
Investigating SAR Insights into Royleanones for P-gp Modulation †

 ,
,  ,
,  , ,
, ,  ,
,  ,
,  , , ,
, , , 
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Methods
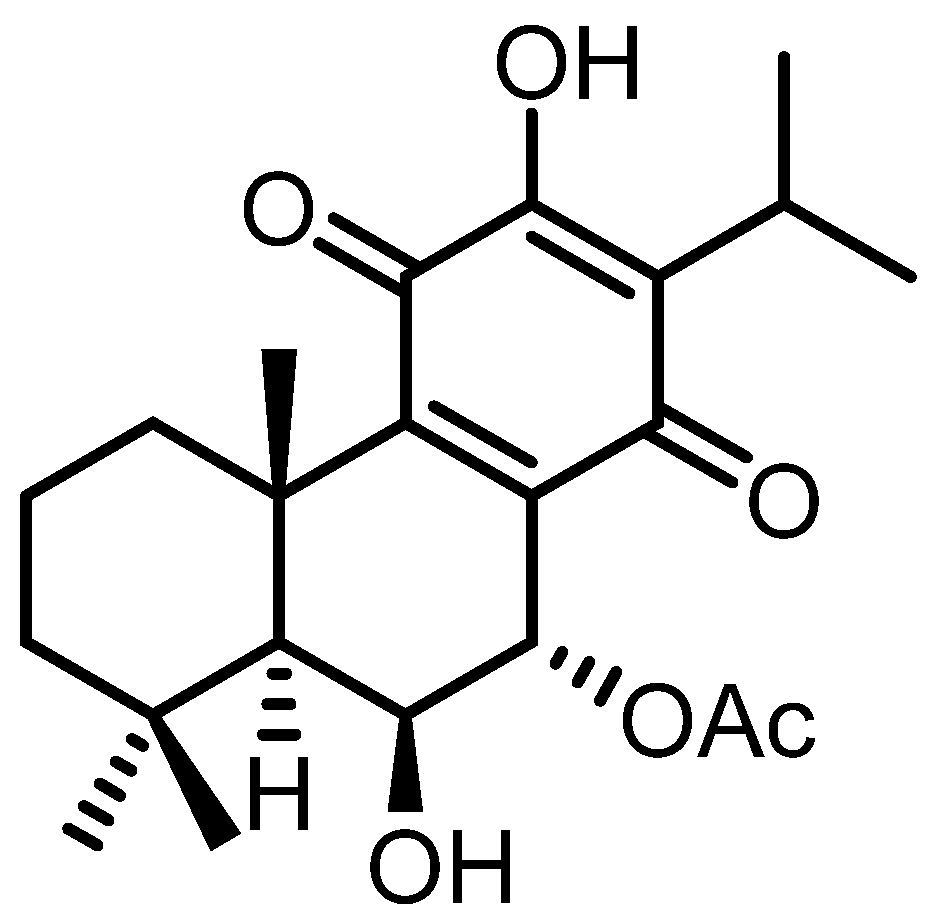
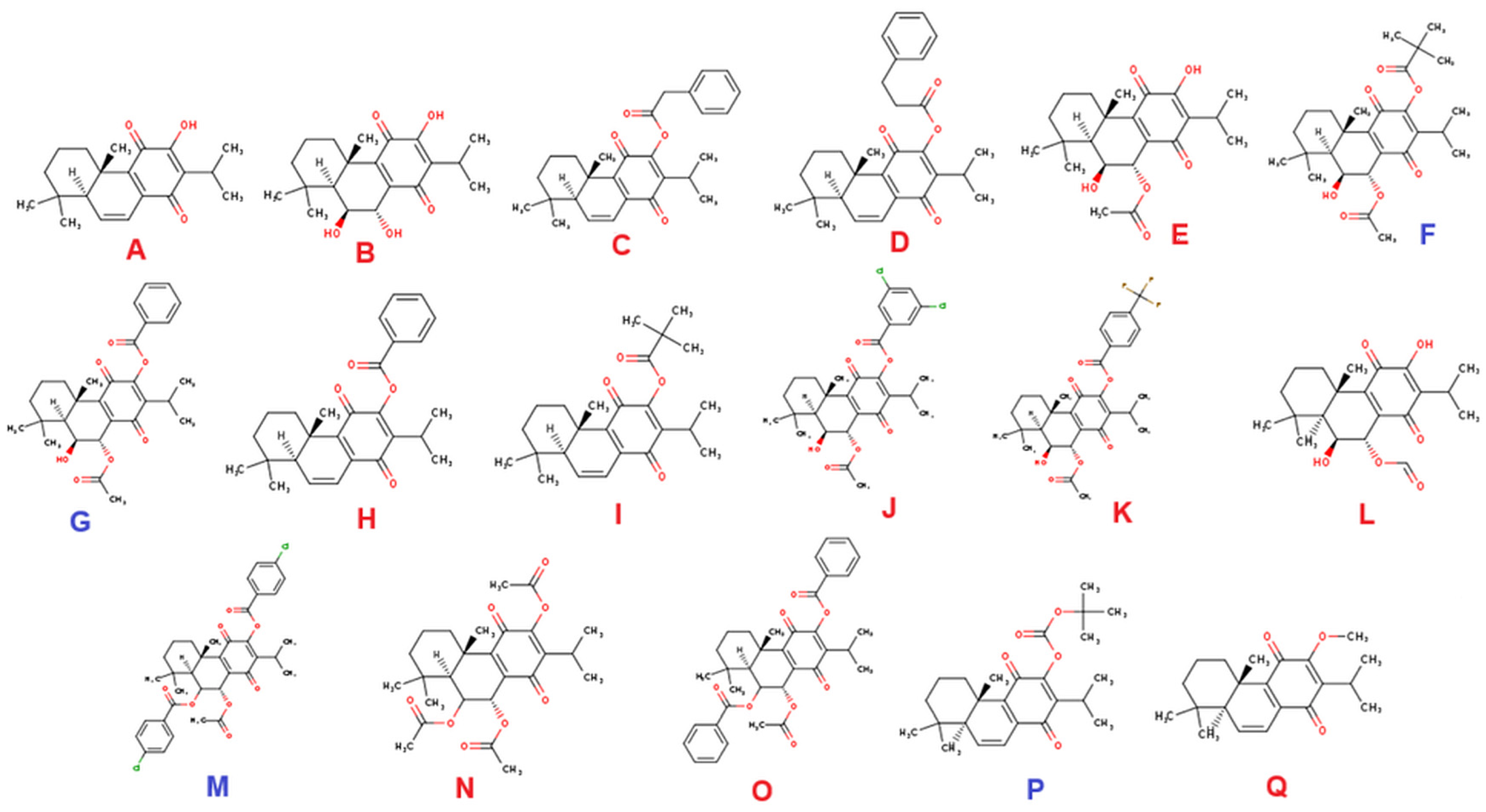
3. Results and Discussion
+ 0044 × (D7) + 0.0049 × (D8) + 0.0055 × (D9) + 0.0065 × (D10) − 0.2095 × (D11) + 0.0939 × (D12)
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Wang, X.; Zhang, H.; Chen, X. Drug resistance and combating drug resistance in cancer. Cancer Drug Resist. 2019, 2, 141–160. [Google Scholar] [CrossRef] [PubMed]
- Bin Emran, T.; Shahriar, A.; Mahmud, A.R.; Rahman, T.; Abir, M.H.; Siddiquee, M.F.-R.; Ahmed, H.; Rahman, N.; Nainu, F.; Wahyudin, E.; et al. Multidrug Resistance in Cancer: Understanding Molecular Mechanisms, Immunoprevention and Therapeutic Approaches. Front. Oncol. 2022, 12, 891652. [Google Scholar] [CrossRef] [PubMed]
- Illmer, T.; Schaich, M.; Platzbecker, U.; Freiberg-Richter, J.; Oelschlägel, U.; von Bonin, M.; Pursche, S.; Bergemann, T.; Ehninger, G.; Schleyer, E. P-glycoprotein-mediated drug efflux is a resistance mechanism of chronic myelogenous leukemia cells to treatment with imatinib mesylate. Leukemia 2004, 18, 401–408. [Google Scholar] [CrossRef]
- Pennock, G.D.; Dalton, W.S.; Roeske, W.R.; Appleton, C.P.; Mosley, K.; Plezia, P.; Miller, T.P.; Salmon, S.E. Systemic Toxic Effects Associated With High-Dose Verapamil Infusion and Chemotherapy Administration. JNCI J. Natl. Cancer Inst. 1991, 83, 105–110. [Google Scholar] [CrossRef] [PubMed]
- Pusztai, L.; Wagner, P.; Ibrahim, N.; Rivera, E.; Theriault, R.; Booser, D.; Symmans, F.W.; Wong, F.; Blumenschein, G.; Fleming, D.R.; et al. Phase II study of tariquidar, a selective P-glycoprotein inhibitor, in patients with chemotherapy-resistant, advanced breast carcinoma. Cancer 2005, 104, 682–691. [Google Scholar] [CrossRef] [PubMed]
