, B.; et al. Variant in the Glucokinase Regulatory Protein (GCKR) Gene Is Associated with Fatty Liver in Obese Children and Adolescents. Hepatology 2012, 55, 781–789. [Google Scholar] [CrossRef]
- Ma, Y.; Belyaeva, O.V.; Brown, P.M.; Fujita, K.; Valles, K.; Karki, S.; de Boer, Y.S.; Koh, C.; Chen, Y.; Du, X.; et al. 17-Beta Hydroxysteroid Dehydrogenase 13 Is a Hepatic Retinol Dehydrogenase Associated with Histological Features of Nonalcoholic Fatty Liver Disease. Hepatology 2019, 69, 1504–1519. [Google Scholar] [CrossRef]
- Abul-Husn, N.S.; Cheng, X.; Li, A.H.; Xin, Y.; Schurmann, C.; Stevis, P.; Liu, Y.; Kozlitina, J.; Stender, S.; Wood, G.C.; et al. A Protein-Truncating HSD17B13 Variant and Protection from Chronic Liver Disease. N. Engl. J. Med. 2018, 378, 1096–1106. [Google Scholar] [CrossRef] [PubMed]
- Berger, S.L.; Kouzarides, T.; Shiekhattar, R.; Shilatifard, A. An Operational Definition of Epigenetics. Genes Dev. 2009, 23, 781–783. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Chen, H.; Zhang, X.; Liu, W.; Ding, Y.; Huang, D.; Zhai, J.; Wei, W.; Wen, J.; Chen, D.; et al. METTL3 Drives NAFLD-Related Hepatocellular Carcinoma and Is a Therapeutic Target for Boosting Immunotherapy. Cell Rep. Med. 2023, 4, 101144. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Zhu, L.; Liang, C.; Huang, X.; Liu, Z.; Huo, J.; Zhang, Y.; Zhang, Y.; Chen, L.; Xu, H.; et al. Targeting N6-Methyladenosine Reader YTHDF1 with siRNA Boosts Antitumor Immunity in NASH-HCC by Inhibiting EZH2-IL-6 Axis. J. Hepatol. 2023, 79, 1185–1200. [Google Scholar] [CrossRef]
- Chan, W.K.; Chuah, K.H.; Rajaram, R.B.; Lim, L.L.; Ratnasingam, J.; Vethakkan, S.R. Metabolic Dysfunction-Associated Steatotic Liver Disease (MASLD): A State-of-the-Art Review. J. Obes. Metab. Syndr. 2023, 32, 197–213. [Google Scholar] [CrossRef]
- European Association for the Study of the Liver (EASL); European Association for the Study of Diabetes (EASD); European Association for the Study of Obesity (EASO). EASL–EASD–EASO Clinical Practice Guidelines for the Management of Non-Alcoholic Fatty Liver Disease. J. Hepatol. 2016, 64, 1388–1402. [Google Scholar] [CrossRef]
- Castro, A.V.B.; Kolka, C.M.; Kim, S.P.; Bergman, R.N. Obesity, Insulin Resistance and Comorbidities: Mechanisms of Association. Arq. Bras. Endocrinol. Metabol. 2014, 58, 600–609. [Google Scholar] [CrossRef]
- Rampanelli, E.; Ochodnicky, P.; Vissers, J.P.; Butter, L.M.; Claessen, N.; Calcagni, A.; Kors, L.; Gethings, L.A.; Bakker, S.J.; de Borst, M.H.; et al. Excessive Dietary Lipid Intake Provokes an Acquired Form of Lysosomal Lipid Storage Disease in the Kidney. J. Pathol. 2018, 246, 470–484. [Google Scholar] [CrossRef]
- Ye, J. Mechanisms of Insulin Resistance in Obesity. Front. Med. 2013, 7, 14–24. [Google Scholar] [CrossRef]
- Hutchison, A.L.; Tavaglione, F.; Romeo, S.; Charlton, M. Endocrine Aspects of Metabolic Dysfunction-Associated Steatotic Liver Disease (MASLD): Beyond Insulin Resistance. J. Hepatol. 2023, 79, 1524–1541. [Google Scholar] [CrossRef]
- Hamdy, O. The Role of Adipose Tissue as an Endocrine Gland. Curr. Diab. Rep. 2005, 5, 317–319. [Google Scholar] [CrossRef] [PubMed]
- Virtue, S.; Vidal-Puig, A. Adipose Tissue Expandability, Lipotoxicity and the Metabolic Syndrome—An Allostatic Perspective. Biochim. Biophys. Acta BBA—Mol. Cell Biol. Lipids 2010, 1801, 338–349. [Google Scholar] [CrossRef] [PubMed]
- Yaskolka Meir, A.; Tene, L.; Cohen, N.; Shelef, I.; Schwarzfuchs, D.; Gepner, Y.; Zelicha, H.; Rein, M.; Bril, N.; Serfaty, D.; et al. Intrahepatic Fat, Abdominal Adipose Tissues, and Metabolic State: Magnetic Resonance Imaging Study. Diabetes Metab. Res. Rev. 2017, 33, e2888. [Google Scholar] [CrossRef] [PubMed]
- Rayner, J.J.; Banerjee, R.; Holloway, C.J.; Lewis, A.J.M.; Peterzan, M.A.; Francis, J.M.; Neubauer, S.; Rider, O.J. The Relative Contribution of Metabolic and Structural Abnormalities to Diastolic Dysfunction in Obesity. Int. J. Obes. 2018, 42, 441–447. [Google Scholar] [CrossRef]
- Murakami, T.; Michelagnoli, S.; Longhi, R.; Gianfranceschi, G.; Pazzucconi, F.; Calabresi, L.; Sirtori, C.R.; Franceschini, G. Triglycerides Are Major Determinants of Cholesterol Esterification/Transfer and HDL Remodeling in Human Plasma. Arterioscler. Thromb. Vasc. Biol. 1995, 15, 1819–1828. [Google Scholar] [CrossRef]
- Shao, B.; Heinecke, J.W. Quantifying HDL Proteins by Mass Spectrometry: How Many Proteins Are There and What Are Their Functions? Expert Rev. Proteom. 2018, 15, 31–40. [Google Scholar] [CrossRef]
- Cohen, D.; Fisher, E. Lipoprotein Metabolism, Dyslipidemia, and Nonalcoholic Fatty Liver Disease. Semin. Liver Dis. 2013, 33, 380–388. [Google Scholar]
- Caturano, A.; Galiero, R.; Loffredo, G.; Vetrano, E.; Medicamento, G.; Acierno, C.; Rinaldi, L.; Marrone, A.; Salvatore, T.; Monda, M.; et al. Effects of a Combination of Empagliflozin Plus Metformin vs. Metformin Monotherapy on NAFLD Progression in Type 2 Diabetes: The IMAGIN Pilot Study. Biomedicines 2023, 11, 322. [Google Scholar] [CrossRef]
- Stehouwer, C.D.A.; Henry, R.M.A.; Ferreira, I. Arterial Stiffness in Diabetes and the Metabolic Syndrome: A Pathway to Cardiovascular Disease. Diabetologia 2008, 51, 527–539. [Google Scholar] [CrossRef]
- Marušić, M.; Paić, M.; Knobloch, M.; Liberati Pršo, A.M. NAFLD, Insulin Resistance, and Diabetes Mellitus Type 2. Can. J. Gastroenterol. Hepatol. 2021, 2021, 6613827. [Google Scholar] [CrossRef]
- Safar, M.E.; Asmar, R.; Benetos, A.; Blacher, J.; Boutouyrie, P.; Lacolley, P.; Laurent, S.; London, G.; Pannier, B.; Protogerou, A.; et al. Interaction Between Hypertension and Arterial Stiffness: An Expert Reappraisal. Hypertension 2018, 72, 796–805. [Google Scholar] [CrossRef] [PubMed]
- Rizzoni, D.; Porteri, E.; Guelfi, D.; Muiesan, M.L.; Valentini, U.; Cimino, A.; Girelli, A.; Rodella, L.; Bianchi, R.; Sleiman, I.; et al. Structural Alterations in Subcutaneous Small Arteries of Normotensive and Hypertensive Patients With Non–Insulin-Dependent Diabetes Mellitus. Circulation 2001, 103, 1238–1244. [Google Scholar] [CrossRef] [PubMed]
- Day, C.P.; James, O.F.W. Steatohepatitis: A Tale of Two “Hits”? Gastroenterology 1998, 114, 842–845. [Google Scholar] [CrossRef] [PubMed]
