
A Combined 2D- and 3D-QSAR Study, Design and Synthesis of Some Monocarbonyl Curcumin Analogs as Potential Inhibitors of MDA-MB-231 Breast Cancer Cells †

Abstract
1. Introduction
2. Materials and Methods
2.1. Chemistry
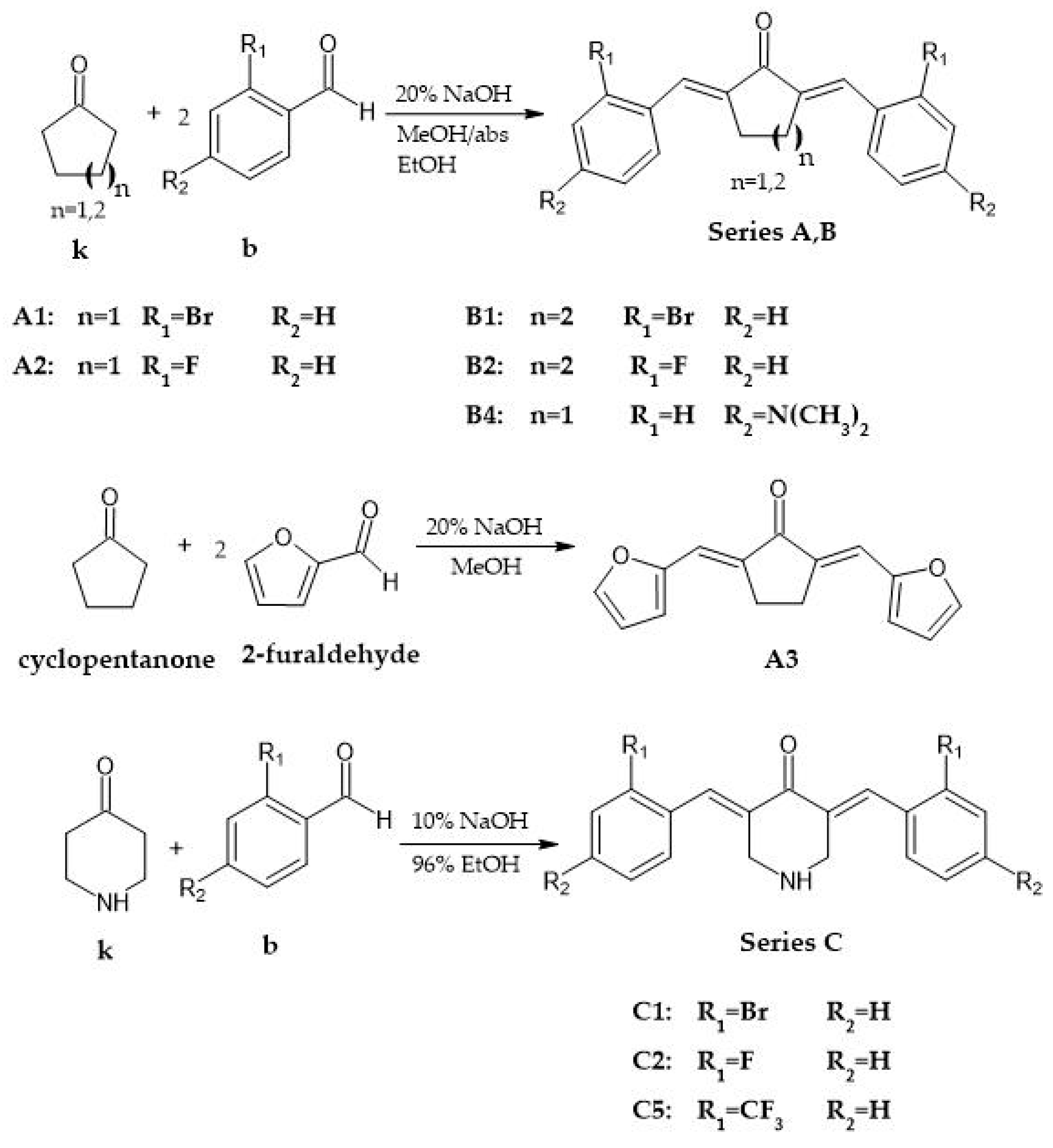
2.2. General Procedure for the Synthesis of Analogs with Cyclopentanone and Cyclohexanone Cores
2.3. General Procedure for the Synthesis of Analogs with 4-Piperidone Cores
