
Study of Acetylcholinesterase and Butyrylcholinesterase (AChE/BuChE) Inhibition Using Molecular Modelling Methods †

Abstract
:1. Introduction
2. Materials and Methods
3. Results and Discussion
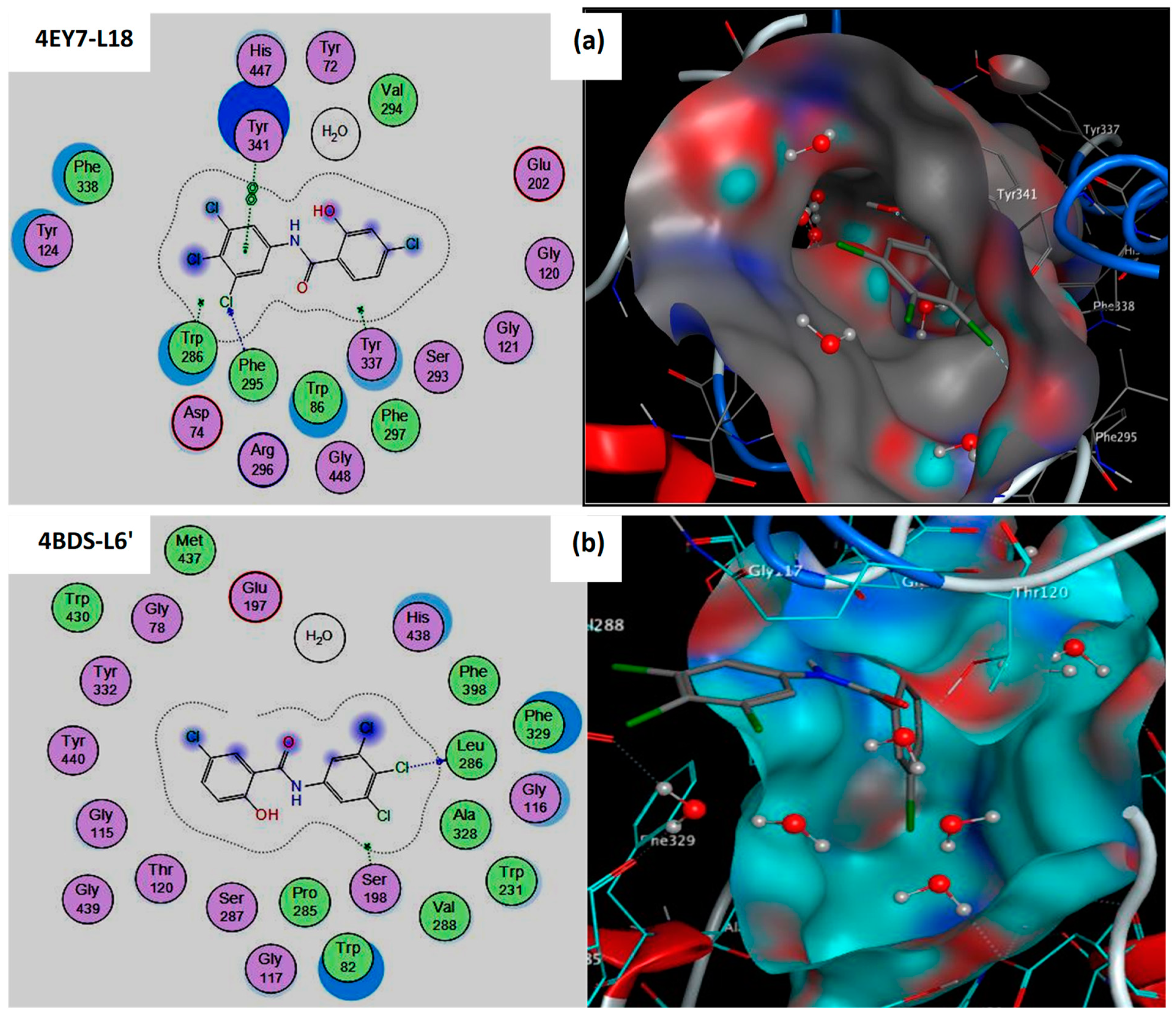
3.1. Interaction between Compounds and Targets (AChE/BuChE)
3.2. QSAR Modeling
3.3. Evaluation of ADME Properties
4. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Spaio, T.B.; Savall, A.S.; Gutierrez, M.E.; Pinton, S. Neurotrophic factors in Alzheimer’s and Parkinson’s diseases: Implications for pathogenesis and therapy. Neural Regen. Res. 2017, 12, 549. [Google Scholar]
- Mohamed, T.; Yeung, J.C.; Vasefi, M.S.; Beazely, M.A.; Rao, P.P. Development and evaluation of multifunctional agents for potential treatment of Alzheimer’s disease: Application to a pyrimidine-2,4-diamine template. Bioorganic Med. Chem. Lett. 2012, 22, 4707–4712. [Google Scholar] [CrossRef] [PubMed]
- Luo, W.; Li, Y.P.; He, Y.; Huang, S.L.; Tan, J.H.; Ou, T.M.; Li, D.; Gu, L.Q.; Huang, Z.S. Design, synthesis and evaluation of novel tacrine-multialkoxybenzene hybrids as dual inhibitors for cholinesterases and amyloid beta aggregation. Bioorganic Med. Chem. 2011, 19, 763–770. [Google Scholar] [CrossRef] [PubMed]
- Guevara-Salazar, J.A.; Espinoza-Fonseca, M.; Beltrán, H.I.; Correa-Basurto, J.; Quintana Zavala, D.; Trujillo-Ferrara, J.G. The electronic influence on the active site-directed inhibition of acetylcholinesterase by N-aryl-substituted succinimides. J. Mex. Chem. Soc. 2007, 51, 222–227. [Google Scholar]
- Kola, I.; Landis, J. Can the pharmaceutical industry reduce attrition rates? Nat. Rev. Drug Discov. 2004, 3, 711–716. [Google Scholar] [CrossRef] [PubMed]
- Molecular Operating Environment (MOE); Version 2014.09; Chemical Computing Group Inc.: Montreal, QC, Canada, 2014.
- Ibrahim, M.T.; Uzairu, A.; Shallangwa, G.A.; Ibrahim, A. In-silico studies of someoxadiazoles derivatives as anti-diabetic compounds. J. King Saud. Univ. Sci. 2020, 32, 423–432. [Google Scholar] [CrossRef]