- Buzzetti, E.; Pinzani, M.; Tsochatzis, E.A. The Multiple-Hit Pathogenesis of Non-Alcoholic Fatty Liver Disease (NAFLD). Metabolism 2016, 65, 1038–1048. [Google Scholar] [CrossRef]
- Schwärzler, J.; Grabherr, F.; Grander, C.; Adolph, T.E.; Tilg, H. The Pathophysiology of MASLD: An Immunometabolic Perspective. Expert Rev. Clin. Immunol. 2024, 20, 375–386. [Google Scholar] [CrossRef]
- Hotamisligil, G.S. Inflammation and Metabolic Disorders. Nature 2006, 444, 860–867. [Google Scholar] [CrossRef]
- Mladenić, K.; Lenartić, M.; Marinović, S.; Polić, B.; Wensveen, F.M. The “Domino Effect” in MASLD: The Inflammatory Cascade of Steatohepatitis. Eur. J. Immunol. 2024, 54, 2149641. [Google Scholar] [CrossRef]
- Duan, Y.; Pan, X.; Luo, J.; Xiao, X.; Li, J.; Bestman, P.L.; Luo, M. Association of Inflammatory Cytokines with Non-Alcoholic Fatty Liver Disease. Front. Immunol. 2022, 13, 880298. [Google Scholar] [CrossRef]
- Norton, L.; Shannon, C.; Gastaldelli, A.; DeFronzo, R.A. Insulin: The Master Regulator of Glucose Metabolism. Metabolism 2022, 129, 155142. [Google Scholar] [CrossRef]
- Begriche, K.; Igoudjil, A.; Pessayre, D.; Fromenty, B. Mitochondrial Dysfunction in NASH: Causes, Consequences and Possible Means to Prevent It. Mitochondrion 2006, 6, 1–28. [Google Scholar] [CrossRef]
- Bansal, S.K.; Bansal, M.B. Pathogenesis of MASLD and MASH—Role of Insulin Resistance and Lipotoxicity. Aliment. Pharmacol. Ther. 2024, 59, S10–S22. Available online: https://onlinelibrary.wiley.com/doi/10.1111/apt.17930 (accessed on 3 December 2024). [CrossRef] [PubMed]
- Li, Y.; Yang, P.; Ye, J.; Xu, Q.; Wu, J.; Wang, Y. Updated Mechanisms of MASLD Pathogenesis. Lipids Health Dis. 2024, 23, 117. [Google Scholar] [CrossRef] [PubMed]
- Polimeni, L. Oxidative Stress: New Insights on the Association of Non-Alcoholic Fatty Liver Disease and Atherosclerosis. World J. Hepatol. 2015, 7, 1325–1335. [Google Scholar] [CrossRef] [PubMed]
- Sjøgaard-Frich, L.M.; Henriksen, M.S.; Lam, S.M.; Birkbak, F.J.; Czaplinska, D.; Flinck, M.; Pedersen, S.F. NHE1 Regulation in NAFLD In Vitro Contributes to Hepatocyte Injury and HSC Crosstalk. J. Endocrinol. 2024, 263, e240099. [Google Scholar] [CrossRef]
- Sunny, N.E.; Parks, E.J.; Browning, J.D.; Burgess, S.C. Excessive Hepatic Mitochondrial TCA Cycle and Gluconeogenesis in Humans with Nonalcoholic Fatty Liver Disease. Cell Metab. 2011, 14, 804–810. [Google Scholar] [CrossRef]
- Radosavljevic, T.; Brankovic, M.; Samardzic, J.; Djuretić, J.; Vukicevic, D.; Vucevic, D.; Jakovljevic, V. Altered Mitochondrial Function in MASLD: Key Features and Promising Therapeutic Approaches. Antioxidants 2024, 13, 906. [Google Scholar] [CrossRef]
- Ajoolabady, A.; Kaplowitz, N.; Lebeaupin, C.; Kroemer, G.; Kaufman, R.J.; Malhi, H.; Ren, J. Endoplasmic Reticulum Stress in Liver Diseases. Hepatology 2023, 77, 619–639. [Google Scholar] [CrossRef]
- Almanza, A.; Carlesso, A.; Chintha, C.; Creedican, S.; Doultsinos, D.; Leuzzi, B.; Luís, A.; McCarthy, N.; Montibeller, L.; More, S.; et al. Endoplasmic Reticulum Stress Signaling—From Basic Mechanisms to Clinical Applications. FEBS J. 2019, 286, 241–278. [Google Scholar] [CrossRef]
- Feldstein, A.E.; Werneburg, N.W.; Canbay, A.; Guicciardi, M.E.; Bronk, S.F.; Rydzewski, R.; Burgart, L.J.; Gores, G.J. Free Fatty Acids Promote Hepatic Lipotoxicity by Stimulating TNF-α Expression via a Lysosomal Pathway. Hepatology 2004, 40, 185–194. [Google Scholar] [CrossRef]
- Baratta, F.; Pastori, D.; Del Ben, M.; Polimeni, L.; Labbadia, G.; Di Santo, S.; Piemonte, F.; Tozzi, G.; Violi, F.; Angelico, F. Reduced Lysosomal Acid Lipase Activity in Adult Patients with Non-Alcoholic Fatty Liver Disease. EBioMedicine 2015, 2, 750–754. [Google Scholar] [CrossRef]
- Frietze, K.K.; Brown, A.M.; Das, D.; Franks, R.G.; Cunningham, J.L.; Hayward, M.; Nickels, J.T., Jr. Lipotoxicity Reduces DDX58/Rig-1 Expression and Activity Leading to Impaired Autophagy and Cell Death. Autophagy 2022, 18, 142–160. [Google Scholar] [CrossRef] [PubMed]
- Kvansakul, M.; Hinds, M.G. The Bcl-2 Family: Structures, Interactions and Targets for Drug Discovery. Apoptosis 2015, 20, 136–150. [Google Scholar] [CrossRef] [PubMed]
- Alkhouri, N.; Carter-Kent, C.; Feldstein, A.E. Apoptosis in Nonalcoholic Fatty Liver Disease: Diagnostic and Therapeutic Implications. Expert Rev. Gastroenterol. Hepatol. 2011, 5, 201–212. [Google Scholar] [CrossRef] [PubMed]
- Flores-Romero, H.; Hohorst, L.; John, M.; Albert, M.; King, L.E.; Beckmann, L.; Szabo, T.; Hertlein, V.; Luo, X.; Villunger, A.; et al. BCL-2-Family Protein tBID Can Act as a BAX-Like Effector of Apoptosis. EMBO J. 2022, 41, e108690. [Google Scholar] [CrossRef]
- Hartman, L.; Czyz, M. Pro-Apoptotic Activity of BH3-Only Proteins and BH3 Mimetics: From Theory to Potential Cancer Therapy. Anticancer Agents Med. Chem. 2012, 12, 966–981. [Google Scholar] [CrossRef]
- Peleman, C.; Hellemans, S.; Veeckmans, G.; Arras, W.; Zheng, H.; Koeken, I.; Van San, E.; Hassannia, B.; Walravens, M.; Kayirangwa, E.; et al. Ferroptosis is a Targetable Detrimental Factor in Metabolic Dysfunction-Associated Steatotic Liver Disease. Cell Death Differ. 2024, 31, 1113–1126. [Google Scholar] [CrossRef]
- Qi, J.; Kim, J.W.; Zhou, Z.; Lim, C.W.; Kim, B. Ferroptosis Affects the Progression of Nonalcoholic Steatohepatitis via the Modulation of Lipid Peroxidation-Mediated Cell Death in Mice. Am. J. Pathol. 2020, 190, 68–81. [Google Scholar] [CrossRef]
- Tsurusaki, S.; Tsuchiya, Y.; Koumura, T.; Nakasone, M.; Sakamoto, T.; Matsuoka, M.; Imai, H.; Yuet-Yin Kok, C.; Okochi, H.; Nakano, H.; et al. Hepatic Ferroptosis Plays an Important Role as the Trigger for Initiating Inflammation in Nonalcoholic Steatohepatitis. Cell Death Dis. 2019, 10, 449. [Google Scholar] [CrossRef]
- Tong, J.; Lan, X.T.; Zhang, Z.; Liu, Y.; Sun, D.Y.; Wang, X.J.; Ou-Yang, S.X.; Zhuang, C.L.; Shen, F.M.; Wang, P.; et al. Ferroptosis Inhibitor Liproxstatin-1 Alleviates Metabolic Dysfunction-Associated Fatty Liver Disease in Mice: Potential Involvement of PANoptosis. Acta Pharmacol. Sin. 2023, 44, 1014–1028. [Google Scholar] [CrossRef]
- Kubes, P.; Jenne, C. Immune Responses in the Liver. Annu. Rev. Immunol. 2018, 36, 247–277. [Google Scholar] [CrossRef]
