
High-Throughput Virtual Screening of Compounds with Electrophilic Fragments for New Potential Covalent Inhibitors of Bacterial Proteins †

 ,
,
Abstract
1. Introduction
2. Materials and Methods
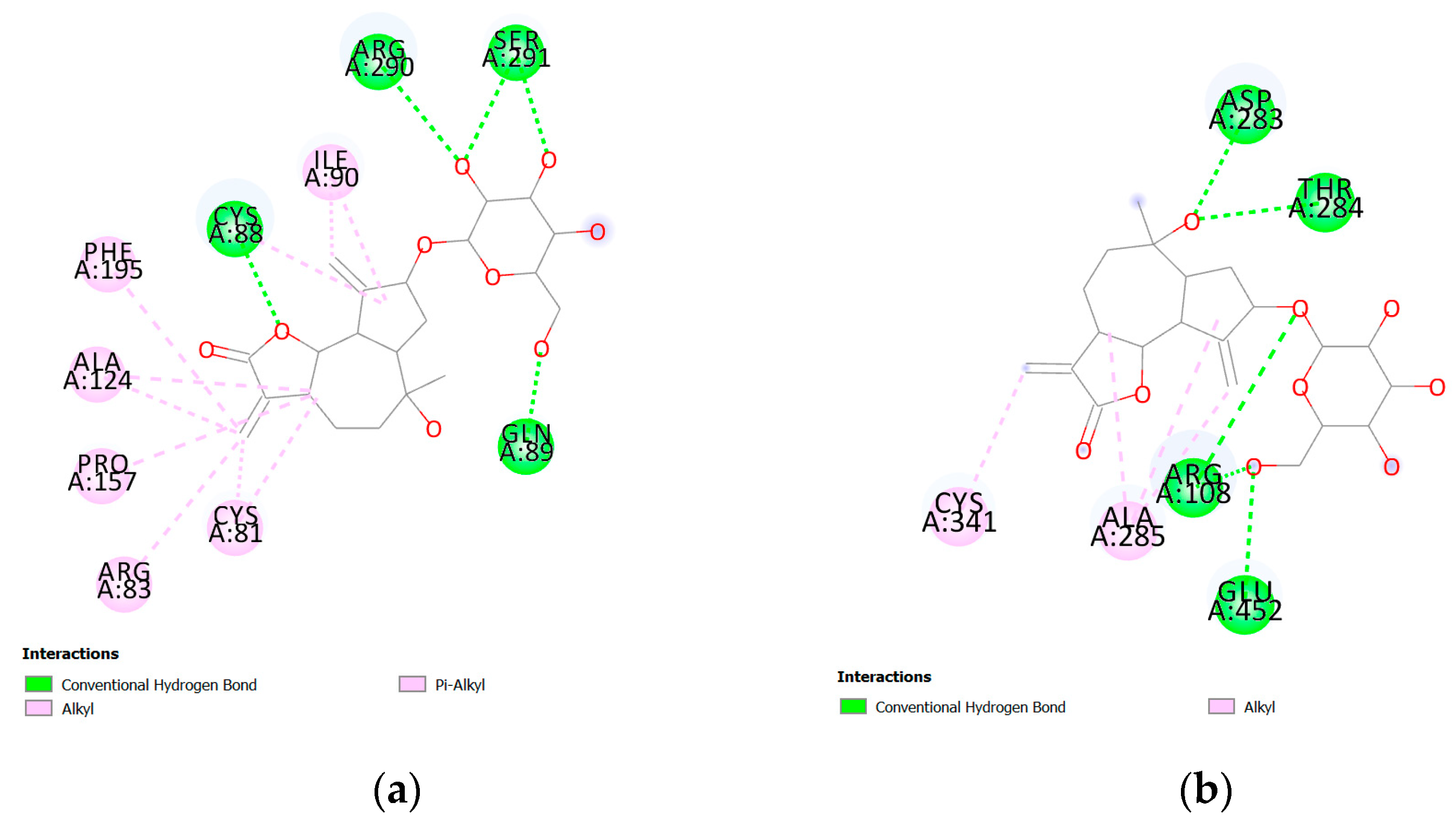
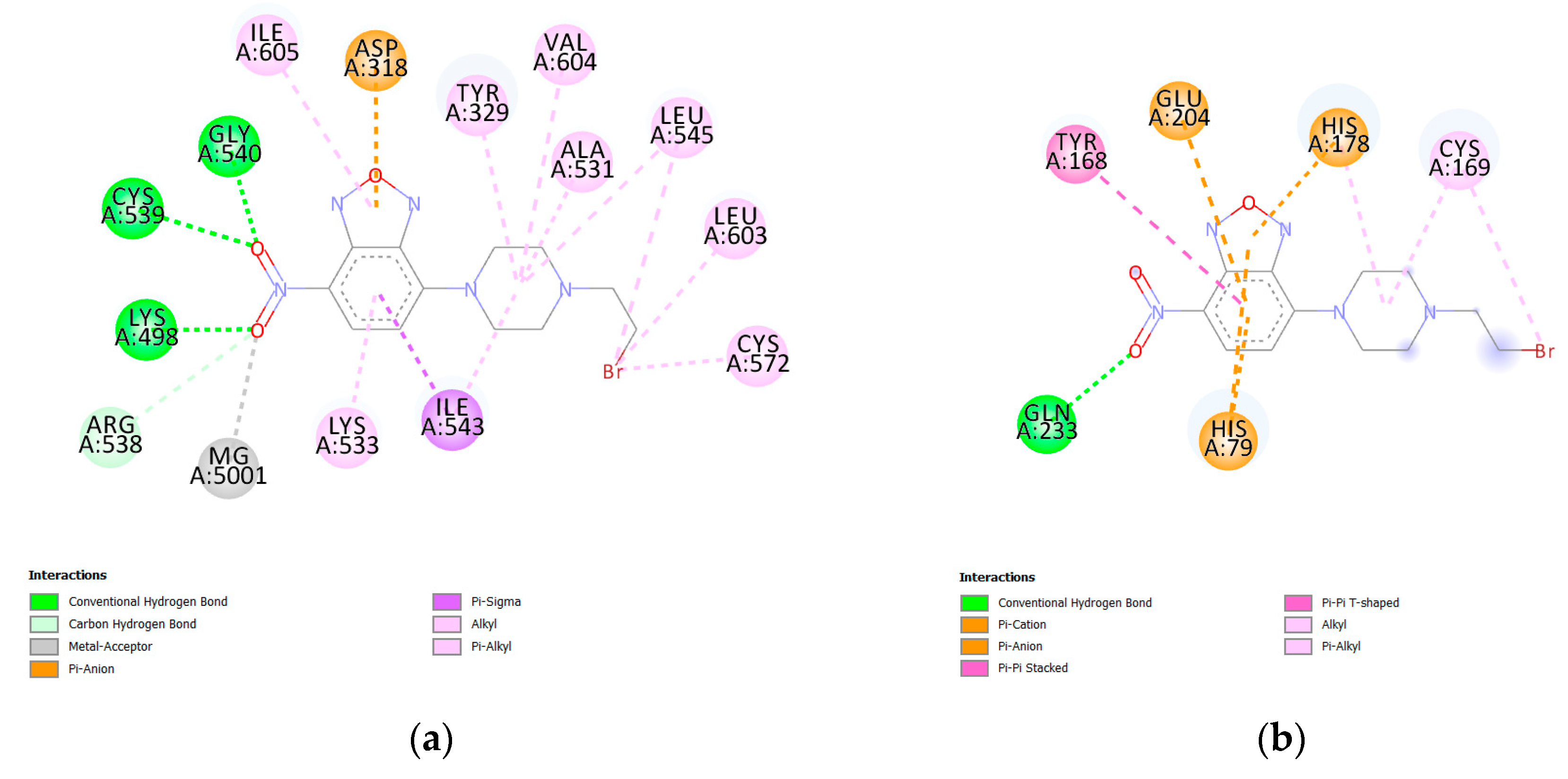
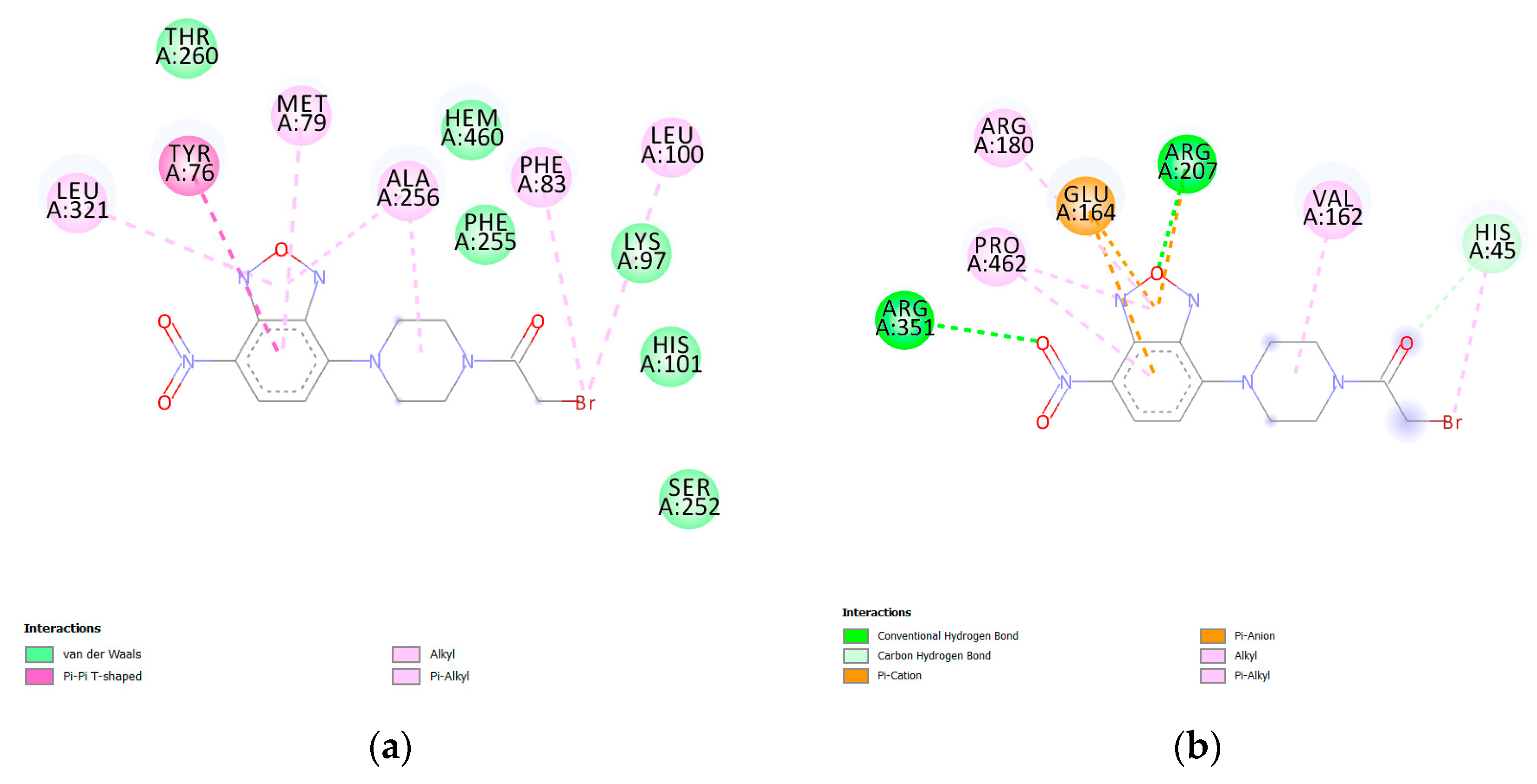
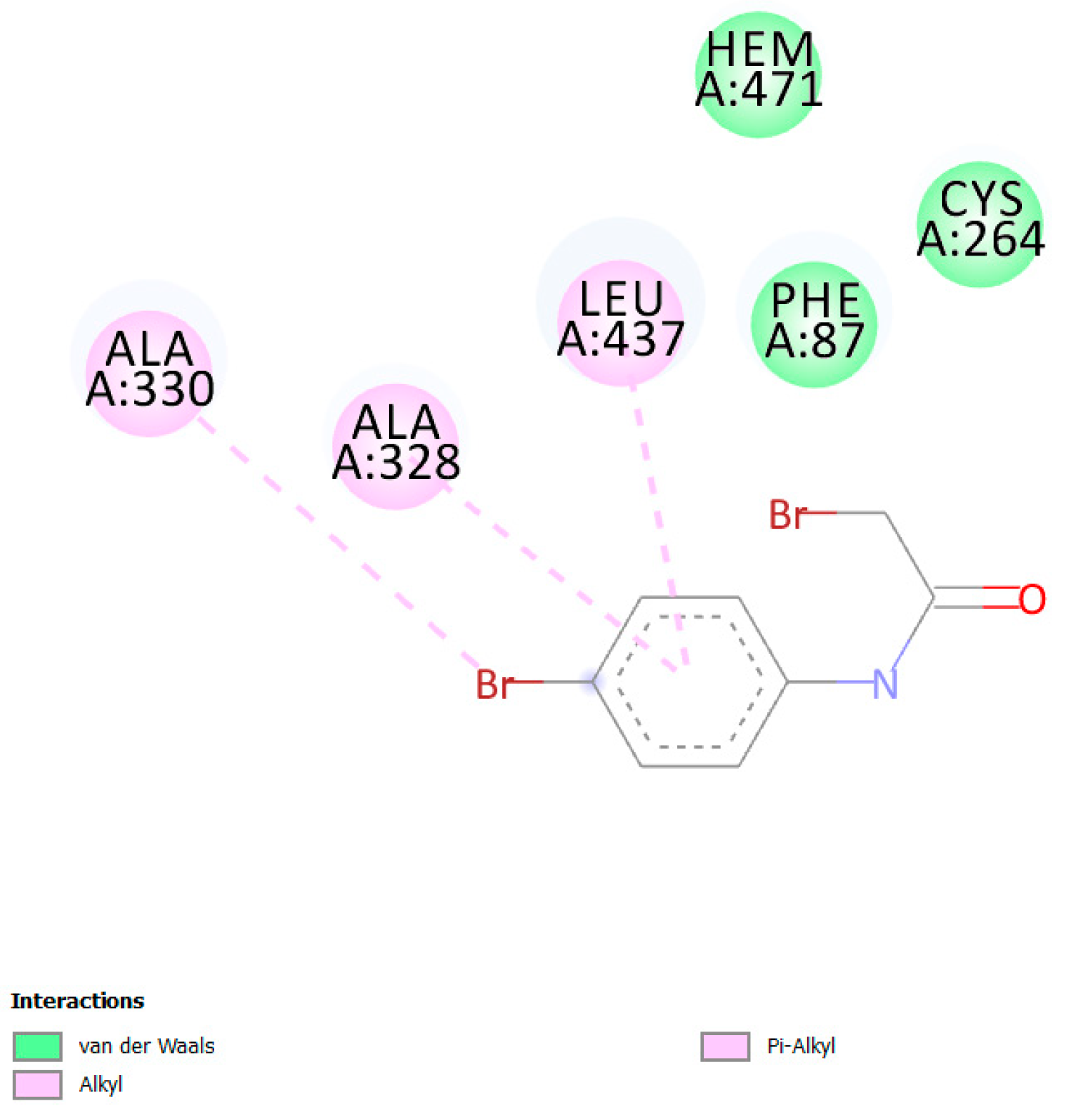
3. Results
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Kibou, Z.; Aissaoui, N.; Daoud, I.; Seijas, J.A.; Vázquez-Tato, M.P.; Khelil, N.K.; Choukchou-Braham, N. Efficient Synthesis of 2-Aminopyridine Derivatives: Antibacterial Activity Assessment and Molecular Docking Studies. Molecules 2022, 27, 3439. [Google Scholar] [CrossRef] [PubMed]
- Faletrov, Y.; Brzostek, A.; Plocinska, R.; Dziadek, J.; Rudaya, E.; Edimecheva, I.; Shkumatov, V. Uptake and Metabolism of Fluorescent Steroids by Mycobacterial Cells. Steroids 2017, 117, 29–37. [Google Scholar] [CrossRef]
- Faletrov, Y.V.; Karpushenkova, V.S.; Zavalinich, V.A.; Yakovets, P.S.; Shkredava, A.D.; Shkumatov, V.M. Interaction of Nitrobenzoxadiazole Derivatives of Piperazine and Aniline with Bovine Serum Albumine in Silico and in Vitro. J. Belarusian State Univ. Chem. 2021, 2, 25–35. [Google Scholar] [CrossRef]
