
Comparative Investigation of (10%Co+0.5%Pd)/TiO2(Al2O3) Catalysts in CO Hydrogenation at Low and High Pressure †


 , , ,
, , , {kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
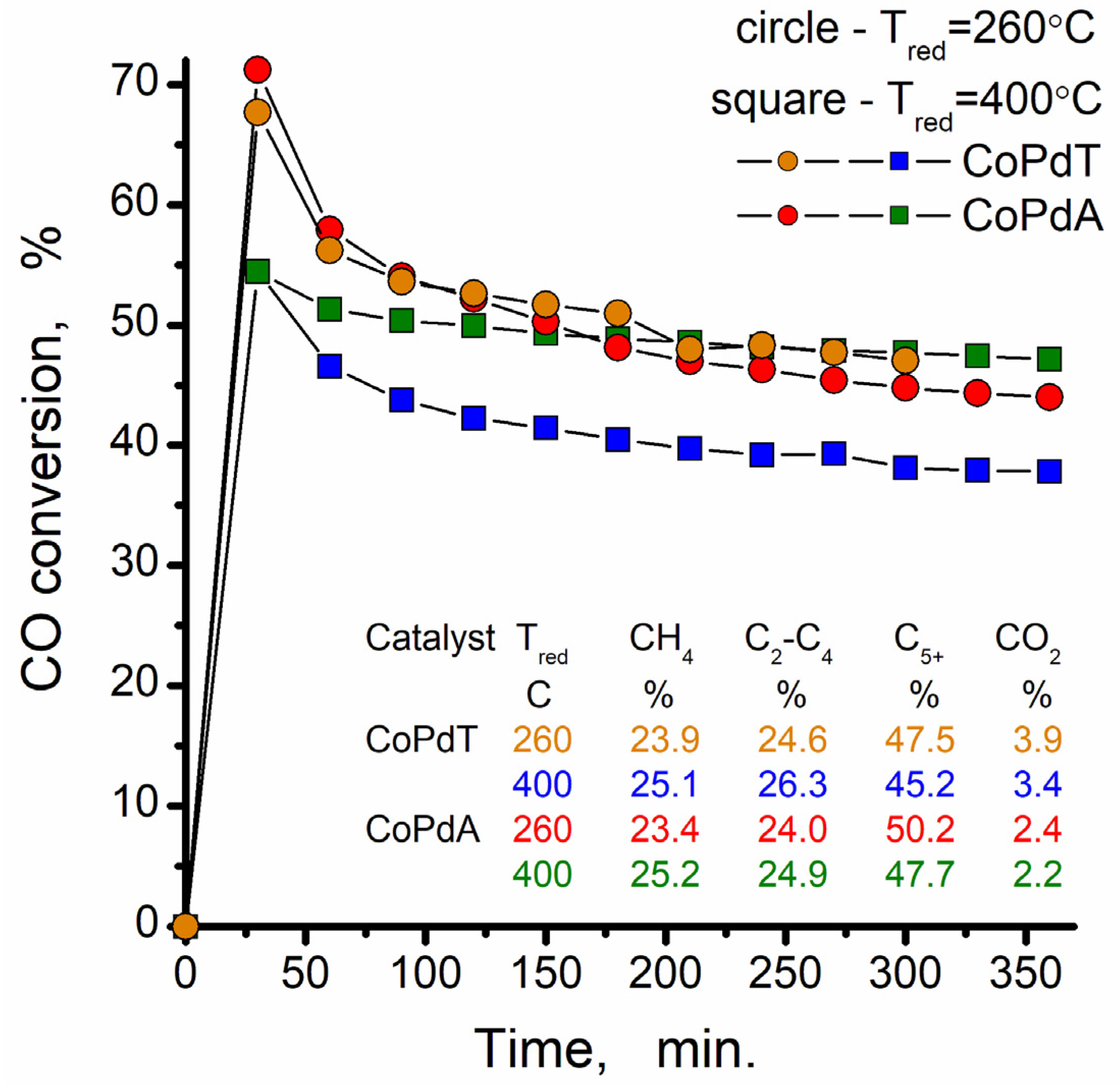
3. Results and Discussion
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhang, Q.; Kang, J.; Wang, Y. Development of novel catalysts for Fischer–Tropsch synthesis: Tuning the product selectivity. ChemCatChem 2010, 2, 1030–1058. [Google Scholar] [CrossRef]
- Riyahin, M.; Atashi, H.; Mohebbi-Kalhori, D. Optimization of reaction condition on the product selectivity of Fischer-Tropsch synthesis over a Co-SiO2/SiC catalyst using a fixed bed reaction. Petroleum Sci. Technol. 2017, 35, 1078–1084. [Google Scholar] [CrossRef]
- Schmidt-Rohr, K. Why combustions are always exothermic, yielding about 418 kJ per mole of O2. J. Chem. Educ. 2015, 92, 2094–2099. [Google Scholar] [CrossRef] [Green Version]
- Murdoch, A. Structural and Compositional Analysis of Co-Pd Model Catalyst Surfaces. Ph.D. Thesis, University of St. Andrews, St. Andrews, UK, 2012. [Google Scholar]
- Davis, B.H.; Iglesia, E. Technology Development for Iron and Cobalt Fischer-Tropsch Catalysts, Final Technical Report DE-FC26-98FT40308; University of California: Berkeley, CA, USA; University of Kentucky Research Foundation: Lexington, KY, USA, 2002. [Google Scholar]
- Arsalanfar, M.; Mirzaei, A.A.; Bozorgzadeh, H.R.; Samimi, A. A review of Fischer–Tropsch synthesis on the cobalt based catalysts. Phys. Chem. Res. 2014, 2, 179–201. [Google Scholar] [CrossRef]
- Rabo, J.A.; Risch, A.P.; Poutsma, M.L. Reactions of carbon monoxide and hydrogen on Co, Ni, Ru, and Pd metals. J. Catal. 1978, 53, 295–311. [Google Scholar] [CrossRef]
- Dinse, A.; Aigner, M.; Ulbrich, M.; Johnson, G.R.; Bell, A.T. Effects of Mn promotion on the activity and selectivity of Co/SiO2 for Fischer-Tropsch synthesis. J. Catal. 2012, 288, 104–114. [Google Scholar] [CrossRef]
- Sari, A.; Zamani, Y.; Taheri, S.A. Intrinsic kinetics of Fischer-Tropsch reactions over an industrial Co-Ru/γ-Al2O3 catalyst in slurry phase reactor. Fuel Proc. Technol. 2009, 90, 1305–1313. [Google Scholar] [CrossRef]
- Gibson, E.J.; Hall, C.C. The Fischer-Tropsch synthesis with cobalt catalysts: The effect of process conditions on the composition of the reaction products. J. Appl. Chem. 1954, 4, 49–61. [Google Scholar] [CrossRef]
- Zhou, W.; Chen, J.-G.; Fang, K.-G.; Sun, Y.-H. The deactivation of Co/SiO2 catalyst for Fischer-Tropsch synthesis at different ratios of H2 to CO. Fuel Proc. Thechnol. 2006, 87, 609–616. [Google Scholar] [CrossRef]