- Hammerich, L.; Tacke, F. Hepatic Inflammatory Responses in Liver Fibrosis. Nat. Rev. Gastroenterol. Hepatol. 2023, 20, 633–646. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Zhang, H.; Wang, Y.; Brown, Z.J.; Xia, Y.; Huang, Z.; Shen, C.; Hu, Z.; Beane, J.; Ansa-Addo, E.A.; et al. Regulatory T-Cell and Neutrophil Extracellular Trap Interaction Contributes to Carcinogenesis in Non-Alcoholic Steatohepatitis. J. Hepatol. 2021, 75, 1271–1283. [Google Scholar] [CrossRef] [PubMed]
- Dudek, M.; Pfister, D.; Donakonda, S.; Filpe, P.; Schneider, A.; Laschinger, M.; Hartmann, D.; Hüser, N.; Meiser, P.; Bayerl, F.; et al. Author Correction: Auto-Aggressive CXCR6+ CD8 T Cells Cause Liver Immune Pathology in NASH. Nature 2021, 593, E14. [Google Scholar] [CrossRef] [PubMed]
- Leigh, S.J.; Morris, M.J. Diet, Inflammation and the Gut Microbiome: Mechanisms for Obesity-Associated Cognitive Impairment. Biochim. Biophys. Acta BBA—Mol. Basis Dis. 2020, 1866, 165767. [Google Scholar] [CrossRef]
- Tilg, H.; Adolph, T.E.; Trauner, M. Gut-Liver Axis: Pathophysiological Concepts and Clinical Implications. Cell Metab. 2022, 34, 1700–1718. [Google Scholar] [CrossRef]
- Cinti, S. Anatomy and Physiology of the Nutritional System. Mol. Asp. Med. 2019, 68, 101–107. [Google Scholar] [CrossRef]
- Kiela, P.R.; Ghishan, F.K. Physiology of Intestinal Absorption and Secretion. Best Pract. Res. Clin. Gastroenterol. 2016, 30, 145–159. [Google Scholar] [CrossRef]
- Mowat, A.M.; Agace, W.W. Regional Specialization Within the Intestinal Immune System. Nat. Rev. Immunol. 2014, 14, 667–685. [Google Scholar] [CrossRef]
- Shaker, A.; Rubin, D.C. Intestinal Stem Cells and Epithelial–Mesenchymal Interactions in the Crypt and Stem Cell Niche. Transl. Res. 2010, 156, 180–187. [Google Scholar] [CrossRef]
- Zheng, Z.; Wang, B. The Gut-Liver Axis in Health and Disease: The Role of Gut Microbiota-Derived Signals in Liver Injury and Regeneration. Front. Immunol. 2021, 12, 775526. [Google Scholar] [CrossRef]
- Chopyk, D.M.; Grakoui, A. Contribution of the Intestinal Microbiome and Gut Barrier to Hepatic Disorders. Gastroenterology 2020, 159, 849–863. [Google Scholar] [CrossRef] [PubMed]
- Cani, P.D.; Bibiloni, R.; Knauf, C.; Waget, A.; Neyrinck, A.M.; Delzenne, N.M.; Burcelin, R. Changes in Gut Microbiota Control Metabolic Endotoxemia-Induced Inflammation in High-Fat Diet–Induced Obesity and Diabetes in Mice. Diabetes 2008, 57, 1470–1481. [Google Scholar] [CrossRef] [PubMed]
- Violi, F.; Cammisotto, V.; Bartimoccia, S.; Pignatelli, P.; Carnevale, R.; Nocella, C. Gut-Derived Low-Grade Endotoxaemia, Atherothrombosis, and Cardiovascular Disease. Nat. Rev. Cardiol. 2023, 20, 24–37. [Google Scholar] [CrossRef] [PubMed]
- Vancamelbeke, M.; Vermeire, S. The Intestinal Barrier: A Fundamental Role in Health and Disease. Expert Rev. Gastroenterol. Hepatol. 2017, 11, 821–834. [Google Scholar] [CrossRef]
- Hansson, G.C. Mucins and the Microbiome. Annu. Rev. Biochem. 2020, 89, 769–793. [Google Scholar] [CrossRef]
- Bansil, R.; Turner, B.S. The Biology of Mucus: Composition, Synthesis, and Organization. Adv. Drug Deliv. Rev. 2018, 124, 3–15. [Google Scholar] [CrossRef]
- Cornick, S.; Tawiah, A.; Chadee, K. Roles and Regulation of the Mucus Barrier in the Gut. Tissue Barriers 2015, 3, e982426. [Google Scholar] [CrossRef]
- Vanuytsel, T.; Tack, J.; Farre, R. The Role of Intestinal Permeability in Gastrointestinal Disorders and Current Methods of Evaluation. Front. Nutr. 2021, 8, 717925. [Google Scholar] [CrossRef]
- González-Mariscal, L.; Betanzos, A.; Nava, P.; Jaramillo, B.E. Tight Junction Proteins. Prog. Biophys. Mol. Biol. 2003, 81, 1–44. [Google Scholar] [CrossRef]
- Hyun, J.; Romero, L.; Riveron, R.; Flores, C.; Kanagavelu, S.; Chung, K.D.; Alonso, A.; Sotolongo, J.; Ruiz, J.; Manukyan, A.; et al. Human Intestinal Epithelial Cells Express Interleukin-10 through Toll-Like Receptor 4-Mediated Epithelial-Macrophage Crosstalk. J. Innate Immun. 2015, 7, 87–101. [Google Scholar] [CrossRef]
- Buckley, A.; Turner, J.R. Cell Biology of Tight Junction Barrier Regulation and Mucosal Disease. Cold Spring Harb. Perspect. Biol. 2018, 10, a029314. [Google Scholar] [CrossRef] [PubMed]
- Pietrzak, B.; Tomela, K.; Olejnik-Schmidt, A.; Mackiewicz, A.; Schmidt, M. Secretory IgA in Intestinal Mucosal Secretions as an Adaptive Barrier Against Microbial Cells. Int. J. Mol. Sci. 2020, 21, 9254. [Google Scholar] [CrossRef] [PubMed]
- Yatsunenko, T.; Rey, F.E.; Manary, M.J.; Trehan, I.; Dominguez-Bello, M.G.; Contreras, M.; Magris, M.; Hidalgo, G.; Baldassano, R.N.; Anokhin, A.P.; et al. Human Gut Microbiome Viewed Across Age and Geography. Nature 2012, 486, 222–227. [Google Scholar] [CrossRef] [PubMed]
- Rowland, I.; Gibson, G.; Heinken, A.; Scott, K.; Swann, J.; Thiele, I.; Tuohy, K. Gut Microbiota Functions: Metabolism of Nutrients and Other Food Components. Eur. J. Nutr. 2018, 57, 1–24. [Google Scholar] [CrossRef]
- Jiang, L.; Fan, J.G. The Role of the Gut Microbiome in Chronic Liver Diseases: Present Insights and Future Outlook. Hepatobiliary Pancreat. Dis. Int. 2023, 22, 441–443. [Google Scholar] [CrossRef]
- Woese, C.R.; Fox, G.E. Phylogenetic Structure of the Prokaryotic Domain: The Primary Kingdoms. Proc. Natl. Acad. Sci. USA 1977, 74, 5088–5090. [Google Scholar] [CrossRef]
- Rosenberg, E.; Zilber-Rosenberg, I. Microbes Drive Evolution of Animals and Plants: The Hologenome Concept. mBio 2016, 7, e01395-15. [Google Scholar] [CrossRef]
- Davies, J. In a Map for Human Life, Count the Microbes, Too. Science 2001, 291, 2316. [Google Scholar] [CrossRef]
- The NIH HMP Working Group; Peterson, J.; Garges, S.; Giovanni, M.; McInnes, P.; Wang, L.; Schloss, J.A.; Bonazzi, V.; McEwen, J.E.; Wetterstrand, K.A.; et al. The NIH Human Microbiome Project. Genome Res. 2009, 19, 2317–2323. [Google Scholar]
- Relman, D. The Meaning and Impact of the Human Genome Sequence for Microbiology. Trends Microbiol. 2001, 9, 206–208. [Google Scholar] [CrossRef]
- El-Sayed, A.; Aleya, L.; Kamel, M. Microbiota’s Role in Health and Diseases. Environ. Sci. Pollut. Res. 2021, 28, 36967–36983. [Google Scholar] [CrossRef] [PubMed]
- Manos, J. The Human Microbiome in Disease and Pathology. APMIS 2022, 130, 690–705. [Google Scholar] [CrossRef] [PubMed]
- MetaHIT Consortium; Li, J.; Jia, H.; Cai, X.; Zhong, H.; Feng, Q.; Sunagawa, S.; Arumugam, M.; Kultima, J.R.; Prifti, E.; et al. An Integrated Catalog of Reference Genes in the Human Gut Microbiome. Nat. Biotechnol. 2014, 32, 834–841. [Google Scholar]