- Faletrov, Y.V.; Staravoitava, V.A.; Dudko, A.R.; Shkumatov, V.M. Application of Docking-Based Inverse High Throughput Virtual Screening to Found Phytochemical Covalent Inhibitors of SARS-CoV-2 Main Protease, NSP12 and NSP16. 2022; preprint. [Google Scholar] [CrossRef]
- Sachdeva, S.; Reynolds, K.A. Mycobacterium tuberculosis β-Ketoacyl Acyl Carrier Protein Synthase III (mtFabH) Assay: Principles and Method. In New Antibiotic Targets. Methods In Molecular Medicine™; Champney, W.S., Ed.; Humana Press: Totowa, NJ, USA, 2008; Volume 142, pp. 205–213. [Google Scholar] [CrossRef]
- Zong, Y.; Mazmanian, S.K.; Schneewind, O.; Narayana, S.V.L. The Structure of Sortase B, a Cysteine Transpeptidase That Tethers Surface Protein to the Staphylococcus Aureus Cell Wall. Structure 2004, 12, 105–112. [Google Scholar] [CrossRef] [PubMed]
- Chiang, B.-Y.; Chen, T.-C.; Pai, C.-H.; Chou, C.-C.; Chen, H.-H.; Ko, T.-P.; Hsu, W.-H.; Chang, C.-Y.; Wu, W.-F.; Wang, A.H.-J.; et al. Protein S-Thiolation by Glutathionylspermidine (Gsp). J. Biol. Chem. 2010, 285, 25345–25353. [Google Scholar] [CrossRef]