2.4. Computational Studies
2.4.1. Dataset
2.4.2. Geometry Optimization
2.4.3. 2D-QSAR Methodology
2.4.4. 3D-QSAR Methodology
- Quenched Molecular Dynamics and Alignment
- CoMFA Model
3. Results
3.1. Synthesis
3.2. 2D-QSAR
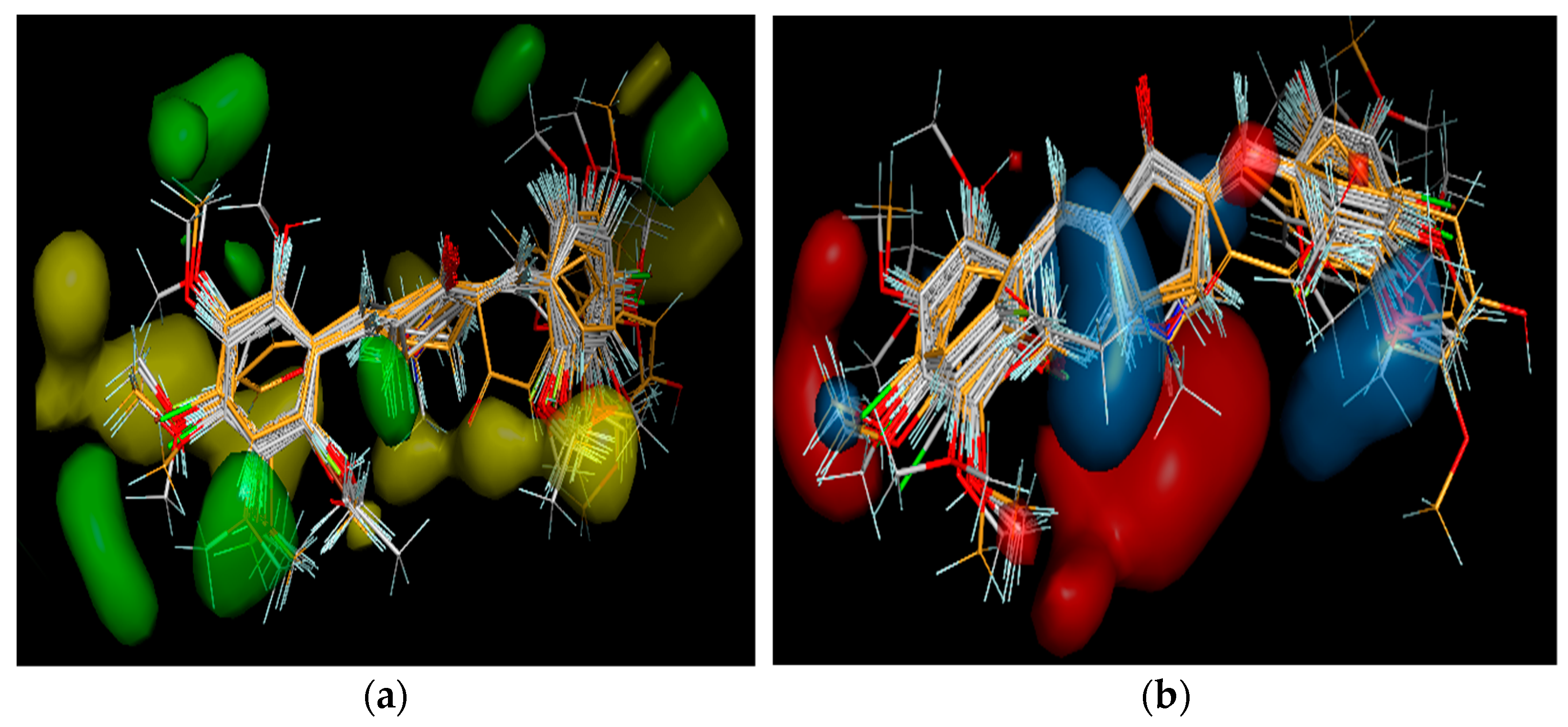
3.3. 3D-QSAR
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Breast Cancer. Available online: https://www.who.int/news-room/fact-sheets/detail/breast-cancer (accessed on 15 August 2021).