- Bäckhed, F.; Roswall, J.; Peng, Y.; Feng, Q.; Jia, H.; Kovatcheva-Datchary, P.; Li, Y.; Xia, Y.; Xie, H.; Zhong, H.; et al. Dynamics and Stabilization of the Human Gut Microbiome during the First Year of Life. Cell Host Microbe 2015, 17, 690–703. [Google Scholar] [CrossRef]
- Lagier, J.C.; Hugon, P.; Khelaifia, S.; Fournier, P.E.; La Scola, B.; Raoult, D. The Rebirth of Culture in Microbiology through the Example of Culturomics to Study Human Gut Microbiota. Clin. Microbiol. Rev. 2015, 28, 237–264. [Google Scholar] [CrossRef]
- Browne, H.P.; Forster, S.C.; Anonye, B.O.; Kumar, N.; Neville, B.A.; Stares, M.D.; Goulding, D.; Lawley, T.D. Culturing of ‘Uncultured’ Human Microbiota Reveals Novel Taxa and Extensive Sporulation. Nature 2016, 533, 543–546. [Google Scholar] [CrossRef]
- Metzker, M.L. Emerging Technologies in DNA Sequencing. Genome Res. 2005, 15, 1767–1776. [Google Scholar] [CrossRef]
- Le Chatelier, E.; Nielsen, T.; Qin, J.; Prifti, E.; Hildebrand, F.; Falony, G.; Almeida, M.; Arumugam, M.; Batto, J.M.; Kennedy, S.; et al. Richness of Human Gut Microbiome Correlates with Metabolic Markers. Nature 2013, 500, 541–546. [Google Scholar] [CrossRef]
- Nardone, G.; Compare, D. The Human Gastric Microbiota: Is It Time to Rethink the Pathogenesis of Stomach Diseases? United Eur. Gastroenterol. J. 2015, 3, 255–260. [Google Scholar] [CrossRef]
- El Aidy, S.; Dinan, T.G.; Cryan, J.F. Gut Microbiota: The Conductor in the Orchestra of Immune–Neuroendocrine Communication. Clin. Ther. 2015, 37, 954–967. [Google Scholar] [CrossRef]
- Eckburg, P.B.; Bik, E.M.; Bernstein, C.N.; Purdom, E.; Dethlefsen, L.; Sargent, M.; Gill, S.R.; Nelson, K.E.; Relman, D.A. Diversity of the Human Intestinal Microbial Flora. Science 2005, 308, 1635–1638. [Google Scholar] [CrossRef] [PubMed]
- Mariat, D.; Firmesse, O.; Levenez, F.; Guimarâes, V.; Sokol, H.; Doré, J.; Corthier, G.; Furet, J.P. The Firmicutes/Bacteroidetes Ratio of the Human Microbiota Changes with Age. BMC Microbiol. 2009, 9, 123. [Google Scholar] [CrossRef] [PubMed]
- Hollister, E.B.; Gao, C.; Versalovic, J. Compositional and Functional Features of the Gastrointestinal Microbiome and Their Effects on Human Health. Gastroenterology 2014, 146, 1449–1458. [Google Scholar] [CrossRef] [PubMed]
- Binda, C.; Lopetuso, L.R.; Rizzatti, G.; Gibiino, G.; Cennamo, V.; Gasbarrini, A. Actinobacteria: A Relevant Minority for the Maintenance of Gut Homeostasis. Dig. Liver Dis. 2018, 50, 421–428. [Google Scholar] [CrossRef]
- Shin, N.R.; Whon, T.W.; Bae, J.W. Proteobacteria: Microbial Signature of Dysbiosis in Gut Microbiota. Trends Biotechnol. 2015, 33, 496–503. [Google Scholar] [CrossRef]
- Puljiz, Z.; Kumric, M.; Vrdoljak, J.; Martinovic, D.; Ticinovic Kurir, T.; Krnic, M.O.; Urlic, H.; Puljiz, Z.; Zucko, J.; Dumanic, P.; et al. Obesity, Gut Microbiota, and Metabolome: From Pathophysiology to Nutritional Interventions. Nutrients 2023, 15, 2236. [Google Scholar] [CrossRef]
- Vallianou, N.; Stratigou, T.; Christodoulatos, G.S.; Dalamaga, M. Understanding the Role of the Gut Microbiome and Microbial Metabolites in Obesity and Obesity-Associated Metabolic Disorders: Current Evidence and Perspectives. Curr. Obes. Rep. 2019, 8, 317–332. [Google Scholar] [CrossRef]
- Usuda, H.; Okamoto, T.; Wada, K. Leaky Gut: Effect of Dietary Fiber and Fats on Microbiome and Intestinal Barrier. Int. J. Mol. Sci. 2021, 22, 7613. [Google Scholar] [CrossRef]
- Shi, N.; Li, N.; Duan, X.; Niu, H. Interaction between the Gut Microbiome and Mucosal Immune System. Mil. Med. Res. 2017, 4, 14. [Google Scholar] [CrossRef]
- Schoeler, M.; Caesar, R. Dietary Lipids, Gut Microbiota, and Lipid Metabolism. Rev. Endocr. Metab. Disord. 2019, 20, 461–472. [Google Scholar] [CrossRef]
- Xin, D.; Zong-Shun, L.; Bang-Mao, W.; Lu, Z. Expression of Intestinal Tight Junction Proteins in Patients with Non-Alcoholic Fatty Liver Disease. Hepatogastroenterology 2014, 61, 136–140. [Google Scholar] [PubMed]
- Rahman, K.; Desai, C.; Iyer, S.S.; Thorn, N.E.; Kumar, P.; Liu, Y.; Smith, T.; Neish, A.S.; Li, H.; Tan, S.; et al. Loss of Junctional Adhesion Molecule A Promotes Severe Steatohepatitis in Mice on a Diet High in Saturated Fat, Fructose, and Cholesterol. Gastroenterology 2016, 151, 733–746.e12. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Yin, M.; Gao, J.; Yu, C.; Lin, J.; Wu, A.; Zhu, J.; Xu, C.; Liu, X. Intestinal Barrier Function in the Pathogenesis of Nonalcoholic Fatty Liver Disease. J. Clin. Transl. Hepatol. 2023, 11, 452–458. [Google Scholar] [CrossRef] [PubMed]
- Spadoni, I.; Zagato, E.; Bertocchi, A.; Paolinelli, R.; Hot, E.; Di Sabatino, A.; Caprioli, F.; Bottiglieri, L.; Oldani, A.; Viale, G.; et al. A Gut-Vascular Barrier Controls the Systemic Dissemination of Bacteria. Science 2015, 350, 830–834. [Google Scholar] [CrossRef]
- Mouries, J.; Brescia, P.; Silvestri, A.; Spadoni, I.; Sorribas, M.; Wiest, R.; Mileti, E.; Galbiati, M.; Invernizzi, P.; Adorini, L.; et al. Microbiota-Driven Gut Vascular Barrier Disruption Is a Prerequisite for Non-Alcoholic Steatohepatitis Development. J. Hepatol. 2019, 71, 1216–1228. [Google Scholar] [CrossRef]
- Cui, Y.; Wang, Q.; Chang, R.; Zhou, X.; Xu, C. Intestinal Barrier Function–Non-Alcoholic Fatty Liver Disease Interactions and Possible Role of Gut Microbiota. J. Agric. Food Chem. 2019, 67, 2754–2762. [Google Scholar] [CrossRef]
- Pellicciotta, M.; Rigoni, R.; Falcone, E.L.; Holland, S.M.; Villa, A.; Cassani, B. The Microbiome and Immunodeficiencies: Lessons from Rare Diseases. J. Autoimmun. 2019, 98, 132–148. [Google Scholar] [CrossRef]
- Kubes, P.; Mehal, W.Z. Sterile Inflammation in the Liver. Gastroenterology 2012, 143, 1158–1172. [Google Scholar] [CrossRef]
- McPherson, S.; Henderson, E.; Burt, A.D.; Day, C.P.; Anstee, Q.M. Serum Immunoglobulin Levels Predict Fibrosis in Patients with Non-Alcoholic Fatty Liver Disease. J. Hepatol. 2014, 60, 1055–1062. [Google Scholar] [CrossRef]
- Shalapour, S.; Lin, X.J.; Bastian, I.N.; Brain, J.; Burt, A.D.; Aksenov, A.A.; Vrbanac, A.F.; Li, W.; Perkins, A.; Matsutani, T.; et al. Inflammation-Induced IgA+ Cells Dismantle Anti-Liver Cancer Immunity. Nature 2017, 551, 340–345. [Google Scholar] [CrossRef]