- Tassoni, R.; Blok, A.; Pannu, N.S.; Ubbink, M. New Conformations of Acylation Adducts of Inhibitors of β-Lactamase from Mycobacterium tuberculosis. Biochemistry 2019, 58, 997–1009. [Google Scholar] [CrossRef]
- Pai, C.-H.; Chiang, B.-Y.; Ko, T.-P.; Chou, C.-C.; Chong, C.-M.; Yen, F.-J.; Chen, S.; Coward, J.K.; Wang, A.H.-J.; Lin, C.-H. Dual Binding Sites for Translocation Catalysis by Escherichia coli Glutathionylspermidine Synthetase. EMBO J. 2006, 25, 5970–5982. [Google Scholar] [CrossRef] [PubMed]
- Evdokimov, A.G.; Pokross, M.; Walter, R.L.; Mekel, M.; Barnett, B.L.; Amburgey, J.; Seibel, W.L.; Soper, S.J.; Djung, J.F.; Fairweather, N.; et al. Serendipitous Discovery of Novel Bacterial Methionine Aminopeptidase Inhibitors. Proteins 2006, 66, 538–546. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein PDB | Protein Name | Cysteine | Ebind, kcal/mol |
|---|---|---|---|
| 5EXI | Lipoyl synthase | CYS81 | −10.7 |
| 4ZQR | The catalytic domain of the inosine monophosphate dehydrogenase | CYS341 | −9.4 |
