Abstract
Chromenopirazolopyrimidinone (CPP) was synthesized using microwave radiation in the absence of solvent by a multicomponent reaction of 4-hydroxy-2H-chromen-2-one with 5-amine-1H-pyrazole and benzaldehyde. CPP exists as a mixture of tautomeric forms, the structure of which was examined by means of spectroscopic methods and quantum chemistry. As was shown by the MILCA algorithm, UV-Vis spectra obtained in different solvents of various polarities contain bands of all tautomeric forms. Decomposition of the absorption spectra of CPP in various solvents on the individual components of the absorption showed the presence of two tautomers. The molar ratios of tautomeric forms depend on the polarity of the solvent used.
1. Introduction
The scientific community faces the challenge of creating complex organic compounds from simple substrates using environmental criteria, including the safety and efficiency of the synthetic procedures [1]. The multicomponent reaction, the Biginelli reaction, is a well-known method of the synthesis of azaheterocycles [2].
One option for the multicomponent synthesis conditions is the microwave exposure in the absence of solvent, with the addition of a Lewis acid catalyst [3]. Currently, microwave activation (Microwave Assisted Organic Synthesis, MAOS) is one of the most rapidly growing trends in organic chemistry. First of all, this is due to the multiple (101–103 times) shortening of the chemical reactions.
The combined experimental and theoretical study of the spectral characteristics of molecular systems is one of the most important modern tools for solving problems of current organic chemistry. Such an approach is relevant for a correct description of the composition and structure of the reaction mixtures of biologically active compounds, especially when it comes to mobile tautomeric equilibrium.
Thus, by combining the techniques of FTIR, NMR, UV-Vis spectroscopy, and by comparison of experimental data with the calculated values, chromenopirazolopyrimidinone (CPP), a product of the multicomponent reaction of 4-hydroxy-2H-chromen-2-one with 5-amine-1H-pyrazole and benzaldehyde, was characterized. CPP exists as a mixture of tautomeric forms, the structure of which was examined using an integrated approach of spectroscopic methods and quantum chemistry.
2. Materials and Methods
2.1. Synthesis of 7-Phenyl-7, 12-dihydro-6H-chromeno[4,3-d]pyrazolo[1,5-a]pyrimidin-6-one (CPP)
CPP was synthesized using microwave radiation in the absence of solvent by a multicomponent reaction of 4-hydroxy-2H-chromen-2-one with 5-amine-1H-pyrazole and benzaldehyde. Yield 1.5 g (78%), m.p. 125–127 °C; FTIR, cm−1 3251, 3101, 3099, 3032, 2956, 1660, 1604, 1569, 1518, 1473, 1451, 1409, 1359, 1340, 1230, 1195, 1114, 1076, 1049 1033, 1002, 980, 961, 950, 903, 854, 836, 808, 754, 725, 706, 697, 660, 618, 601, 589, 551, 529, 484, 470, 443, 407. Anal. calc. for C19H13N3O2, %: C, 72.37; H, 4.16; N, 13.33; found: C, 72.42; H, 4.17; N, 13.26.
2.2. NMR Spectra
NMR spectra were recorded on a Varian 400 spectrometer at 20–25 °C. The operating frequency for the 1H NMR spectra was 400 MHz, and for the 13C NMR spectra, it was 100 MHz. The internal standard was TMS, and the solvents were CDCl3 and dimethyl sulfoxide-d6.
2.3. LDI-MS
The laser desorption and ionization (LDI) mass spectrum was collected with an AB Sciex 5800 TOF/TOF (Foster City, CA, USA) in reflector mode with positive ion detection using a solid-state Nd: YAG laser (355 nm) and an accelerating voltage of 20 kV.
2.4. FTIR Spectra
The FTIR spectrum was recorded using a Nicolet 6700 spectrometer (Thermo Scientific, Waltham, MA, USA) with a KBr pellet (wavenumber range of 4000–400 cm−1), with a spectral resolution of 4 cm−1.
2.5. UV-Vis Spectra
The UV-Vis spectra of the CPP solutions were recorded on a Shimadzu-1800 spectrophotometer using 1 cm cuvettes; the scanning step was 1 nm. Solvents and reagents were of special grade. Working solutions were prepared by dissolving exactly weighed samples of the compounds (C = 5 × 10−5 M) in an appropriate solvent.