- Boursier, J.; Mueller, O.; Barret, M.; Machado, M.; Fizanne, L.; Araujo-Perez, F.; Guy, C.D.; Seed, P.C.; Rawls, J.F.; David, L.A.; et al. The Severity of Nonalcoholic Fatty Liver Disease Is Associated with Gut Dysbiosis and Shift in the Metabolic Function of the Gut Microbiota. Hepatology 2016, 63, 764–775. [Google Scholar] [CrossRef] [PubMed]
- Wilcz-Villega, E.M.; McClean, S.; O’Sullivan, M.A. Mast Cell Tryptase Reduces Junctional Adhesion Molecule-A (JAM-A) Expression in Intestinal Epithelial Cells: Implications for the Mechanisms of Barrier Dysfunction in Irritable Bowel Syndrome. Am. J. Gastroenterol. 2013, 108, 1140–1151. [Google Scholar] [CrossRef] [PubMed]
- Potts, R.A.; Tiffany, C.M.; Pakpour, N.; Lokken, K.L.; Tiffany, C.R.; Cheung, K.; Tsolis, R.M.; Luckhart, S. Mast Cells and Histamine Alter Intestinal Permeability During Malaria Parasite Infection. Immunobiology 2016, 221, 468–474. [Google Scholar] [CrossRef] [PubMed]
- Tomita, K.; Teratani, T.; Yokoyama, H.; Suzuki, T.; Irie, R.; Ebinuma, H.; Saito, H.; Hokari, R.; Miura, S.; Hibi, T. Serum Immunoglobulin A Concentration Is an Independent Predictor of Liver Fibrosis in Nonalcoholic Steatohepatitis Before the Cirrhotic Stage. Dig. Dis. Sci. 2011, 56, 3648–3654. [Google Scholar] [CrossRef]
- Fang, J.; Yu, C.H.; Li, X.J.; Yao, J.M.; Fang, Z.Y.; Yoon, S.H.; Yu, W.Y. Gut Dysbiosis in Nonalcoholic Fatty Liver Disease: Pathogenesis, Diagnosis, and Therapeutic Implications. Front. Cell Infect. Microbiol. 2022, 12, 997018. [Google Scholar] [CrossRef]
- Tokuhara, D. Role of the Gut Microbiota in Regulating Non-Alcoholic Fatty Liver Disease in Children and Adolescents. Front. Nutr. 2021, 8, 700058. [Google Scholar] [CrossRef]
- Ghosh, S.; Whitley, C.S.; Haribabu, B.; Jala, V.R. Regulation of Intestinal Barrier Function by Microbial Metabolites. Cell Mol. Gastroenterol. Hepatol. 2021, 11, 1463–1482. [Google Scholar] [CrossRef]
- Brown, K.; DeCoffe, D.; Molcan, E.; Gibson, D.L. Diet-Induced Dysbiosis of the Intestinal Microbiota and the Effects on Immunity and Disease. Nutrients 2012, 4, 1095–1119. [Google Scholar] [CrossRef]
- Khan, M.J.; Gerasimidis, K.; Edwards, C.A.; Shaikh, M.G. Role of Gut Microbiota in the Aetiology of Obesity: Proposed Mechanisms and Review of the Literature. J. Obes. 2016, 2016, 7353642. [Google Scholar] [CrossRef]
- Magne, F.; Gotteland, M.; Gauthier, L.; Zazueta, A.; Pesoa, S.; Navarrete, P.; Balamurugan, R. The Firmicutes/Bacteroidetes Ratio: A Relevant Marker of Gut Dysbiosis in Obese Patients? Nutrients 2020, 12, 1474. [Google Scholar] [CrossRef]
- Duarte, S.B.M.; Stefano, J.T.; Oliveira, C.P. Microbiota and Nonalcoholic Fatty Liver Disease/Nonalcoholic Steatohepatitis (NAFLD/NASH). Ann. Hepatol. 2019, 18, 416–421. [Google Scholar] [CrossRef] [PubMed]
- Yuan, H.; Wu, X.; Wang, X.; Zhou, J.Y.; Park, S. Microbial Dysbiosis Linked to Metabolic Dysfunction-Associated Fatty Liver Disease in Asians: Prevotella copri Promotes Lipopolysaccharide Biosynthesis and Network Instability in the Prevotella Enterotype. Int. J. Mol. Sci. 2024, 25, 2183. [Google Scholar] [CrossRef] [PubMed]
- Kuraji, R.; Ye, C.; Zhao, C.; Gao, L.; Martinez, A.; Miyashita, Y.; Radaic, A.; Kamarajan, P.; Le, C.; Zhan, L.; et al. Nisin Lantibiotic Prevents NAFLD Liver Steatosis and Mitochondrial Oxidative Stress Following Periodontal Disease by Abrogating Oral, Gut and Liver Dysbiosis. NPJ Biofilms Microbiomes 2024, 10, 3. [Google Scholar] [CrossRef] [PubMed]
- Kapil, S.; Duseja, A.; Sharma, B.K.; Singla, B.; Chakraborti, A.; Das, A.; Ray, P.; Dhiman, R.K.; Chawla, Y. Small Intestinal Bacterial Overgrowth and Toll-Like Receptor Signaling in Patients with Non-Alcoholic Fatty Liver Disease. J. Gastroenterol. Hepatol. 2016, 31, 213–221. [Google Scholar] [CrossRef]
- Ganesan, R.; Gupta, H.; Jeong, J.J.; Sharma, S.P.; Won, S.M.; Oh, K.K.; Yoon, S.J.; Kim, D.J.; Suk, K.T. A Metabolomics Approach to the Validation of Predictive Metabolites and Phenotypic Expression in Non-Alcoholic Fatty Liver Disease. Life Sci. 2023, 322, 121626. [Google Scholar] [CrossRef]
- Zeng, F.; Su, X.; Liang, X.; Liao, M.; Zhong, H.; Xu, J.; Gou, W.; Zhang, X.; Shen, L.; Zheng, J.S.; et al. Gut Microbiome Features and Metabolites in Non-Alcoholic Fatty Liver Disease Among Community-Dwelling Middle-Aged and Older Adults. BMC Med. 2024, 22, 104. [Google Scholar] [CrossRef]
- Mardinoglu, A.; Ural, D.; Zeybel, M.; Yuksel, H.H.; Uhlén, M.; Borén, J. The Potential Use of Metabolic Cofactors in Treatment of NAFLD. Nutrients 2019, 11, 1578. [Google Scholar] [CrossRef]
- Quesada-Vázquez, S.; Bone, C.; Saha, S.; Triguero, I.; Colom-Pellicer, M.; Aragonès, G.; Hildebrand, F.; Del Bas, J.M.; Caimari, A.; Beraza, N.; et al. Microbiota Dysbiosis and Gut Barrier Dysfunction Associated with Non-Alcoholic Fatty Liver Disease Are Modulated by a Specific Metabolic Cofactors’ Combination. Int. J. Mol. Sci. 2022, 23, 13675. [Google Scholar] [CrossRef]
- Hayashi, T.; Yamashita, T.; Takahashi, T.; Tabata, T.; Watanabe, H.; Gotoh, Y.; Shinohara, M.; Kami, K.; Tanaka, H.; Matsumoto, K.; et al. Uncovering the Role of Gut Microbiota in Amino Acid Metabolic Disturbances in Heart Failure Through Metagenomic Analysis. Front. Cardiovasc. Med. 2021, 8, 789325. [Google Scholar] [CrossRef]
- Niu, Y.C.; Feng, R.N.; Hou, Y.; Li, K.; Kang, Z.; Wang, J.; Sun, C.H.; Li, Y. Histidine and Arginine Are Associated with Inflammation and Oxidative Stress in Obese Women. Br. J. Nutr. 2012, 108, 57–61. [Google Scholar] [CrossRef]
- Quesada-Vázquez, S.; Castells-Nobau, A.; Latorre, J.; Oliveras-Cañellas, N.; Puig-Parnau, I.; Tejera, N.; Tobajas, Y.; Baudin, J.; Hildebrand, F.; Beraza, N.; et al. Potential Therapeutic Implications of Histidine Catabolism by the Gut Microbiota in NAFLD Patients with Morbid Obesity. Cell Rep. Med. 2023, 4, 101341. [Google Scholar] [CrossRef] [PubMed]
- Huus, K.E.; Petersen, C.; Finlay, B.B. Diversity and Dynamism of IgA−Microbiota Interactions. Nat. Rev. Immunol. 2021, 21, 514–525. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.L.; Schnabl, B. The Gut–Liver Axis and Gut Microbiota in Health and Liver Disease. Nat. Rev. Microbiol. 2023, 21, 719–733. [Google Scholar] [CrossRef] [PubMed]
