1. Introduction
The Thorpe Reaction (
Figure 1) was reported in 1904 [
1] by the British researcher Sir Jocelyn Thorpe (1872–1940). This reaction is described as a self-condensation of nitriles in the presence of a basic catalyst producing cyanoenamines (β-enaminonitriles). Sir Thorpe realized this reaction using a hot alcoholic dissolution to which was added the nitrile and a catalytic quantity of sodium ethoxide (EtONa). He informed that the product obtained was a cyanoimine [
1]. However, posteriorly spectrophotometric studies [
2,
3] confirmed that cyanoimine tautomerizes rapidly to the corresponding cyanoenamine.
Nowadays, stronger bases such as lithium bis(trimethylsilyl)amide (LHMDS) or other alkoxides have been shown to improve reaction yields [
3,
4,
5]. Similarly, other solvents have been used, such as THF and DME, obtaining remarkable results [
3,
4,
5]. Furthermore, the reaction has even been acceptably evaluated as solvent-free; that is, using the same starting nitrile as the solvent [
3]. Additionally, it is known that the reaction is stereospecific. It shows E/Z stereoisomerism, although in most cases the E stereoisomer is preferred. This reaction has also been conducted using nitriles with different α-substituents (-R in
Figure 1) such as alkyl (-Alkyl), aryl (-Ar), alkoxide (-OAr), and halogen (-X) groups [
3,
4,
5]. All these cases present different reaction yields, so the groups in the alpha position are expected to play an important stereoelectronic role in the reaction.
The importance of the Thorpe reaction lies in the high versatility that cyanoenamines exhibit. Due to cyanoenamines being easy to functionalize then, this reaction is widely used to synthetize cyclic, aromatic, and open-chain nitrogen derivatives [
6,
7], whose structures have an extensive branch of applications. In general, there are privileged scaffolds that can be synthesized using β-enaminonitriles such as derivatives of cytosine [
4,
5], pyrimidine [
8,
9], indole [
10], and others [
4,
5,
6,
7,
8,
9,
10].
Since its discovery, the mechanism of the Thorpe reaction [
5,
7,
10,
11] has been described as ionic and by four steps (
Figure 2). Firstly, the deprotonation of the α-hydrogen in the nitrile is promoted because of the basic catalyst. An intermediate carbanion is formed, and it attacks nucleophilically the carbon of a nitrile group in a second molecule, forming the conjugate base of the corresponding imine as an intermediate. Then, a proton exchange occurs that neutralizes the imine and regenerates the catalyst. Finally, tautomerization ensues to form the cyanoenamine. While it has never been clarified how this last step takes place, it is believed that the tautomerization is favored towards the cyanoenamine because it presents a π-conjugated system capable of causing effective electronic delocalization which supplies greater stability compared to cyanoimine.
Nevertheless, no convincing evidence has been reported that the Thorpe reaction effectively follows this mechanism. If the reaction mechanism is determined, it will allow the development of the experimental synthesis through a more economical, efficient, and safe route. Therefore, it can now be clearly recognized that a rigorous demonstration of how the Thorpe reaction occurs is needed. Thus, in this work, DFT calculations at level ωB97XD/def2-svpd were performed to determine three different proposals that explain step-by-step the mechanism of the Thorpe reaction. Likewise, the effects caused by the solvent (EtOH, THF, DME, and EtCN) and the α-substituent groups (-CH3 and -F) in the reaction coordinate were evaluated. We find that the reaction arises optimally in THF or DME instead of EtOH, and the presence of electron-withdrawing groups (EWG) in the α position improves the thermodynamics and kinetics of the reaction.
2. Methods
Theoretical calculations were performed using the suite of programs Gaussian. Structures of reactants, intermediates, transition states, and products were constructed in a three-dimensional form in GaussView. Afterward, computational calculations were performed using Gaussian09 without any geometric constraint. We calculated geometry optimizations and vibrational frequencies for all the structures involved in the three proposal mechanisms. Transition states were verified by the vibrational frequencies results. The functional ωB97X-D and the basis set def2-svpd were used. Numerical accuracy was improved using the base def2-tzvpd. Solvation effect was calculated employing the SMD model. The reaction mechanisms were firstly determined at standard temperature (25 °C) using propionitrile, sodium ethoxide, and ethanol as reagent, catalyst, and solvent, respectively. Once the energy requirements for each mechanism were calculated, we chose the one with the lowest energy, following the Transition State Theory (TST). Then, we studied the solvent and substituent effects caused by other solvents instead of EtOH (THF, DME, and EtCN) and different groups (-F) in the alpha position. These results were compared to data when EtOH is used as solvent and a methyl group (-CH3) is in the alpha position. Finally, energy profiles were constructed, extracting thermodynamic data from ChemCraft.