- Mirzaei, A.A.; Shirzadi, B.; Atashi, H.; Mansouri, M. Modeling and operating conditions optimization of Fischer-Tropsch synthesis in a fixed-bed reactor. J. Ind. Eng. Chem. 2012, 18, 1515–1521. [Google Scholar] [CrossRef]
- Kwack, S.-H.; Park, M.-J.; Bae, J.W.; Ha, K.-S.; Jun, K.-W. Development of a kinetic model of the Fischer-Tropsch synthesis reaction with a cobalt-based catalyst. React. Kinet. Mech. Catal. 2011, 104, 483–502. [Google Scholar] [CrossRef]
- Jalama, K. Chapter 39—Effect of operating pressure on Fischer-Tropsch synthesis kinetics over titania-supported cobalt catalyst. In Proceedings of the Transactions on Engineering Technologies, World Congress on Engineering and Computer Science, San Francisco, CA, USA, 21–23 October 2015; Ao, S.-I., Kim, H.K., Amouzegar, M.A., Eds.; Springer: Singapore, 2017; pp. 555–562. [Google Scholar] [CrossRef]
- de la Pena O’Shea, V.A.; Alvarez-Galavan, M.C.; Campos-Martin, J.M.; Fierro, J.L.G. Strong dependence on pressure of the performance of a Co/SiO2 catalyst in Fischer-Tropsch slurry reactor synthesis. Catal. Lett. 2005, 100, 105–110. [Google Scholar] [CrossRef]
- Geerlings, J.J.C.; Wilson, J.H.; Kramer, G.J.; Kuipers, H.P.C.E.; Hoek, A.; Huisman, H.M. Fischer-Tropsch technology—From active site to commercial process. Appl. Catal. A Gen. 1999, 186, 27–40. [Google Scholar] [CrossRef]
- Yates, I.C.; Sattarfield, C.N. Hydrocarbon selectivity from cobalt Fischer-Tropsch catalysts. Energy Fuels 1992, 6, 308–314. [Google Scholar] [CrossRef]
- Yan, Z.; Wang, Z.; Bukur, D.B.; Goodman, D.W. Fischer-Tropsch synthesis on a model Co/SiO2 catalyst. J. Catal. 2009, 268, 196–200. [Google Scholar] [CrossRef]
- Aben, P.C. Palladium areas in supported catalysts: Determination of palladium surface areas in supported catalysts by means of hydrogen chemisorption. J. Catal. 1968, 10, 224. [Google Scholar] [CrossRef]
- Reuel, R.C.; Bartholomew, C.H. The stoichiometries of H2 and CO adsorptions on cobalt: Effects of support and preparation. J. Catal. 1984, 85, 63. [Google Scholar] [CrossRef]
- Zowtiak, J.M.; Bartholomew, C.H. The kinetics of H2 adsorption on and desorption from cobalt and the effects of support thereon. J. Catal. 1983, 83, 107. [Google Scholar] [CrossRef]
- Anderson, J.R. Structure of Metallic Catalysts; Mir: Moscow, Russia, 1978. (In Russian) [Google Scholar]
- Shirley, D.A. High-resolution X-ray photoemission spectrum of the valence bands of gold. Phys. Rev. B 1972, 5, 4709–4714. [Google Scholar] [CrossRef] [Green Version]
- Scofield, J.H. Hertree-Slater subshell photoionization cross-sections at 1254 and 1487 eV. J. Electron Spectrosc. Relat. Phenom. 1976, 8, 129–137. [Google Scholar] [CrossRef]