- Ohtani, N.; Kamiya, T.; Kawada, N. Recent Updates on the Role of the Gut-Liver Axis in the Pathogenesis of NAFLD/NASH, HCC, and Beyond. Hepatol. Commun. 2023, 7, 9. Available online: https://journals.lww.com/10.1097/HC9.0000000000000241 (accessed on 3 December 2024). [CrossRef]
- Ma, R.; Shi, G.; Li, Y.; Shi, H. Trimethylamine N-Oxide, Choline and Its Metabolites Are Associated with the Risk of Non-Alcoholic Fatty Liver Disease. Br. J. Nutr. 2024, 131, 1915–1923. [Google Scholar] [CrossRef]
- Martínez-Montoro, J.I.; Martín-Núñez, G.M.; González-Jiménez, A.; Garrido-Sánchez, L.; Moreno-Indias, I.; Tinahones, F.J. Interactions between the Gut Microbiome and DNA Methylation Patterns in Blood and Visceral Adipose Tissue in Subjects with Different Metabolic Characteristics. J. Transl. Med. 2024, 22, 1089. [Google Scholar] [CrossRef]
- Shirai, Y.; Yoshiji, H.; Noguchi, R.; Kaji, K.; Aihara, Y.; Douhara, A.; Moriya, K.; Namisaki, T.; Kawaratani, H.; Fukui, H. Cross Talk between Toll-Like Receptor-4 Signaling and Angiotensin-II in Liver Fibrosis Development in the Rat Model of Non-Alcoholic Steatohepatitis. J. Gastroenterol. Hepatol. 2013, 28, 723–730. [Google Scholar] [CrossRef]
- Li, J.; Deng, X.; Bai, T.; Wang, S.; Jiang, Q.; Xu, K. Resolvin D1 Mitigates Non-Alcoholic Steatohepatitis by Suppressing the TLR4-MyD88-Mediated NF-κB and MAPK Pathways and Activating the Nrf2 Pathway in Mice. Int. Immunopharmacol. 2020, 88, 106961. [Google Scholar] [CrossRef]
- American Diabetes Association. 5. Lifestyle Management: Standards of Medical Care in Diabetes—2019. Diabetes Care 2019, 42, S46–S60. [Google Scholar] [CrossRef]
- Plauth, M.; Bernal, W.; Dasarathy, S.; Merli, M.; Plank, L.D.; Schütz, T.; Bischoff, S.C. ESPEN Guideline on Clinical Nutrition in Liver Disease. Clin. Nutr. 2019, 38, 485–521. [Google Scholar] [CrossRef]
- Murphy, E.F.; Cotter, P.D.; Healy, S.; Marques, T.M.; O’Sullivan, O.; Fouhy, F.; Clarke, S.F.; O’Toole, P.W.; Quigley, E.M.; Stanton, C.; et al. Composition and Energy Harvesting Capacity of the Gut Microbiota: Relationship to Diet, Obesity and Time in Mouse Models. Gut 2010, 59, 1635–1642. [Google Scholar] [CrossRef] [PubMed]
- Turnbaugh, P.J.; Bäckhed, F.; Fulton, L.; Gordon, J.I. Diet-Induced Obesity Is Linked to Marked but Reversible Alterations in the Mouse Distal Gut Microbiome. Cell Host Microbe 2008, 3, 213–223. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, T.; Oishi, K. Influence of Coffee (Coffea arabica) and Galacto-Oligosaccharide Consumption on Intestinal Microbiota and the Host Responses. FEMS Microbiol. Lett. 2013, 343, 161–168. [Google Scholar] [CrossRef] [PubMed]
- Molloy, J.W.; Calcagno, C.J.; Williams, C.D.; Jones, F.J.; Torres, D.M.; Harrison, S.A. Association of Coffee and Caffeine Consumption with Fatty Liver Disease, Nonalcoholic Steatohepatitis, and Degree of Hepatic Fibrosis. Hepatology 2012, 55, 429–436. [Google Scholar] [CrossRef]
- Shen, L. Letter: Gut Microbiota Modulation Contributes to Coffee’s Benefits for Non-Alcoholic Fatty Liver Disease. Aliment. Pharmacol. Ther. 2014, 39, 1441–1442. [Google Scholar] [CrossRef]
- Clarke, S.F.; Murphy, E.F.; O’Sullivan, O.; Lucey, A.J.; Humphreys, M.; Hogan, A.; Hayes, P.; O’Reilly, M.; Jeffery, I.B.; Wood-Martin, R.; et al. Exercise and Associated Dietary Extremes Impact on Gut Microbial Diversity. Gut 2014, 63, 1913–1920. [Google Scholar] [CrossRef]
- Monda, V.; Villano, I.; Messina, A.; Valenzano, A.; Esposito, T.; Moscatelli, F.; Viggiano, A.; Cibelli, G.; Chieffi, S.; Monda, M.; et al. Exercise Modifies the Gut Microbiota with Positive Health Effects. Oxid. Med. Cell. Longev. 2017, 2017, 3831972. [Google Scholar] [CrossRef]
- Ortiz-Alvarez, L.; Xu, H.; Martinez-Tellez, B. Influence of Exercise on the Human Gut Microbiota of Healthy Adults: A Systematic Review. Clin. Transl. Gastroenterol. 2020, 11, e00126. [Google Scholar] [CrossRef]
- Reijnders, D.; Goossens, G.H.; Hermes, G.D.A.; Neis, E.P.J.G.; van der Beek, C.M.; Most, J.; Holst, J.J.; Lenaerts, K.; Kootte, R.S.; Nieuwdorp, M.; et al. Effects of Gut Microbiota Manipulation by Antibiotics on Host Metabolism in Obese Humans: A Randomized Double-Blind Placebo-Controlled Trial. Cell Metab. 2016, 24, 63–74. [Google Scholar] [CrossRef]
- Chong, C.Y.L.; Orr, D.; Plank, L.D.; Vatanen, T.; O’Sullivan, J.M.; Murphy, R. Randomized Double-Blind Placebo-Controlled Trial of Inulin with Metronidazole in Non-Alcoholic Fatty Liver Disease (NAFLD). Nutrients 2020, 12, 937. [Google Scholar] [CrossRef]
- Kakiyama, G.; Pandak, W.M.; Gillevet, P.M.; Hylemon, P.B.; Heuman, D.M.; Daita, K.; Takei, H.; Muto, A.; Nittono, H.; Ridlon, J.M.; et al. Modulation of the Fecal Bile Acid Profile by Gut Microbiota in Cirrhosis. J. Hepatol. 2013, 58, 949–955. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Razik, A.; Mousa, N.; Shabana, W.; Refaey, M.; Elzehery, R.; Elhelaly, R.; Zalata, K.; Abdelsalam, M.; Eldeeb, A.A.; Awad, M.; et al. Rifaximin in Nonalcoholic Fatty Liver Disease: Hit Multiple Targets with a Single Shot. Eur. J. Gastroenterol. Hepatol. 2018, 30, 1237–1246. [Google Scholar] [CrossRef] [PubMed]
- Gangarapu, V.; Ince, A.T.; Baysal, B.; Kayar, Y.; Kılıç, U.; Gök, Ö.; Uysal, Ö.; Şenturk, H. Efficacy of Rifaximin on Circulating Endotoxins and Cytokines in Patients with Nonalcoholic Fatty Liver Disease. Eur. J. Gastroenterol. Hepatol. 2015, 27, 840–845. [Google Scholar] [CrossRef] [PubMed]
- Cobbold, J.F.L.; Atkinson, S.; Marchesi, J.R.; Smith, A.; Wai, S.N.; Stove, J.; Shojaee-Moradie, F.; Jackson, N.; Umpleby, A.M.; Fitzpatrick, J.; et al. Rifaximin in Non-Alcoholic Steatohepatitis: An Open-Label Pilot Study. Hepatol. Res. Off. J. Jpn. Soc. Hepatol. 2018, 48, 69–77. [Google Scholar] [CrossRef]
- Escouto, G.S.; Port, G.Z.; Tovo, C.V.; Fernandes, S.A.; Peres, A.; Dorneles, G.P.; Houde, V.P.; Varin, T.V.; Pilon, G.; Marette, A.; et al. Probiotic Supplementation, Hepatic Fibrosis, and the Microbiota Profile in Patients with Nonalcoholic Steatohepatitis: A Randomized Controlled Trial. J. Nutr. 2023, 153, 1984–1993. [Google Scholar] [CrossRef]
- Manzhalii, E.; Virchenko, O.; Falalyeyeva, T.; Beregova, T.; Stremmel, W. Treatment Efficacy of a Probiotic Preparation for Non-Alcoholic Steatohepatitis: A Pilot Trial. J. Dig. Dis. 2017, 18, 698–703. [Google Scholar] [CrossRef]