3. Results and Discussion
3.1. Reaction Mechanism
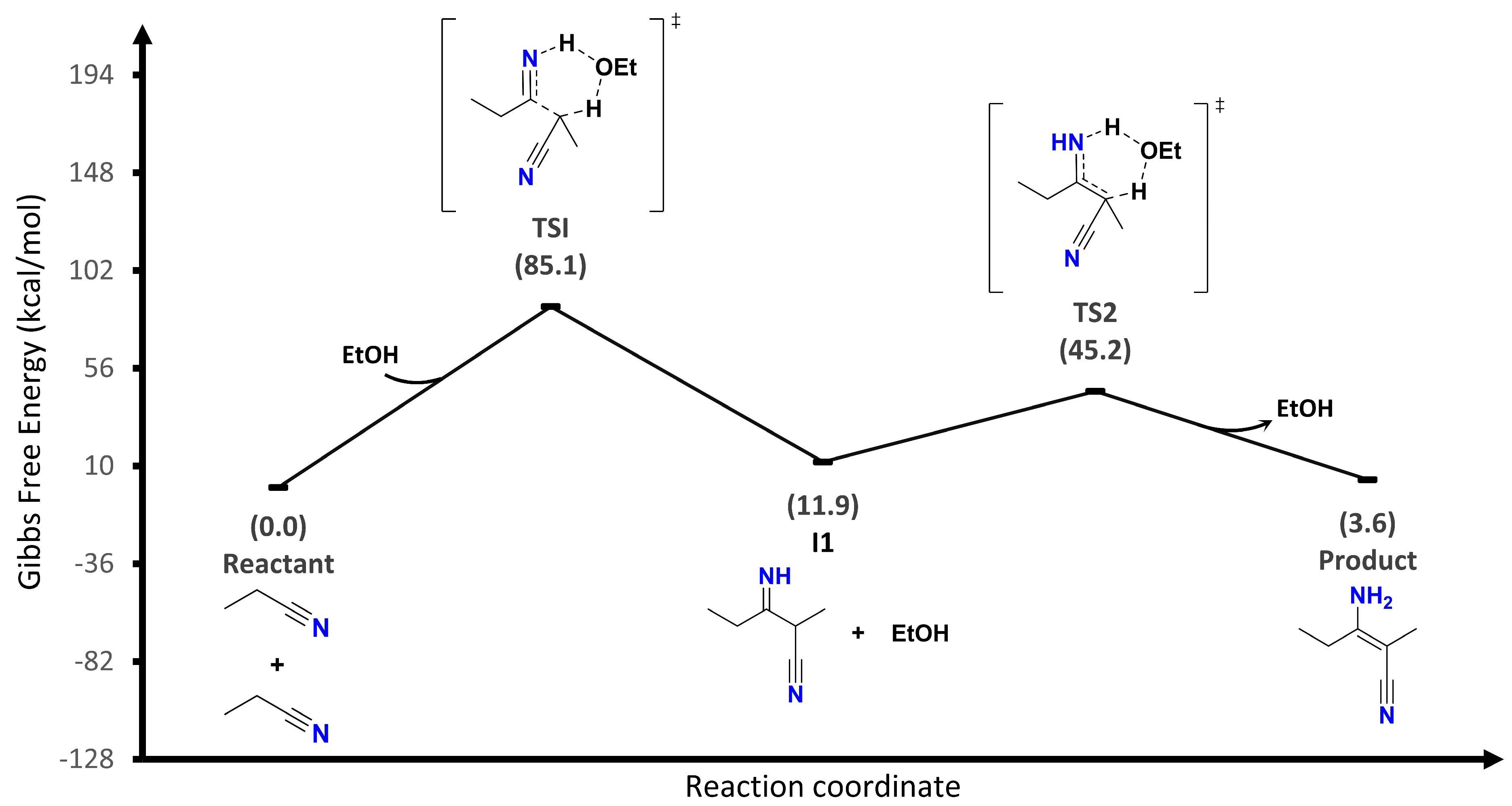
The first proposal (
Figure 3) is a concerted reaction mechanism excluding the alkoxide participation as the catalyst. It starts with the formation of an imine (I1) followed by its tautomerization to an enamine (product) involving two cyclic transition states. For this proposal, the limiting step of the reaction is the first transition state (TS1) whose energy barrier is 85.1 kcal/mol. This is a very high energy value that, in the laboratory, requires very aggressive temperature conditions.