- Brooijmans, N. Chapter: Docking methods, ligand design, and validating data sets in the structural genomics era. In Structural Bioinformatics; Gu, J., Bourne, P.E., Eds.; Wiley: New York, NY, USA, 2009; pp. 635–663. [Google Scholar]
- Imberty, A.; Hardman, K.D.; Carver, J.P.; Perez, S. Molecular modeling of protein-carbohydrate interactions. Docking of monosaccharides in the binding site of concanavalin A. Glycobiology 1991, 1, 631–642. [Google Scholar] [CrossRef] [PubMed]
- Daoud, I.; Melkemi, N.; Salah, T.; Ghalem, S. Combined QSAR, molecular docking and molecular dynamics study on new Acetylcholinesterase and Butyrylcholinesterase inhibitors. Comput. Biol. Chem. 2018, 74, 304–326. [Google Scholar] [CrossRef] [PubMed]
- Kherachi, R.; Daoud, I.; Melkemi, N.; Kenouche, S.; Mettai, M.; Mesli, F. Investigation of spirooxindole-pyrrolidine derivatives as acetylcholinesterase inhibitors using molecular docking/dynamics simulations, bioisosteric replacement, MEP, and ADME/Tox properties. In Biologia; Springer: Berlin/Heidelberg, Germany, 2023; pp. 1–21. [Google Scholar]
- Nikseresht, A.; Ghasemi, S.; Parak, S. [Cu3(BTC)2]: A metal–organic framework as an environment-friendly and economically catalyst for the synthesis of tacrine analogues by Friedlander reaction under conventional and ultrasound irradiation. Polyhedron 2018, 151, 112–117. [Google Scholar] [CrossRef]
- Baba-Ahmed, I.; Kibou, Z.; Daoud, I.; Belhadj, F.; Lahcen, B.; Daich, A.; Choukchou-Braham, N. Synthesis, molecular docking and ADME-TOX studies of new tacrine analogs as promising for alzheimer’s disease therapy. Curr. Org. Chem. 2022, 26, 1218–1233. [Google Scholar] [CrossRef]

{kind=link}
| Compds | IC50 Value (µM) | S-Score (kcal/mol) | RMSD (Å) | Bonds between Atoms of Compounds and Active Site Residues | ||||
|---|---|---|---|---|---|---|---|---|
| Atom of Compound | Involved Receptor Atoms | Involved Receptor Residues | Type of Interaction Bond | Distance (Å) | ||||
| AChE | ||||||||
| L6 | 51.06 ± 0.49 | −7.368 | 2.115 | Cl18 | N | PHE295 | H-acceptor | 3.37 |
| 6-ring | 6-ring | TYR341 | pi-pi | 3.98 | ||||