- Barcelos, S.T.A.; Silva-Sperb, A.S.; Moraes, H.A.; Longo, L.; De Moura, B.C.; Michalczuk, M.T.; Uribe-Cruz, C.; Cerski, C.T.S.; da Silveira, T.R.; Dall’Alba, V.; et al. Oral 24-Week Probiotics Supplementation Did Not Decrease Cardiovascular Risk Markers in Patients with Biopsy Proven NASH: A Double-Blind Placebo-Controlled Randomized Study. Ann. Hepatol. 2023, 28, 100769. [Google Scholar] [CrossRef]
- Tarantino, G.; Finelli, C. Systematic Review on Intervention with Prebiotics/Probiotics in Patients with Obesity-Related Nonalcoholic Fatty Liver Disease. Future Microbiol. 2015, 10, 889–902. [Google Scholar] [CrossRef]
- Carpi, R.Z.; Barbalho, S.M.; Sloan, K.P.; Laurindo, L.F.; Gonzaga, H.F.; Grippa, P.C.; Zutin, T.L.M.; Girio, R.J.S.; Repetti, C.S.F.; Detregiachi, C.R.P.; et al. The Effects of Probiotics, Prebiotics and Synbiotics in Non-Alcoholic Fat Liver Disease (NAFLD) and Non-Alcoholic Steatohepatitis (NASH): A Systematic Review. Int. J. Mol. Sci. 2022, 23, 8805. [Google Scholar] [CrossRef]
- Kanchanasurakit, S.; Kositamongkol, C.; Lanoi, K.; Nunta, M.; Saetuan, T.; Chaiyakunapruk, N.; Saokaew, S.; Phisalprapa, P. Effects of Synbiotics, Probiotics, and Prebiotics on Liver Enzymes of Patients with Non-Alcoholic Fatty Liver Disease: A Systematic Review and Network Meta-Analysis. Front. Nutr. 2022, 9, 880014. [Google Scholar] [CrossRef]
- Malaguarnera, M.; Vacante, M.; Antic, T.; Giordano, M.; Chisari, G.; Acquaviva, R.; Mastrojeni, S.; Malaguarnera, G.; Mistretta, A.; Li Volti, G.; et al. Bifidobacterium longum with Fructo-Oligosaccharides in Patients with Non Alcoholic Steatohepatitis. Dig. Dis. Sci. 2012, 57, 545–553. [Google Scholar] [CrossRef] [PubMed]
- Asgharian, A.; Askari, G.; Esmailzade, A.; Feizi, A.; Mohammadi, V. The Effect of Symbiotic Supplementation on Liver Enzymes, C-Reactive Protein and Ultrasound Findings in Patients with Non-Alcoholic Fatty Liver Disease: A Clinical Trial. Int. J. Prev. Med. 2016, 7, 59. [Google Scholar] [PubMed]
- Mofidi, F.; Poustchi, H.; Yari, Z.; Nourinayyer, B.; Merat, S.; Sharafkhah, M.; Malekzadeh, R.; Hekmatdoost, A. Synbiotic Supplementation in Lean Patients with Non-Alcoholic Fatty Liver Disease: A Pilot, Randomised, Double-Blind, Placebo-Controlled, Clinical Trial. Br. J. Nutr. 2017, 117, 662–668. [Google Scholar] [CrossRef] [PubMed]
- Vaughn, B.P.; Rank, K.M.; Khoruts, A. Fecal Microbiota Transplantation: Current Status in Treatment of GI and Liver Disease. Clin. Gastroenterol. Hepatol. Off. Clin. Pract. J. Am. Gastroenterol. Assoc. 2019, 17, 353–361. [Google Scholar] [CrossRef]
- Routy, B.; Lenehan, J.G.; Miller, W.H.; Jamal, R.; Messaoudene, M.; Daisley, B.A.; Hes, C.; Al, K.F.; Martinez-Gili, L.; Punčochář, M.; et al. Fecal Microbiota Transplantation Plus Anti-PD-1 Immunotherapy in Advanced Melanoma: A Phase I Trial. Nat. Med. 2023, 29, 2121–2132. [Google Scholar] [CrossRef]
- Belvoncikova, P.; Maronek, M.; Gardlik, R. Gut Dysbiosis and Fecal Microbiota Transplantation in Autoimmune Diseases. Int. J. Mol. Sci. 2022, 23, 10729. [Google Scholar] [CrossRef]
- Craven, L.; Rahman, A.; Nair Parvathy, S.; Beaton, M.; Silverman, J.; Qumosani, K.; Hramiak, I.; Hegele, R.; Joy, T.; Meddings, J.; et al. Allogenic Fecal Microbiota Transplantation in Patients with Nonalcoholic Fatty Liver Disease Improves Abnormal Small Intestinal Permeability: A Randomized Control Trial. Am. J. Gastroenterol. 2020, 115, 1055–1065. [Google Scholar] [CrossRef]
- Xue, L.F.; Luo, W.H.; Wu, L.H.; He, X.X.; Xia, H.H.X.; Chen, Y. Fecal Microbiota Transplantation for the Treatment of Nonalcoholic Fatty Liver Disease. Explor. Res. Hypothesis Med. 2019, 4, 12–18. [Google Scholar] [CrossRef]
- Bajaj, J.S.; Fagan, A.; Gavis, E.A.; Kassam, Z.; Sikaroodi, M.; Gillevet, P.M. Long-Term Outcomes of Fecal Microbiota Transplantation in Patients with Cirrhosis. Gastroenterology 2019, 156, 1921–1923.e3. [Google Scholar] [CrossRef]
- Vrieze, A.; Van Nood, E.; Holleman, F.; Salojärvi, J.; Kootte, R.S.; Bartelsman, J.F.; Dallinga-Thie, G.M.; Ackermans, M.T.; Serlie, M.J.; Oozeer, R.; et al. Transfer of Intestinal Microbiota from Lean Donors Increases Insulin Sensitivity in Individuals with Metabolic Syndrome. Gastroenterology 2012, 143, 913–916.e7. [Google Scholar] [CrossRef]
- de Groot, P.; Scheithauer, T.; Bakker, G.J.; Prodan, A.; Levin, E.; Khan, M.T.; Herrema, H.; Ackermans, M.; Serlie, M.J.M.; de Brauw, M.; et al. Donor Metabolic Characteristics Drive Effects of Faecal Microbiota Transplantation on Recipient Insulin Sensitivity, Energy Expenditure and Intestinal Transit Time. Gut 2020, 69, 502–512. [Google Scholar] [CrossRef] [PubMed]
- Brandt, L.J.; Aroniadis, O.C. An Overview of Fecal Microbiota Transplantation: Techniques, Indications, and Outcomes. Gastrointest. Endosc. 2013, 78, 240–249. [Google Scholar] [CrossRef] [PubMed]
- Persky, S.E.; Brandt, L.J. Treatment of Recurrent Clostridium difficile-Associated Diarrhea by Administration of Donated Stool Directly Through a Colonoscope. Am. J. Gastroenterol. 2000, 95, 3283–3285. [Google Scholar] [PubMed]
- Allegretti, J.R.; Kassam, Z.; Mullish, B.H.; Chiang, A.; Carrellas, M.; Hurtado, J.; Marchesi, J.R.; McDonald, J.A.K.; Pechlivanis, A.; Barker, G.F.; et al. Effects of Fecal Microbiota Transplantation with Oral Capsules in Obese Patients. Clin. Gastroenterol. Hepatol. Off. Clin. Pract. J. Am. Gastroenterol. Assoc. 2020, 18, 855–863.e2. [Google Scholar] [CrossRef]
- Yang, J.; Tang, X.; Liang, Z.; Chen, M.; Sun, L. Taurocholic Acid Promotes Hepatic Stellate Cell Activation via S1PR2/p38 MAPK/YAP Signaling under Cholestatic Conditions. Clin. Mol. Hepatol. 2023, 29, 465–481. [Google Scholar] [CrossRef]
- Mancinelli, R.; Ceci, L.; Kennedy, L.; Francis, H.; Meadows, V.; Chen, L.; Carpino, G.; Kyritsi, K.; Wu, N.; Zhou, T.; et al. The Effects of Taurocholic Acid on Biliary Damage and Liver Fibrosis Are Mediated by Calcitonin-Gene-Related Peptide Signaling. Cells 2022, 11, 1591. [Google Scholar] [CrossRef]
- Duan, D.; Wang, M.; Han, J.; Li, M.; Wang, Z.; Zhou, S.; Xin, W.; Li, X. Advances in Multi-Omics Integrated Analysis Methods Based on the Gut Microbiome and Their Applications. Front. Microbiol. 2025, 15, 1509117. [Google Scholar] [CrossRef]