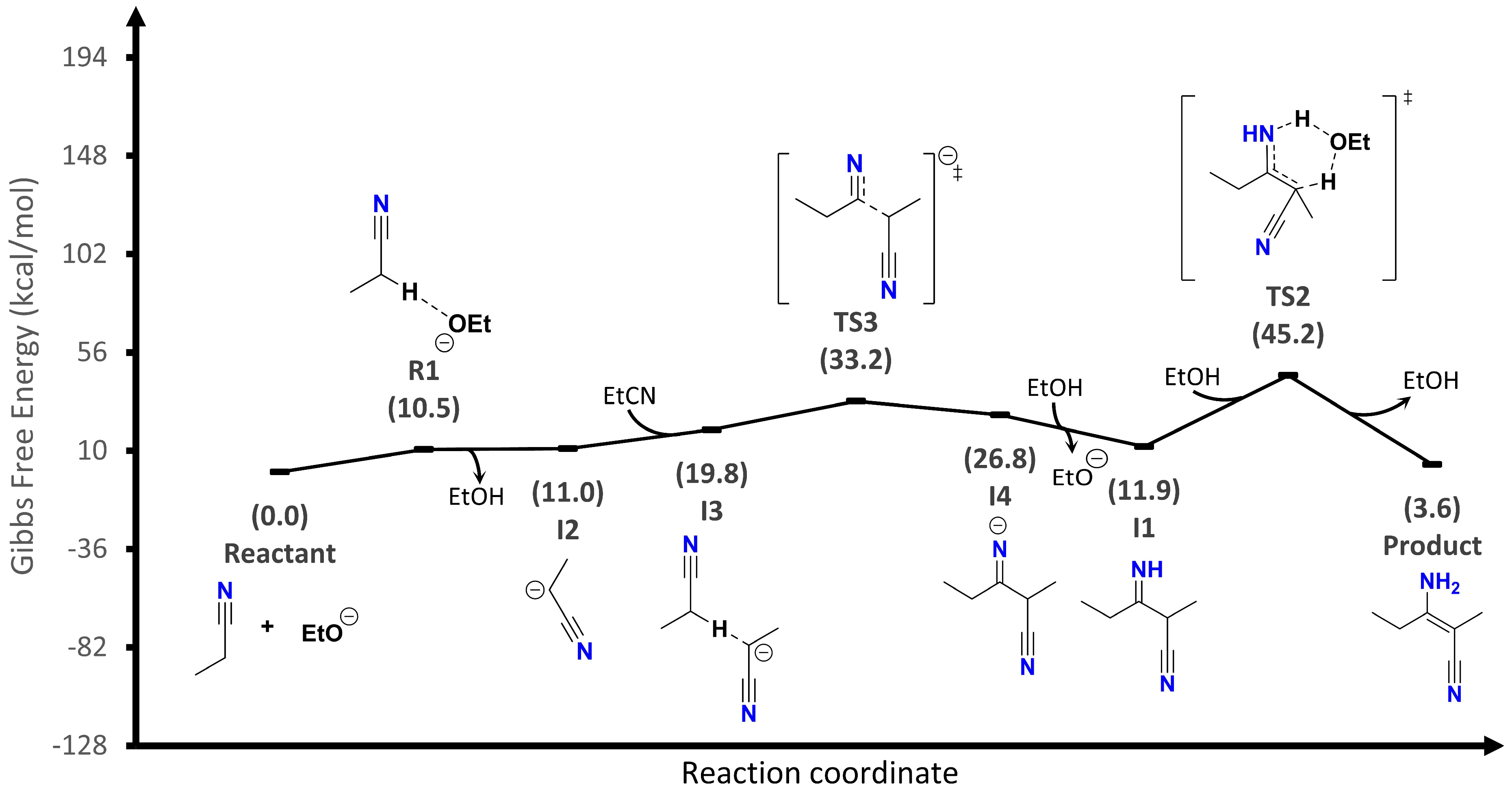
The second proposal (
Figure 4) is an ionic reaction mechanism including the alkoxide participation as the catalyst. In this mechanism, before deprotonation, an adduct (R1) is formed between the ethoxide and the α-hydrogen of the nitrile. Then, a carbanion (I2) is immediately produced, which creates a new adduct (I3) with a second nitrile molecule. From this last adduct, the first transition state (TS3) is reached, and the imine anion (I4) is generated. After that, imine (I1) is neutralized, and it is proposed that tautomerization follows the second transition state (TS2), as in the first proposal. Now, the limiting step of the reaction is the TS2 formation whose energy barrier is 45.2 kcal/mol. This is a milder energy value, although it still requires heating for the reaction to result. This proposal shows the importance of the basic catalyst in the reaction, since the determining step energy has decreased by half compared to the first proposed mechanism.
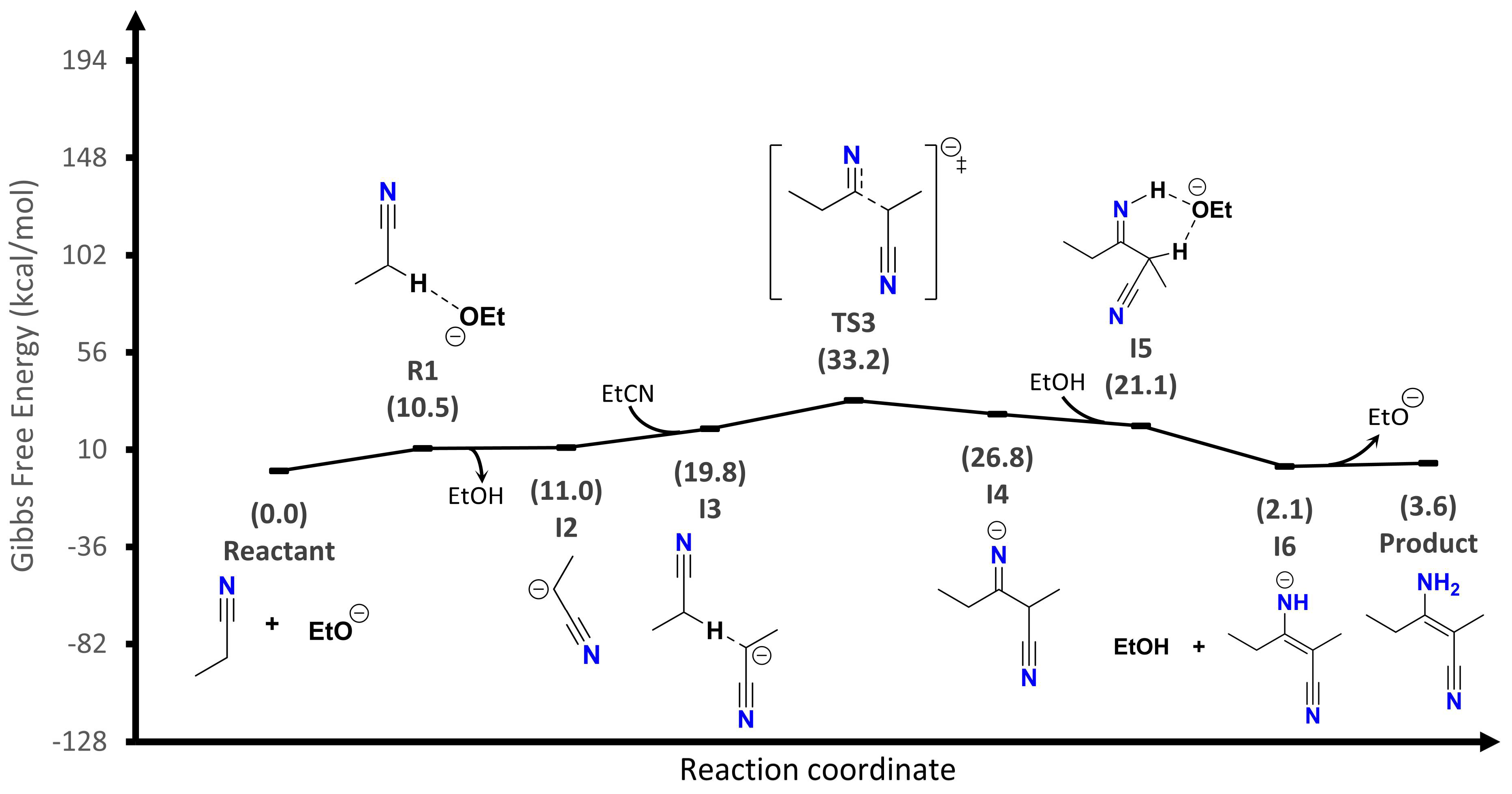
The third proposal (
Figure 5) is another ionic mechanism. This proposal is like the second mechanism until the imine anion (I4) is produced. Following this, the generation of a new adduct (I5) is suggested. It spontaneously converts to an amide (I6) by tautomerization. The exchange of a proton with the conjugate acid (EtOH) generates the corresponding neutral cyanoenamine and regenerates the basic catalyst used. Here, the limiting step of the reaction is the TS3 formation, whose energy barrier is 33.2 kcal/mol.