- Zheng, S.; Liu, Y.; Li, J.; Shi, B. Deuterium tracer study of pressure effect on product distribution in the cobalt-catalyzed Fischer-Tropsch synthesis. Appl. Catal. A Gen. 2007, 330, 63–68. [Google Scholar] [CrossRef]
- Guskos, N.; Typek, J.; Maryniak, M.; Żolnierkiewicz, G.; Podsiadly, M.; Arabczyk, W.; Lendzion-Bielun, Z.; Narkiewicz, U. Effect of calcination and structural additives on the EPR spectra of nanocrystalline cobalt oxides. Mater. Sci.-Pol. 2006, 24, 4. [Google Scholar]
- Popova, N.M.; Babenkova, L.V.; Savel’eva, G.A. Adsorption and Interaction of the Simplest Gases with Metals from VIII Group; Nauka: Alma-Ata, Kazakhstan, 1979. (In Russian) [Google Scholar]
- Potoczna-Petru, D.; Jablonski, J.M.; Okal, J.; Krajczyk, L. Influence of oxidation-reduction treatment on the microstructure of Co/SiO2 catalyst. Appl. Catal. A Gen. 1998, 175, 113–120. [Google Scholar] [CrossRef]
- Singh, B.; Patial, J.; Sharma, P.; Chandra, S.; Maity, S.; Lingaiah, N. A comparative study on basicity based on supported K-salt catalysts for isomerization of 1-methoxy-4-(2-propene-1-yl) benzene. Indian J. Chem. Technol. 2010, 17, 446–450. [Google Scholar]
- Pan, X.; Yang, M.-Q.; Fu, X.; Zhang, N.; Xu, Y.-J. Defective TiO2 with oxygen vacancies: Synthesis, properties and photocatalytic applications. Nanoscale 2013, 5, 3601–3614. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Chen, J.; Ren, J.; Sun, Y. Chemical treatment of γ-Al2O3 and its influence on the properties of Co-based catalysts for Fischer–Tropsch synthesis. Appl. Catal. A 2003, 243, 121. [Google Scholar] [CrossRef]
- Tsubaki, N.; Sun, S.; Fujimoto, K. Different functions of the noble metals added to cobalt catalysts for Fischer–Tropsch synthesis. J. Catal. 2001, 199, 236. [Google Scholar] [CrossRef]
- Kadinov, G.; Bonev, C.; Todorova, S.; Palazov, A. IR spectroscopy study of CO adsorption and of the interaction between CO and hydrogen on alumina supported cobalt. J. Chem. Soc. Faraday Trans. 1998, 94, 3027–3031. [Google Scholar] [CrossRef]
- Singh, J.A.; Yang, N.; Liu, X.; Tsai, C.; Stone, K.H.; Johnson, B.; Koh, A.L.; Bent, S.F. Understanding the active sites of CO hydrogenation on Pt-Co catalysts prepared using atomic layer deposition. J. Phys. Chem. C 2018, 122, 2184–2194. [Google Scholar] [CrossRef]
- Xiong, H.; Zhang, Y.; Liew, K.; Li, J. Ruthenium promotion of Co/SBA-15 catalysts with high cobalt loading for Fischer-Tropsch synthesis. Fuel Proc. Technol. 2009, 90, 237–246. [Google Scholar] [CrossRef]
- Tuxen, A.; Carenco, S.; Chintapalli, M.; Chuang, C.-H.; Escudero, C.; Pach, E.; Jiang, P.; Borondics, F.; Beberwyck, B.; Alivisatos, A.P.; et al. Size-dependent dissociation of carbon monoxide on cobalt nanoparticles. J. Am. Chem. Soc. 2013, 135, 2273–2278. [Google Scholar] [CrossRef]