2.6. Decomposition of UV-Vis Spectra
The computer program of the MILCA algorithm and examples of solving practical tasks are available as independent executables with a MATLAB interface (The MathWorks, Natick, MA, USA).
2.7. Quantum Chemical Calculations
Quantum chemical calculations were performed using density functional theory (DFT) using the hybrid functional Lee–Yang–Parr three-parameter Becke B3LYP, split-valence basis set functions 6-311G, with the inclusion of p-orbitals of the hydrogen atom and d-orbitals for more heavy atoms, as well as the addition of polarization functions (B3LYP/6-311G++ (d,p)).
3. Results and Discussion
3.1. Synthesis of CPP
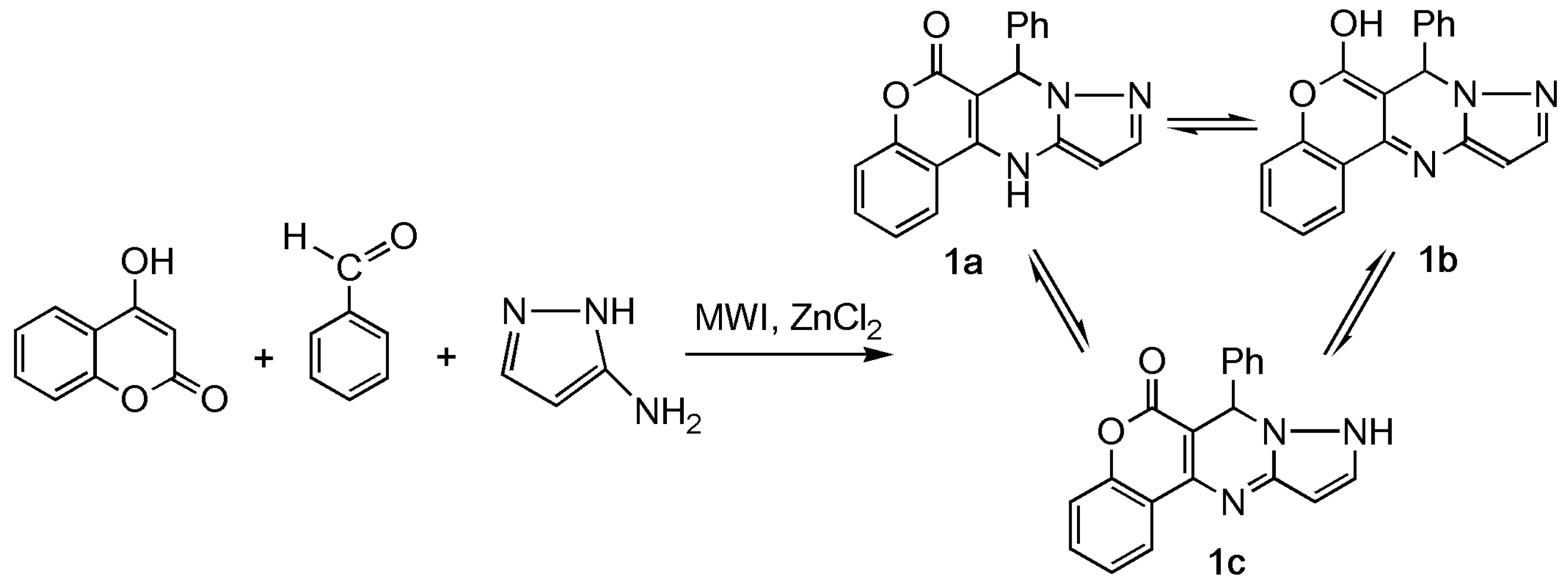
Using microwave radiation and the absence of solvent for a similar reaction results in a product, CPP, that can exist in three forms—as 7-phenyl-7,12-dihydro-6H-chromeno[4,3-d]pyrazolo[1,5-a]pyrimidin-6-one (1a), as 7-phenyl-7H-chromeno[4,3-d]pyrazolo[1,5-a]pyrimidin-6-ol (1b), and as 7-phenyl-7,9-dihydro-6H-chromeno[4,3-d]pyrazolo[1,5-a]pyrimidin-6-one (1c). (Scheme 1).
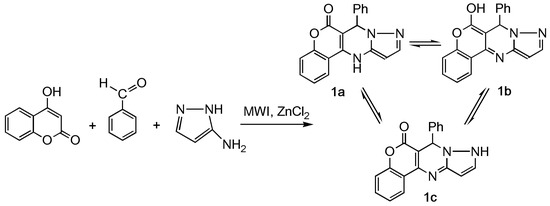
Scheme 1.