| 1M1M | Beta-ketoacyl-acyl carrier protein synthase III | CYS123 | −9.2 |
| 2AJ9 | Beta-ketoacyl-acyl carrier protein synthase III | CYS122 | −9.1 |
| 2AHB | Beta-ketoacyl-acyl carrier protein synthase III | CYS122 | −9.0 |
| Protein PDB | Protein Name | Cysteine | Ebind, kcal/mol |
|---|---|---|---|
| 6h27 | S70C BlaC from Mycobacterium tuberculosis | CYS70 | −7.3 |
| 3a2z | E. coli Gsp amidase Cys59 sulfenic acid | CYS59 | −7.0 |
| 7ock | E. coli K-12 MAT | CYS96 | −6.8 |
| 1qxa | Crystal structure of Staphylococcus aureus Sortase B | CYS223 | −6.5 |
| Protein PDB | Protein Name | Cysteine | Ebind, kcal/mol |
|---|---|---|---|
| 2ioa | E. coli Bifunctional glutathionylspermidine synthetase/amidase | CYS572 | −8.3 |
| 2gg7 | E. coli K-12 methionine aminopeptidase | CYS169 | −7.0 |
| Protein PDB | Protein Name | Cysteine | Ebind, kcal/mol |
|---|---|---|---|
| 1u13 | Crystal structure of C37L/C151T/C442A-triple mutant of CYP51 from Mycobacterium tuberculosis | HIS101 | −9.0 |
| 5nj5 | E. coli Microcin-processing metalloprotease TldD/E | HIS45 | −8.4 |
| 3NC3 | Bacillus subtilis CYP134A1 structure with a closed substrate binding loop | HIS351 | −8.3 |