- Garcia, C.; Isca, V.M.S.; Pereira, F.; Monteiro, C.M.; Ntungwe, E.; Sousa, F.; Dinic, J.; Holmstedt, S.; Roberto, A.; Díaz-Lanza, A.; et al. Royleanone Derivatives from Plectranthus spp. as a Novel Class of P-Glycoprotein Inhibitors. Front. Pharmacol. 2020, 11, 557789. [Google Scholar] [CrossRef]
- Isca, V.M.S.; Ferreira, R.J.; Garcia, C.; Monteiro, C.M.; Dinic, J.; Holmstedt, S.; André, V.; Pesic, M.; dos Santos, D.J.V.A.; Candeias, N.R.; et al. Molecular Docking Studies of Royleanone Diterpenoids from Plectranthus spp. as P-Glycoprotein Inhibitors. ACS Med. Chem. Lett. 2020, 11, 839–845. [Google Scholar] [CrossRef] [PubMed]
- Matias, D.; Nicolai, M.; Saraiva, L.; Pinheiro, R.; Faustino, C.; Lanza, A.D.; Reis, C.P.; Stankovic, T.; Dinic, J.; Pesic, M.; et al. Cytotoxic Activity of Royleanone Diterpenes from Plectranthus madagascariensis Benth. ACS Omega 2019, 4, 8094–8103. [Google Scholar] [CrossRef]
- Sitarek, P.; Toma, M.; Ntungwe, E.; Kowalczyk, T.; Skała, E.; Wieczfinska, J.; Śliwiński, T.; Rijo, P. Insight the Biological Activities of Selected Abietane Diterpenes Isolated from Plectranthus spp. Biomolecules 2020, 10, 194. [Google Scholar] [CrossRef] [PubMed]
- Śliwiński, T.; Sitarek, P.; Skała, E.; Isca, V.M.S.; Synowiec, E.; Kowalczyk, T.; Bijak, M.; Rijo, P. Diterpenoids from Plectranthus spp. as Potential Chemotherapeutic Agents via Apoptosis. Pharmaceuticals 2020, 13, 123. [Google Scholar] [CrossRef] [PubMed]
- Garcia, C.; Silva, C.O.; Monteiro, C.M.; Nicolai, M.; Viana, A.; Andrade, J.M.; Barasoain, I.; Stankovic, T.; Quintana, J.; Hernández, I.; et al. Anticancer Properties of the Abietane Diterpene 6,7-dehydroroyleanone Obtained by Optimized Extraction. Futur. Med. Chem. 2018, 10, 1177–1189. [Google Scholar] [CrossRef] [PubMed]
- Martins, J.P.A.; de Oliveira, M.A.R.; de Queiroz, M.S.O. Web-4D-QSAR: A web-based application to generate 4D-QSAR descriptors. J. Comput. Chem. 2018, 39, 917–924. [Google Scholar] [CrossRef] [PubMed]
- Fortin, F.A.; De Rainville, F.M.; Gardner, M.A.; Parizeau, M.; Gagńe, C. DEAP: Evolutionary algorithms made easy. J. Mach. Learn. Res. 2012, 13, 2171–2175. [Google Scholar]