- Robles-Escajeda, E.; Das, U.; Ortega, N.M.; Parra, K.; Francia, G.; Dimmock, J.R.; Varela-Ramirez, A.; Aguilera, R.J. A novel curcumin-like dienone induces apoptosis in triple-negative breast cancer cells. Cell. Oncol. 2016, 39, 265–277. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.-D.; Liu, X.-E.; Huang, D.-S. Curcumin induces apoptosis of triple-negative breast cancer cells by inhibition of EGFR expression. Mol. Med. Rep. 2012, 6, 1267–1270. [Google Scholar] [CrossRef] [PubMed]
- Nagaraju, G.P.; Aliya, S.; Zafar, S.F.; Basha, R.; Diaz, R.; El-Rayes, B.F. The impact of curcumin on breast cancer. Integr. Biol. 2012, 4, 996–1007. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Yu, J.; Cui, R.; Lin, J.; Ding, X. Curcumin in Treating Breast Cancer: A Review. J. Lab. Autom. 2016, 21, 723–731. [Google Scholar] [CrossRef] [PubMed]
- Labbozzetta, M.; Notarbartolo, M.; Poma, P.; Maurici, A.; Inguglia, L.; Marchetti, P.; Rizzi, M.; Baruchello, R.; Simoni, D.; D’Alessandro, N. Curcumin as a Possible Lead Compound against Hormone-Independent, Multidrug-Resistant Breast Cancer. Ann. N. Y. Acad. Sci. 2009, 1155, 278–283. [Google Scholar] [CrossRef] [PubMed]
- Hu, S.; Xu, Y.; Meng, L.; Huang, L.; Sun, H. Curcumin inhibits proliferation and promotes apoptosis of breast cancer cells. Exp. Ther. Med. 2018, 16, 1266–1272. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; Zhang, M.; Dai, E.; Luo, Y. Molecular targets of curcumin in breast cancer (Review). Mol. Med. Rep. 2019, 19, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Mock, C.D.; Jordan, B.C.; Selvam, C. Recent advances of curcumin and its analogues in breast cancer prevention and treatment. RSC Adv. 2015, 5, 75575–75588. [Google Scholar] [CrossRef] [PubMed]
- Shetty, D.; Kim, Y.J.; Shim, H.; Snyder, J.P. Eliminating the Heart from the Curcumin Molecule: Monocarbonyl Curcumin Mimics (MACs). Molecules 2014, 20, 249–292. [Google Scholar] [CrossRef] [PubMed]
- Zamrus, S.N.H.; Akhtar, M.N.; Yeap, S.K.; Quah, C.K.; Loh, W.-S.; Alitheen, N.B.; Zareen, S.; Tajuddin, S.N.; Hussin, Y.; Shah, S.A.A. Design, synthesis and cytotoxic effects of curcuminoids on HeLa, K562, MCF-7 and MDA-MB-231 cancer cell lines. Chem. Central J. 2018, 12, 31. [Google Scholar] [CrossRef] [PubMed]
- Adams, B.K.; Ferstl, E.M.; Davis, M.C.; Herold, M.; Kurtkaya, S.; Camalier, R.F.; Hollingshead, M.G.; Kaur, G.; Sausville, E.A.; Rickles, F.R.; et al. Synthesis and biological evaluation of novel curcumin analogs as anti-cancer and anti-angiogenesis agents. Bioorganic Med. Chem. 2004, 12, 3871–3883. [Google Scholar] [CrossRef] [PubMed]
- Dolezal, R.; Korabecny, J.; Malinak, D.; Honegr, J.; Musilek, K.; Kuca, K. Ligand-based 3D QSAR analysis of reactivation potency of mono- and bis-pyridinium aldoximes toward VX-inhibited rat acetylcholinesterase. J. Mol. Graph. Model. 2015, 56, 113–129. [Google Scholar] [CrossRef] [PubMed]
- Xu, G.; Chu, Y.; Jiang, N.; Yang, J.; Li, F. The Three Dimensional Quantitative Structure Activity Relationships (3D-QSAR) and Docking Studies of Curcumin Derivatives as Androgen Receptor Antagonists. Int. J. Mol. Sci. 2012, 13, 6138–6155. [Google Scholar] [CrossRef] [PubMed]
- Sippl, W. 3D-QSAR—Applications, Recent Advances, and Limitations. In Recent Advances in QSAR Studies; Springer: Dordrecht, The Netherlands, 2009; pp. 103–125. [Google Scholar] [CrossRef]
- Xue, C.; Cui, S.; Liu, M.; Hu, Z.; Fan, B. 3D QSAR studies on antimalarial alkoxylated and hydroxylated chalcones by CoMFA and CoMSIA. Eur. J. Med. Chem. 2004, 39, 745–753. [Google Scholar] [CrossRef] [PubMed]