- Thakral, N.; Desalegn, H.; Diaz, L.A.; Cabrera, D.; Loomba, R.; Arrese, M.; Arab, J.P. A Precision Medicine Guided Approach to the Utilization of Biomarkers in MASLD. Semin. Liver Dis. 2024, 44, 273–286. [Google Scholar] [CrossRef]
| Intestinal Section | Predominant Bacteria | Main Functions | Ref. |
|---|---|---|---|
| Stomach | Streptococcus, Neisseria, Lactobacillus | Acid resistance, limited diversity | [129] |
| Small Intestine | Aerobic Gram-positive (duodenum), anaerobes (ileum) | Nutrient absorption, gradual transition | [130,131] |
| Large Intestine | Firmicutes, Bacteroidetes. | Fermentative role; Fermentation of complex carbohydrates, SCFA synthesis | [132,133] |
| Function | Description | Ref. |
|---|---|---|
| Nutrient Metabolism | Fermentation of dietary fibers, producing SCFAs (acetate, butyrate, propionate) essential for energy homeostasis and inflammatory modulation. Synthesis of essential vitamins, such as vitamin K and B-group vitamins. | [114,137] |
| Intestinal Barrier Integrity | Modulation of tight junction proteins and increased mucin production to prevent pathogen translocation. | [138] |
| Immune Modulation | Regulation of immune development and tolerance through interactions with dendritic cells, regulatory T cells, and SIgA production. | [136,139] |
| Production of Bioactive Metabolites | Beyond SCFAs, the microbiota produces metabolites such as TMAO and phenolic compounds, influencing systemic metabolism, including liver functions. | [140] |
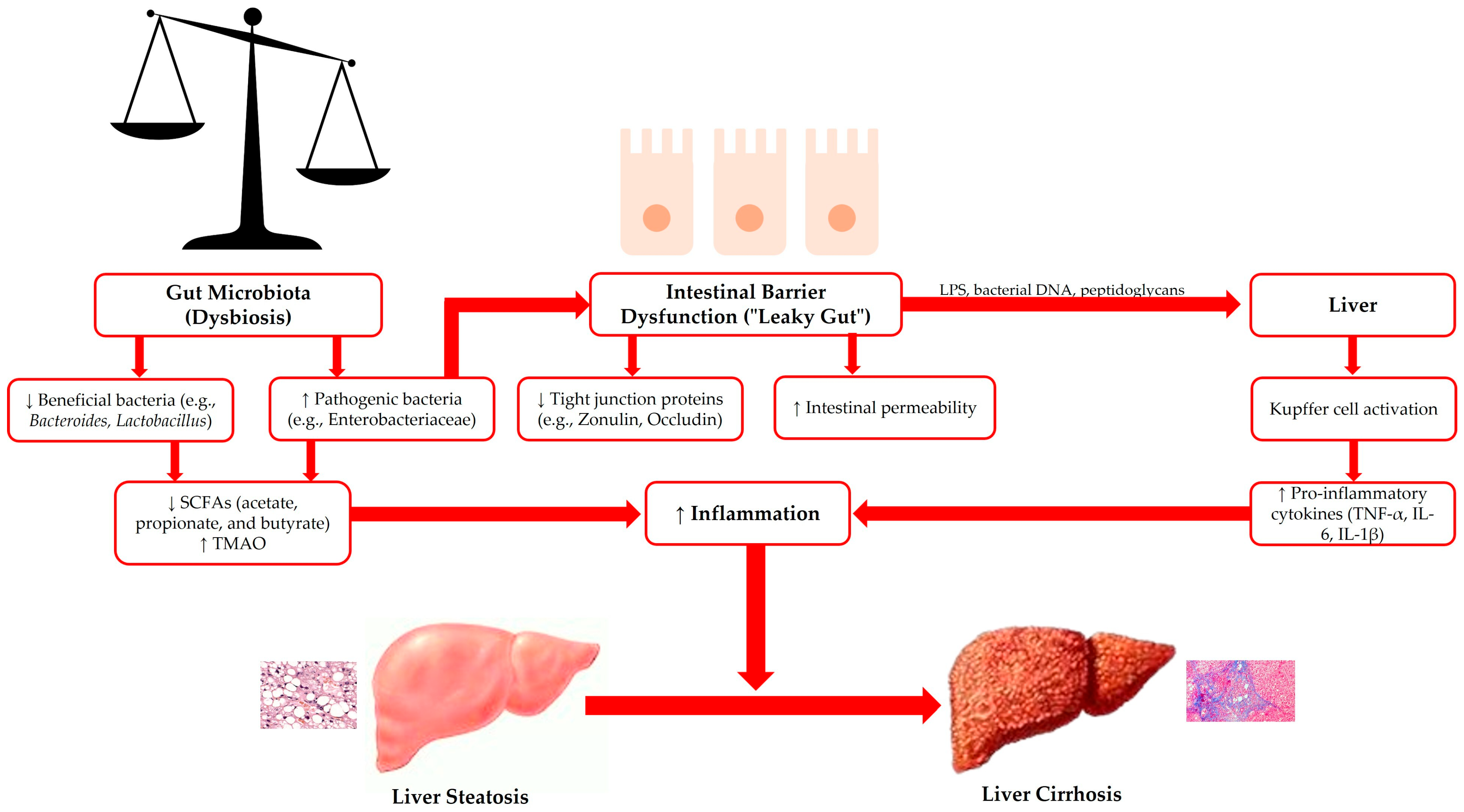
| Type of Damage | Description | Consequences on MASLD | Ref. |
|---|---|---|---|
| Mechanical | Alterations in tight junctions, increased permeability | Bacterial translocation, activation of Kupffer cells | [141,142,143,144,145] |
| Immunological | Reduced production of SIgA, increased pro-inflammatory cytokines | Systemic inflammation, liver damage | [146,147,148,149,150,151,152,153,154] |
| Microbial | Decreased microbial diversity, increased pathogenic bacteria | Dysbiosis, activation of the gut–liver inflammatory cycle | [155,156,157,158,159,160,161] |
| Therapeutic Approach | Intervention and Key Findings | Mechanism and Outcomes | Ref. |
|---|---|---|---|
| Dietary Interventions | Hypocaloric Diet and Exercise: Achieving 7–10% weight loss via a 500–1000 kcal/day deficit improves hepatic outcomes. Mediterranean Diet (MD): Rich in fiber, monounsaturated/polyunsaturated fats, and bioactive compounds; reduces hepatic steatosis and improves metabolic parameters. Moderate Coffee Intake: Associated with reduced oxidative stress and modulation of gut microbiota. | Promotes a negative energy balance and improves metabolic profiles. MD’s anti-inflammatory and antioxidant properties support hepatic and gut health, including a favorable gut microbiota composition (e.g., increased Akkermansia muciniphila). | [177,178,179,180,181,182,183,184,185] |
| Exercise | Regular physical activity in both human and animal studies. Observed increase in microbial diversity, particularly beneficial species (e.g., Akkermansia muciniphila), and modulation of gut taxa. | Enhances insulin sensitivity, lowers LDL cholesterol, and reduces hepatic fat accumulation and serum triglycerides. Modulates the gut microbiota by decreasing the Firmicutes/Bacteroidetes ratio and influencing specific taxa. | [186,187,188] |
| Antibiotics | Use of norfloxacin and neomycin in preclinical studies improves hepatic function by altering microbiota and reducing bacterial translocation. Combination of metronidazole and inulin shows significant reductions in serum ALT. Rifaximin therapy (1100 mg/day for 6 months) improves liver enzymes and inflammatory markers, although results are variable. | Alters gut microbiota composition, reducing harmful bacterial translocation. Rifaximin modulates fecal bile acid profiles, improves insulin resistance, and reduces pro-inflammatory cytokines, though its efficacy may vary across studies. | [189,190,191,192,193,194] |
| Probiotics, Prebiotics, and Synbiotics | Probiotics: Supplementation improves the AST-to-platelet ratio and may reduce hepatic inflammation (varied durations). Prebiotics: Oligofructose (16 g/day for 8 weeks) reduces AST levels, though effects on triglycerides are minimal. Synbiotics: Combined treatments have shown improvements in BMI, liver enzymes (AST, ALT, GGT), NAFLD fibrosis score, and inflammatory markers. | Modulate gut microbiota to reduce hepatic inflammation and oxidative stress. Enhance growth of beneficial bacteria, thereby improving metabolic parameters and reducing liver fat accumulation. Some trials report mixed results, underlining the need for further research. | [195,196,197,198,199,200,201,202,203] |
| Fecal Microbiota Transplantation (FMT) | FMT administered via oral capsules, nasogastric tubes, enemas, or colonoscopy. Single or multiple infusions have shown improvement in small intestinal permeability, modest reductions in hepatic steatosis, and long-term benefits in cirrhosis (e.g., reduced hepatic encephalopathy episodes). | Restores intestinal eubiosis and improves barrier integrity. Reduces systemic inflammation by modulating the gut–liver axis. Donor characteristics (lean vs. obese) are crucial for efficacy, influencing outcomes such as insulin sensitivity. | [204,205,206,207,208,209,210,211,212,213,214,215,216] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Acierno, C.; Nevola, R.; Rinaldi, L.; Sasso, F.C.; Adinolfi, L.E.; Caturano, A. The Intestinal Thread of Fate: How the Microbiota Shapes the Story of Liver Disease. Livers 2025, 5, 17. https://doi.org/10.3390/livers5020017
Acierno C, Nevola R, Rinaldi L, Sasso FC, Adinolfi LE, Caturano A. The Intestinal Thread of Fate: How the Microbiota Shapes the Story of Liver Disease. Livers. 2025; 5(2):17. https://doi.org/10.3390/livers5020017
Chicago/Turabian StyleAcierno, Carlo, Riccardo Nevola, Luca Rinaldi, Ferdinando Carlo Sasso, Luigi Elio Adinolfi, and Alfredo Caturano. 2025. "The Intestinal Thread of Fate: How the Microbiota Shapes the Story of Liver Disease" Livers 5, no. 2: 17. https://doi.org/10.3390/livers5020017
APA StyleAcierno, C., Nevola, R., Rinaldi, L., Sasso, F. C., Adinolfi, L. E., & Caturano, A. (2025). The Intestinal Thread of Fate: How the Microbiota Shapes the Story of Liver Disease. Livers, 5(2), 17. https://doi.org/10.3390/livers5020017