Synthesis of 7-phenyl-7, 12-dihydro-6H-chromeno[4,3-d]pyrazolo[1,5-a]pyrimidin-6-one (CPP) 1a–c.
Theoretically, tautomeric system 1 can be represented by three forms: a, b, c. Forms a and b are realized through keto-enamine-enol-imine tautomerism, and the c form is the result of pyrazole moiety tautomerism.
3.2. NMR Spectroscopic Data
The 1H NMR spectrum indicates the presence of CPP in two tautomeric forms: a and b due to the implementation of keto-enamine-enol-imine tautomerism.
In the 1H NMR spectrum of the mixture of tautomers a and b, the singlets NH and OH groups are at 10.58 ppm and 12.31 ppm, respectively. The tertiary proton H7′ singlet appears in 4.97 ppm, and the proton H7 appears at 5.23 ppm The aromatic portion includes 22H and is presented in a doublet 8.31 ppm (H1,1′, J = 8 Hz); 7.60 (t., N3,3′, J = 8 Hz); 7.42–7.32 (m., N2,2′, N4,4′); 7.28–7.25 (m. N10,10′ N11,11′); 7.24–7.06 (m., 10 Hz).
To confirm the presence in the 1H NMR spectrum signals of tautomeric forms 1a, we used the nuclear Overhauser effect (NOE 1D), which discovers a spatial interaction ortho-proton H1 chromenone fragment and NH group proton of the pyrimidine moiety.
The HSQC spectrum definitively confirmed the existing of two tautomeric forms 1a and b in the DMSO-d6 solution. It was evidenced by the presence of C7/C7 H7 and ′/H7′ cross peaks.
3.3. LDI-MS Data
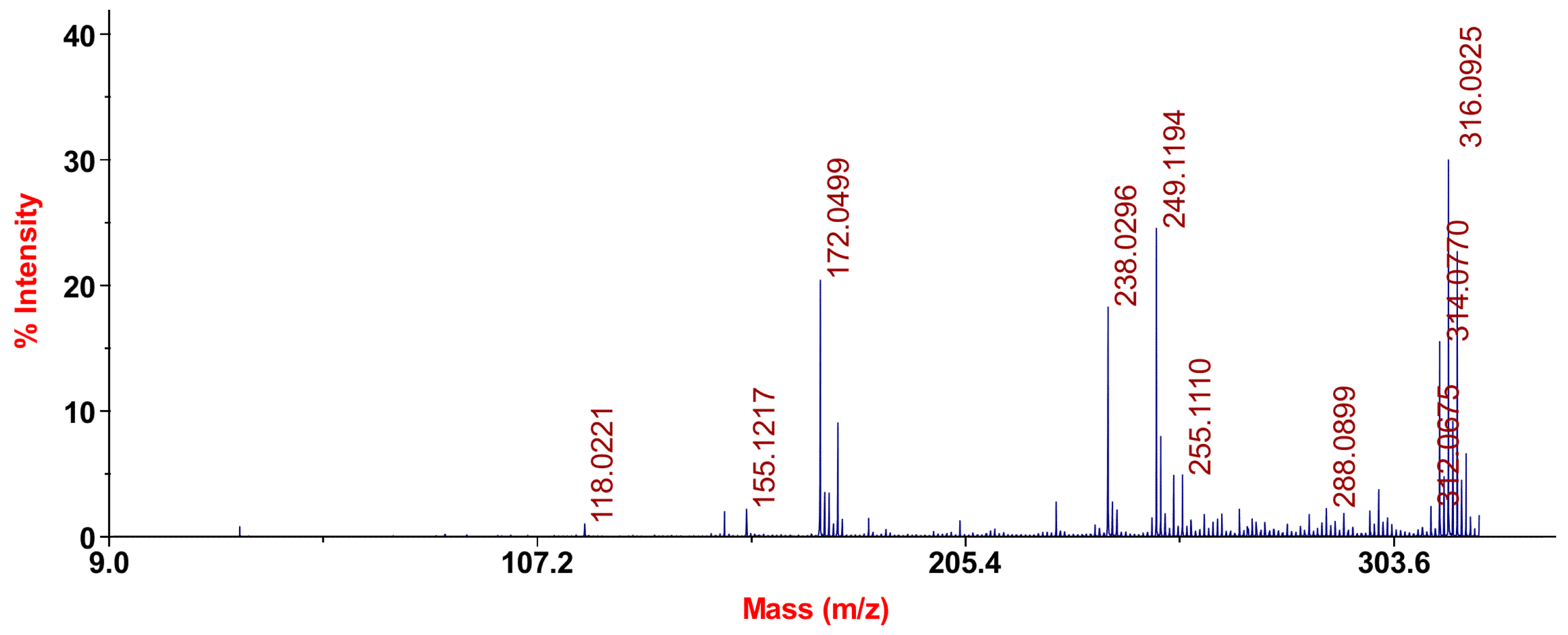
The considered tautomeric mixture of 1a-c was recorded as a mass spectrum by laser desorption and ionization (LDI). The LDI method is different from the “classical” methods of ionization, such as electron impact or chemical ionization, with a higher probability of the occurrence of photochemical processes, resulting in the ion dimer molecule peregruppirovochnye and other more complex cluster ions. In general, LDI mass spectrometry is more complex; however, it can more accurately interpret the structure (Figure 1).