- Tosco, P.; Balle, T. Open3DQSAR: A new open-source software aimed at high-throughput chemometric analysis of molecular interaction fields. J. Mol. Model. 2010, 17, 201–208. [Google Scholar] [CrossRef] [PubMed]
- Papa, E.; Doucet, J.P.; Doucet-Panaye, A. Computational approaches for the prediction of the selective uptake of magnetofluorescent nanoparticles into human cells. RSC Adv. 2016, 6, 68806–68818. [Google Scholar] [CrossRef]
- Achutha, A.S.; Pushpa, V.L.; Suchitra, S. Theoretical Insights into the Anti-SARS-CoV-2 Activity of Chloroquine and Its Analogs and In Silico Screening of Main Protease Inhibitors. J. Proteome Res. 2020, 19, 4706–4717. [Google Scholar] [CrossRef] [PubMed]
- Tadayon, M.; Garkani-Nejad, Z. In silico study combining QSAR, docking and molecular dynamics simulation on 2,4-disubstituted pyridopyrimidine derivatives. J. Recept. Signal Transduct. 2019, 39, 167–174. [Google Scholar] [CrossRef] [PubMed]
- Krishnasamy, C.; Raghuraman, A.; Kier, L.B.; Desai, U.R. Application of Molecular Connectivity and Electro-Topological Indices in Quantitative Structure-Activity Analysis of Pyrazole Derivatives as Inhibitors of Factor Xa and Thrombin. Chem. Biodivers. 2008, 5, 2609–2620. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Statistical Parameters | Values |
|---|---|
| R | 0.881 |
| R2 | 0.775 |
| Adjusted R2 | 0.700 |
| SE | 0.267 |
| F | 10.336 |
| Significance F | 1.4 × 10−5 |
| R2 pred | 0.7827 |
| Statistical Parameters | Values |
|---|---|
| R | 0.979 |
| R2 FFDSEL | 0.959 |
| Q2loo FFDSEL | 0.401 |
| SDEP | 0.371 |
| R2 pred | 0.907 |
| SDEP pred | 0.167 |
| Analog | Predicted pIC50 Values (µmol/L) |
|---|---|
| (3E,5E)-3,5-bis(2-fluorobenzylidene)-4-piperidone | 5.46 |
| (3E, 5E)-3,5-bis(2-bromobenzylidene)-4-piperidone | 5.19 |
| (2E, 6E)-2,6-bis(2-fluorobenzylidene)cyclohexanone | 5.15 |
| (2E, 6E)-2,6-bis(2-bromobenzylidene)cyclohexanone | 5.14 |
| (3E, 5E)-3,5-bis(2-trifluoromethylbenzylidene)-4-piperidone | 5.13 |
| (2E,5E)-2,5-Bis(2-furylmethylene)cyclopentanone | 5.07 |
| (2E, 6E)-2,6-bis(4-dimethylaminobenzylidene)cyclohexanone | 4.54 |
| (2E, 6E)-2,6-bis(2-fluorobenzylidene)cyclopentanone | 4.36 |
| (2E, 6E)-2,6-bis(2-bromobenzylidene)cyclopentanone | 4.29 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Todorovska, I.; Dragarska, K.; Bogdanov, J. A Combined 2D- and 3D-QSAR Study, Design and Synthesis of Some Monocarbonyl Curcumin Analogs as Potential Inhibitors of MDA-MB-231 Breast Cancer Cells. Chem. Proc. 2022, 12, 5. https://doi.org/10.3390/ecsoc-26-13572
Todorovska I, Dragarska K, Bogdanov J. A Combined 2D- and 3D-QSAR Study, Design and Synthesis of Some Monocarbonyl Curcumin Analogs as Potential Inhibitors of MDA-MB-231 Breast Cancer Cells. Chemistry Proceedings. 2022; 12(1):5. https://doi.org/10.3390/ecsoc-26-13572
Chicago/Turabian StyleTodorovska, Ivana, Katerina Dragarska, and Jane Bogdanov. 2022. "A Combined 2D- and 3D-QSAR Study, Design and Synthesis of Some Monocarbonyl Curcumin Analogs as Potential Inhibitors of MDA-MB-231 Breast Cancer Cells" Chemistry Proceedings 12, no. 1: 5. https://doi.org/10.3390/ecsoc-26-13572
APA StyleTodorovska, I., Dragarska, K., & Bogdanov, J. (2022). A Combined 2D- and 3D-QSAR Study, Design and Synthesis of Some Monocarbonyl Curcumin Analogs as Potential Inhibitors of MDA-MB-231 Breast Cancer Cells. Chemistry Proceedings, 12(1), 5. https://doi.org/10.3390/ecsoc-26-13572