| 5njf | E. coli Microcin-processing metalloprotease TldD/E (TldD H262A mutant) | HIS45 | −8.0 |
| 5njc | E. coli Microcin-processing metalloprotease TldD/E (TldD E263A mutant) | HIS45 | −7.7 |
| 2VZM | Crystal structure of E. coli PikC D50N mutant | HIS238 | −7.7 |
| 3LXI | Crystal Structure of E. coli CYP101D1 | HIS400 | −7.4 |
| 4BF4 | PikC D50N mutant from Streptomyces venezuelae | HIS238 | −7.4 |
| 2irv | Crystal structure of E. coli K-12 GlpG, a rhomboid intramembrane serine protease | HIS150 | −7.3 |
| 3zeb | E. coli BL21(DE3) GlpG | HIS141 | −7.3 |
| 5hdi | Mycobacterium tuberculosis cytochrome P450 CYP144A1 | HIS325 | −7.3 |
| 1T2B | Crystal Structure of Citrobacter braakii cytochrome P450cin | HIS391 | −7.1 |
| 4EGM | The X-ray crystal structure of Rhodopseudomonas palustris HaA2 CYP199A4 | HIS202 | −6.9 |
| 3ZC3 | Nostoc sp. PCC 7119 Ferredoxin-NADP reductase (mutation S80A) | HIS42 | −6.8 |