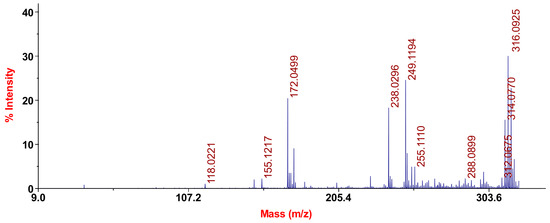
Figure 1.
Mass spectrum of chromenopirazolopyrimidinone (CPP) 1a–c.
For the three tautomeric forms 1a–c, we would expect similar mass-spectrometric patterns as an isomeric structure capable of producing isobaric ions.
The mass spectrum of 1a–c LDI chromenopirazolopyrimidinone has no molecular ion peak, but there are signals of protonated [M + H]+ and deprotonated [M − H]+ forms of molecules with mass-charge numbers m/z 314 and 316, respectively. The instability characteristic of the molecular-ion-fused aromatic systems was often observed in their mass spectra, including rows and heterocyclic compounds.
3.4. FTIR Spectra and Vibrational Analysis
The highest frequency vibrations 3400–3250 cm−1 were assigned to the NH stretching vibrations of forms 1 and 3, and for tautomeric form 2, the highest frequency band was the OH stretching vibration band.
In aromatic heterocycles, C-H ring vibrations are expected to occur in the region of 3000–3030 cm−1, and usually the bands are strong. In our case, these bands appear at 3129 cm−1 (HATR) and 3101 cm−1, while the corresponding scaled calculated frequencies for the third tautomer were at 3131 and 3104 cm−1.
The stretching vibration of the lactone carbonyl group C=O in various cyclic esters differing by the number of atoms in the cycle and the degree of saturation can be observed as a very strong band in the FTIR spectra at 1770–1720 cm−1.
3.5. UV-Vis Spectra and Their Decomposition
To confirm the assumptions about the form of existence synthesized, we recorded the UV spectra of a mixture of the tautomers 1a–c in solvents of different polarities.