- Ferreira, M.M.C. Multivariate QSAR. J. Braz. Chem. Soc. 2002, 13, 742–753. [Google Scholar] [CrossRef]
- Wold, S.; Sjöström, M.; Eriksson, L. PLS-regression: A basic tool of chemometrics. Chemom. Intell. Lab. Syst. 2001, 58, 109–130. [Google Scholar] [CrossRef]
- Martins JP, A.; Ferreira, M. Qsar Modeling: A New Open Source Computational Package To Generate and Validate Qsar Models. Quim Nova. 2013, 36, 554–560. [Google Scholar] [CrossRef]
- Ferreira, R.J.; Ferreira, M.-J.U.; dos Santos, D.J.V.A. Molecular Docking Characterizes Substrate-Binding Sites and Efflux Modulation Mechanisms within P-Glycoprotein. J. Chem. Inf. Model. 2013, 53, 1747–1760. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bangay, G.; Isca, V.M.S.; Brauning, F.Z.; Dinic, J.; Pesic, M.; Palma, B.B.; dos Santos, D.J.V.A.; Díaz-Lanza, A.M.; de Melo, E.B.; Martins, J.P.A.; et al. Investigating SAR Insights into Royleanones for P-gp Modulation. Chem. Proc. 2024, 16, 35. https://doi.org/10.3390/ecsoc-28-20158
Bangay G, Isca VMS, Brauning FZ, Dinic J, Pesic M, Palma BB, dos Santos DJVA, Díaz-Lanza AM, de Melo EB, Martins JPA, et al. Investigating SAR Insights into Royleanones for P-gp Modulation. Chemistry Proceedings. 2024; 16(1):35. https://doi.org/10.3390/ecsoc-28-20158
Chicago/Turabian StyleBangay, Gabrielle, Vera M. S. Isca, Florencia Z. Brauning, Jelena Dinic, Milica Pesic, Bernardo Brito Palma, Daniel J. V. A. dos Santos, Ana M. Díaz-Lanza, Eduardo Borges de Melo, João Paulo Ataide Martins, and et al. 2024. "Investigating SAR Insights into Royleanones for P-gp Modulation" Chemistry Proceedings 16, no. 1: 35. https://doi.org/10.3390/ecsoc-28-20158
APA StyleBangay, G., Isca, V. M. S., Brauning, F. Z., Dinic, J., Pesic, M., Palma, B. B., dos Santos, D. J. V. A., Díaz-Lanza, A. M., de Melo, E. B., Martins, J. P. A., & Rijo, P. (2024). Investigating SAR Insights into Royleanones for P-gp Modulation. Chemistry Proceedings, 16(1), 35. https://doi.org/10.3390/ecsoc-28-20158