| 1P7R | Crystal structure of Pseudomonas putida cytochrome P450CAM | HIS391 | −6.7 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yakovets, P.; Staravoitava, V.; Faletrov, Y.; Shkumatov, V. High-Throughput Virtual Screening of Compounds with Electrophilic Fragments for New Potential Covalent Inhibitors of Bacterial Proteins. Chem. Proc. 2022, 12, 87. https://doi.org/10.3390/ecsoc-26-13574
Yakovets P, Staravoitava V, Faletrov Y, Shkumatov V. High-Throughput Virtual Screening of Compounds with Electrophilic Fragments for New Potential Covalent Inhibitors of Bacterial Proteins. Chemistry Proceedings. 2022; 12(1):87. https://doi.org/10.3390/ecsoc-26-13574
Chicago/Turabian StyleYakovets, Polina, Viktoryia Staravoitava, Yaroslav Faletrov, and Vladimir Shkumatov. 2022. "High-Throughput Virtual Screening of Compounds with Electrophilic Fragments for New Potential Covalent Inhibitors of Bacterial Proteins" Chemistry Proceedings 12, no. 1: 87. https://doi.org/10.3390/ecsoc-26-13574
APA StyleYakovets, P., Staravoitava, V., Faletrov, Y., & Shkumatov, V. (2022). High-Throughput Virtual Screening of Compounds with Electrophilic Fragments for New Potential Covalent Inhibitors of Bacterial Proteins. Chemistry Proceedings, 12(1), 87. https://doi.org/10.3390/ecsoc-26-13574