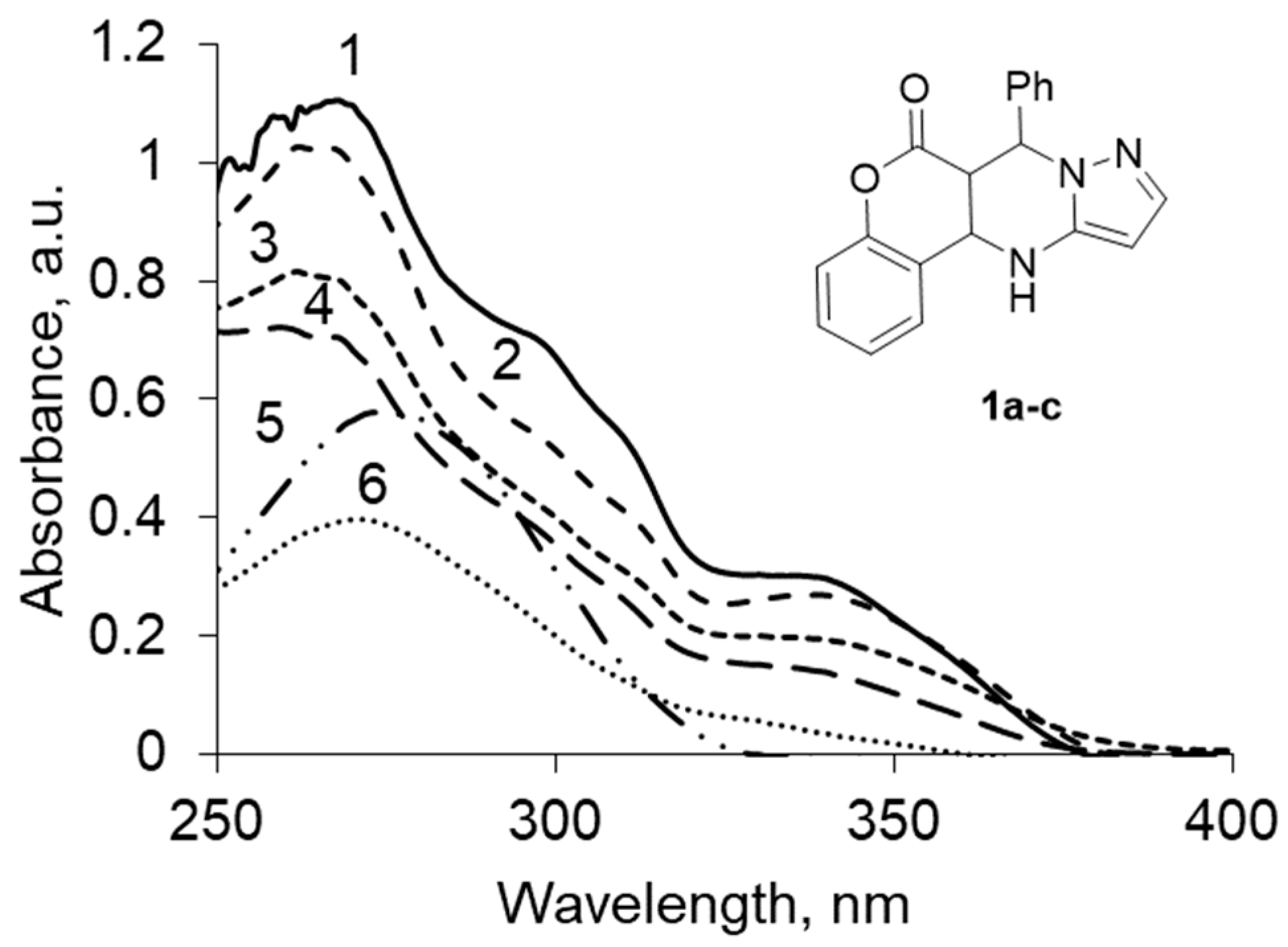
In the UV spectra of the mixture, chromenopirazolopyrimidinone 1a–c exhibits two intense bands with λmax 260–273 and 335–345 nm (Figure 2, Table 1).
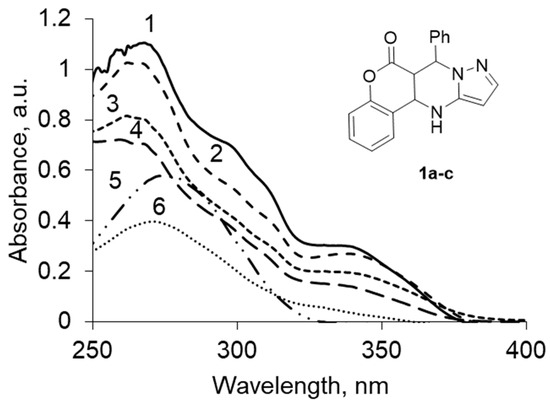
Figure 2.
UV spectrum chromenopirazolopyrimidinone (CPP) 1a–c at (concentration, 5 × 10−5 M) in: (1) dioxane, (2) ethanol, (3) methanol, (4) acetonitrile, (5) hexane, (6) chloroform.

Table 1.
Values of absorption maxima in UV spectra depending on the solvent of the compound 1a–c.
Bands with λmax 260–273 nm (k = 0.39–1.15) are forbidden by the symmetry n-π * transitions observed in the spectra of compounds in which the supporting structure of the unshared electron pair of the heteroatom connected multiple π-bonds with neighboring atoms. Recognize the strip n → π * transition is possible on hypsochromic shift when changing the nonpolar solvent to polar.