Therefore, the most probable reaction mechanism is the one that requires the lowest activation energy to occur. In this way, the third proposed mechanism has the lowest energy, so it is the most probable to exist. Consequently, this proposal is now used to study the solvent and α-substituent effects on the energy profile.
3.2. Solvent Effect
The solvation effect (
Table 1) generated by different solvents (DME, THF, and EtCN), which have been used in different experimental organic reports, was compared to the values when EtOH is used.
The solvents DME and THF decrease the energy value of the reaction-determining step (Step 5,
Table 1) by almost 13 kcal/mol, compared to the values obtained when EtOH is used as a solvent. Thus, these solvents allow the reaction to be carried out with a lower energy requirement. Therefore, they are better options than EtOH. Similarly, it is shown that using the same nitrile reagent as a solvent (EtCN in this study) is a viable option, since it implies a reduction of approximately 10 kcal/mol in the reaction-determining step. However, this depends on the availability of the nitrile as well as its state of aggregation at room temperature.
3.3. Substituent Effect
Additionally, the effect of the substituent in the α position (-R in
Figure 1) of acetonitrile was studied (
Table 2). The results obtained when the substituent is a methyl group, as in EtCN, were compared with respect to a fluorine group (-F) substituted in this position. Because THF is one of the best available solvents for this reaction, as just shown, the next calculations were run in this solvent.
The activation energy for the limiting step (Step 5,
Table 2) has decreased again by around 13 kcal/mol. Additionally, the reaction turned to be exergonic. These results indicate that the reaction requires less energy. It means that it occurs easier when an EWG group, such as fluorine, is placed at the alpha position of the nitrile. Furthermore, calculated energy values show that, under these conditions (R = F,
Table 2), the reaction can take place without thermal heating. This is what is reported in the laboratory experiments, so our calculations are in great agreement.
4. Conclusions
Three proposals to explain the mechanism of the Thorpe reaction have been calculated by computationally applying the DFT. It is concluded that the most probable route is the third proposal. This is an ionic mechanism very similar to the one historically conjectured. Likewise, this work managed to propose two routes that explain how imine–enamine tautomerization arises, something that had not been reported before. Additionally, it was demonstrated that both DME and THF, or the same nitrile reagent if it is possible, are better solvents than EtOH to develop the reaction. It was found that an EWG group, such as fluorine, replaced at α position on the nitrile, highly abate the energy barrier necessary to promote the reaction. Finally, further study is important to explain the stereoselectivity of the reaction, as well as its viability in the presence of different substituents in the α position on nitrile.









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}