- Li, J.; Xu, L.; Keogh, R.A.; Davis, B.H. Effect of boron and ruthenium on the catalytic properties of Co/TiO2 Fischer-Tropsch catalysts. Prepr.-Am. Chem. Society. Div. Pet. Chem. 2000, 45, 253. [Google Scholar]
- Sun, X.; Sartipi, S.; Kapteijn, F.; Gascon, J. Effect of pretreatment atmosphere on the activity and selectivity of Co/mesoHZSM-5 for Fischer–Tropsch synthesis. New J. Chem. 2016, 40, 4167–4177. [Google Scholar] [CrossRef]
- Chen, T.-Y.; Su, J.; Zhang, Z.; Cao, C.; Wang, X.; Si, R.; Liu, X.; Shi, B.; Xu, J.; Han, Y.-F. Structure evolution of Co-CoOx interface for higher alcohol synthesis from syngas over Co/CeO2 catalysts. ACS Catal. 2018, 8, 8606–8617. [Google Scholar] [CrossRef]
- Sanchez-Escribano, V.; Larrubio-Vargas, M.A.; Finocchio, E.; Busca, G. On the mechanisms and the selectivity determining steps in syngas conversion over supported metal catalysts: An IR study. Appl. Catal. A Gen. 2007, 316, 68–74. [Google Scholar] [CrossRef]
- Little, L.H. Infrared Spectra of Adsorbed Species; Academic Press Inc.: London, UK; New York, NY, USA, 1966. [Google Scholar]
- Geerlings, J.J.C.; Zonnevylle, M.C.; de Groot, C.P.M. Studies of the Fischer-Tropsch reaction on Co(0001). Surf. Sci. 1991, 241, 302–314. [Google Scholar] [CrossRef]
- Geerlings, J.J.C.; Zonnevylle, M.C.; de Groot, C.P.M. Structure sensitivity of the Fischer-Tropsch rection on cobalt single crystals. Surf. Sci. 1991, 241, 315–324. [Google Scholar] [CrossRef]
- Szanyi, J.; Kwak, J.H. Dissecting the steps of CO2 reduction: 1. The interaction of CO and CO2 with γ-Al2O3: An in situ FTIR study. Phys. Chem. Chem. Phys. 2014, 16, 15117–15125. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shopska, M.; Caballero, A.; Todorova, S.; Aleksieva, K.; Tenchev, K.; Kolev, H.; Fabian, M.; Kadinov, G. Comparative Investigation of (10%Co+0.5%Pd)/TiO2(Al2O3) Catalysts in CO Hydrogenation at Low and High Pressure. Chem. Proc. 2022, 6, 11. https://doi.org/10.3390/ECCS2021-11105
Shopska M, Caballero A, Todorova S, Aleksieva K, Tenchev K, Kolev H, Fabian M, Kadinov G. Comparative Investigation of (10%Co+0.5%Pd)/TiO2(Al2O3) Catalysts in CO Hydrogenation at Low and High Pressure. Chemistry Proceedings. 2022; 6(1):11. https://doi.org/10.3390/ECCS2021-11105
Chicago/Turabian StyleShopska, Maya, Alfonso Caballero, Silviya Todorova, Katerina Aleksieva, Krassimir Tenchev, Hristo Kolev, Martin Fabian, and Georgi Kadinov. 2022. "Comparative Investigation of (10%Co+0.5%Pd)/TiO2(Al2O3) Catalysts in CO Hydrogenation at Low and High Pressure" Chemistry Proceedings 6, no. 1: 11. https://doi.org/10.3390/ECCS2021-11105