Bands with λmax 335–345 nm (k = 0.13–0.36) are characteristic mostly of the enol form. The bathochromic shift of the absorption bands in the transition from nonpolar to polar solvents can be attributed to the π-π * transitions’ enimine fragment.
In all the tested solvents, keto-enamine form 1a was prevalent, but the ratio trend varied in the tautomeric forms depending on the degree of polarity of the solvent.
3.6. Decomposition of UV-Vis Spectra
Using chemometrics methods, independent components, implemented in the MILCA algorithm, we have shown the possibility of quantitatively studying tautomeric equilibria with overlapping absorption bands of tautomers in an individual solvent. The essence of the self-similar separation of curves is the decomposition of the matrix of spectra of a multicomponent system X of size M × N (N is the number of counts by wavelength, M is the number of spectra of the mixture) into a matrix of spectra of individual components S of size K × N and a matrix of their relative concentrations A of size M × K (K is the number of components in the system) (Equation (1)):
X = A · S.
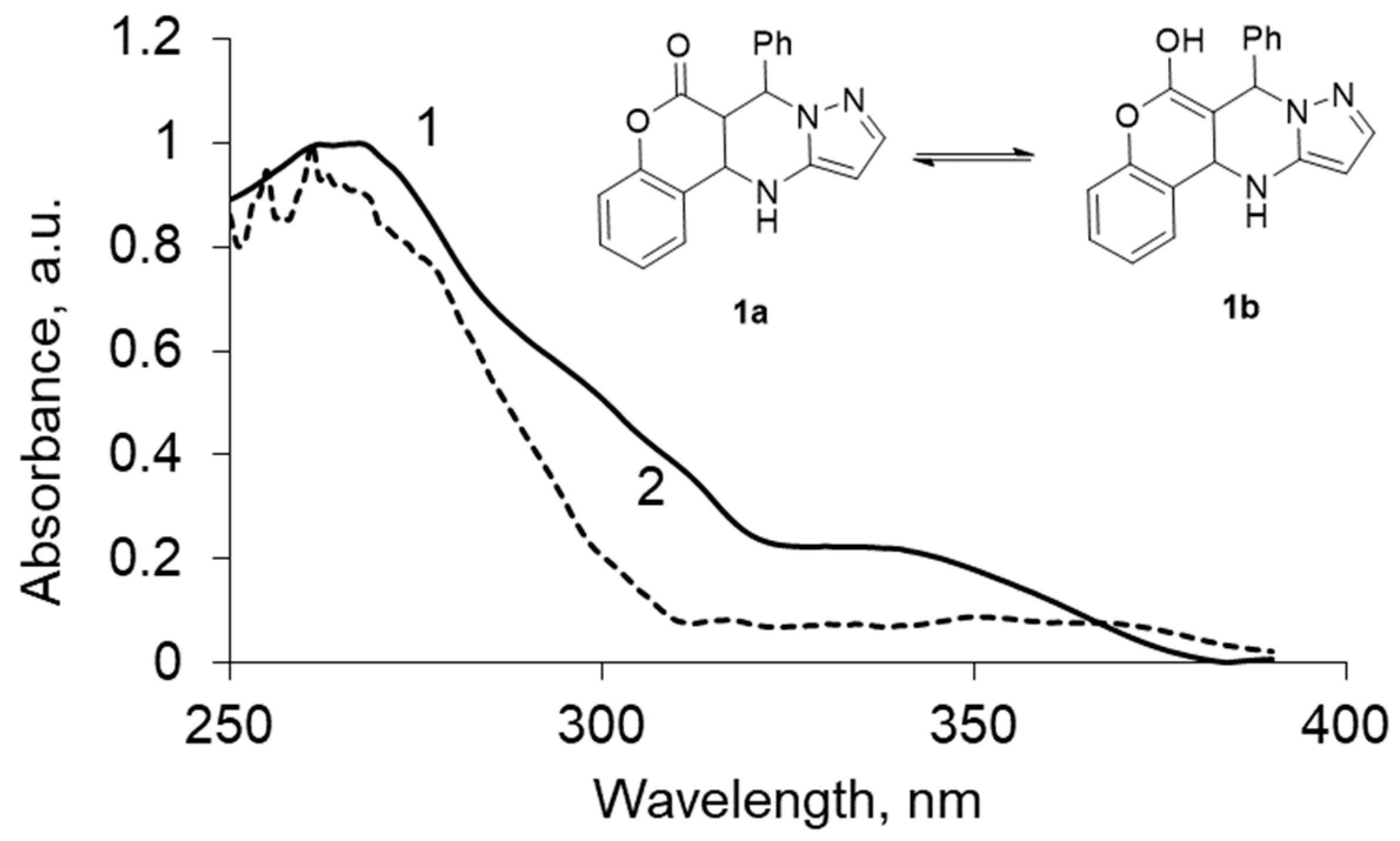
Decomposition of the absorption spectra of compound 1a–c in various solvents into the individual components of the absorption showed the presence of two tautomeric forms (Figure 3), which allowed calculation of the percentage of each of them in various solvents (Table 2).