| L17 | 50.15 ± 0.26 | −7.461 | 0.726 | Cl20 | N | PHE295 | H-acceptor | 3.47 |
| 6-ring | 6-ring | TYR341 | pi-pi | 3.83 | ||||
| L18 | 57.78 ± 4.05 | −7.799 | 1.014 | Cl18 | N | PHE295 | H-acceptor | 3.40 |
| 6-ring | 6-ring | TYR341 | pi-pi | 3.83 | ||||
| Donepezil | 56.10 ± 1.41 | −11.247 | 0.408 | N-14 | O | HOH931 | H-donor | 2.79 |
| C-15 | 6-ring | TYR337 | H-Pi | 4.11 | ||||
| 6-ring | 6-ring | TRP286 | Pi-Pi | 3.73 | ||||
| BuChE | ||||||||
| L4′ | 186.47 ± 15.69 | −5.250 | 4545 | O16 | O | HOH2153 | H-donor | 3.04 |
| Cl22 | O | ALA328 | H-donor | 2.96 | ||||
| Cl27 | OE1 | GLU197 | H-donor | 3.23 | ||||
| 6-ring | O | HOH2055 | pi-H | 3.62 | ||||
| L6′ | 102.72 ± 0.97 | −6.603 | 0.979 | O16 | O | HOH2153 | H-donor | 2.97 |
| Cl22 | OG | SER198 | H-donor | 3.08 | ||||
| L30′ | 140.07 ± 6.20 | −5.590 | 1.930 | O16 | O | HOH2153 | H-donor | 2.93 |
| Br22 | OG | SER198 | H-donor | 3.11 | ||||
| Tacrine | 38.40 ± 1.97 | −6.193 | 0.316 | C1115 | 6-ring | TRP82 | Pi-H | 3.96 |
| 6-ring | 5-ring | TRP82 | pi-pi | 3.80 | ||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hasni, F.; Daoud, I.; Melkemi, N. Study of Acetylcholinesterase and Butyrylcholinesterase (AChE/BuChE) Inhibition Using Molecular Modelling Methods. Chem. Proc. 2023, 14, 77. https://doi.org/10.3390/ecsoc-27-16075
Hasni F, Daoud I, Melkemi N. Study of Acetylcholinesterase and Butyrylcholinesterase (AChE/BuChE) Inhibition Using Molecular Modelling Methods. Chemistry Proceedings. 2023; 14(1):77. https://doi.org/10.3390/ecsoc-27-16075
Chicago/Turabian StyleHasni, Ferdaous, Ismail Daoud, and Nadjib Melkemi. 2023. "Study of Acetylcholinesterase and Butyrylcholinesterase (AChE/BuChE) Inhibition Using Molecular Modelling Methods" Chemistry Proceedings 14, no. 1: 77. https://doi.org/10.3390/ecsoc-27-16075
APA StyleHasni, F., Daoud, I., & Melkemi, N. (2023). Study of Acetylcholinesterase and Butyrylcholinesterase (AChE/BuChE) Inhibition Using Molecular Modelling Methods. Chemistry Proceedings, 14(1), 77. https://doi.org/10.3390/ecsoc-27-16075