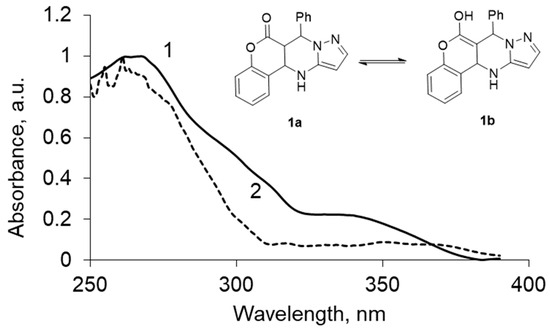
Figure 3.
UV spectrum of the two tautomeric forms obtained by treating the source of the UV spectrum program MILCA, for chromenopirazolopyrimidinone (CPP) 1: (1) keto-enamine form (1a), (2) enol-imine form (1b).
Table 2.
Ratio between keto-enamine and enol-imine forms of 1a–c depending on the solvent.
The analysis is carried out regardless of the number of components, the spectral characteristics of individual compounds, or the quantitative composition of the mixture. Thus, the work of the algorithm consists in isolating the individual spectral contours of the ketone and enol forms and subsequent calculation of the relative content of the enol form in solutions of solvents of different polarities.
3.7. Quantum Chemical Calculations
To compare the thermodynamic stability of all three putative tautomeric forms 1a–c in vacuum and in two solvents, polar methanol and low-polar hexane, we carried out quantum chemical calculations with full geometry optimization. Calculation data indicate that the most thermodynamic stable structure is 1a regardless of the medium. The 1b tautomer is 15–19 kcal/mol less stable; so, one can speculate that in solvent, this form will be significantly less represented in tautomeric equilibrium. These data are in harmony with the UV-Vis spectra and their deconvolution by the MILCA algorithm results. Considering solvents of different polarities, we can conclude that all three tautomers prefer the more polar methanol, where their energies tend to a minimum.
4. Conclusions
Thus, the existence of all three tautomeric forms is possible: in the solid state (mass and IR), form 1c predominates; in solution (NMR, UV), form 1a predominates.
Author Contributions
Conceptualization, V.S.G. and O.A.M.; methodology, O.A.M. and E.M.A.; software, V.S.G.; validation, O.A.M., E.M.A., V.S.G. and A.Y.Y.; formal analysis, E.M.A. and V.S.G.; investigation, O.A.M. and E.M.A.; resources, V.S.G. and A.Y.Y.; writing—original draft preparation, O.A.M. and V.S.G.; writing—review and editing, A.Y.Y.; supervision, A.Y.Y. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented in this study are available on request from the corresponding author.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Ruijter, E.; Scheffellaar, R.; Orru, R.V.A. Multicomponent Reaction Design in the Quest for Molecular Complexity and Diversity. Angew. Chem. Int. Ed. 2011, 50, 6234–6246. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.-H.; Xu, X.-Y.; Liu, H.; Cun, L.-F.; Gong, L.-Z. Highly Enantioselective Organocatalytic Biginelli Reaction. J. Am. Chem. Soc. 2006, 128, 14802–14803. [Google Scholar] [CrossRef] [PubMed]
- Shen, Z.-L.; Xu, X.-P.; Ji, S.-J. Brønsted Base-Catalyzed One-Pot Three-Component Biginelli-Type Reaction: An Efficient Synthesis of 4,5,6-Triaryl-3,4-dihydropyrimidin-2(1H)-one and Mechanistic Study. J. Org. Chem. 2010, 75, 1162–1167. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).